
Pharmacologic ROMK Inhibition Protects Against Myocardial Ischemia Reperfusion Injury

 ,
,
Abstract
1. Introduction
2. Results
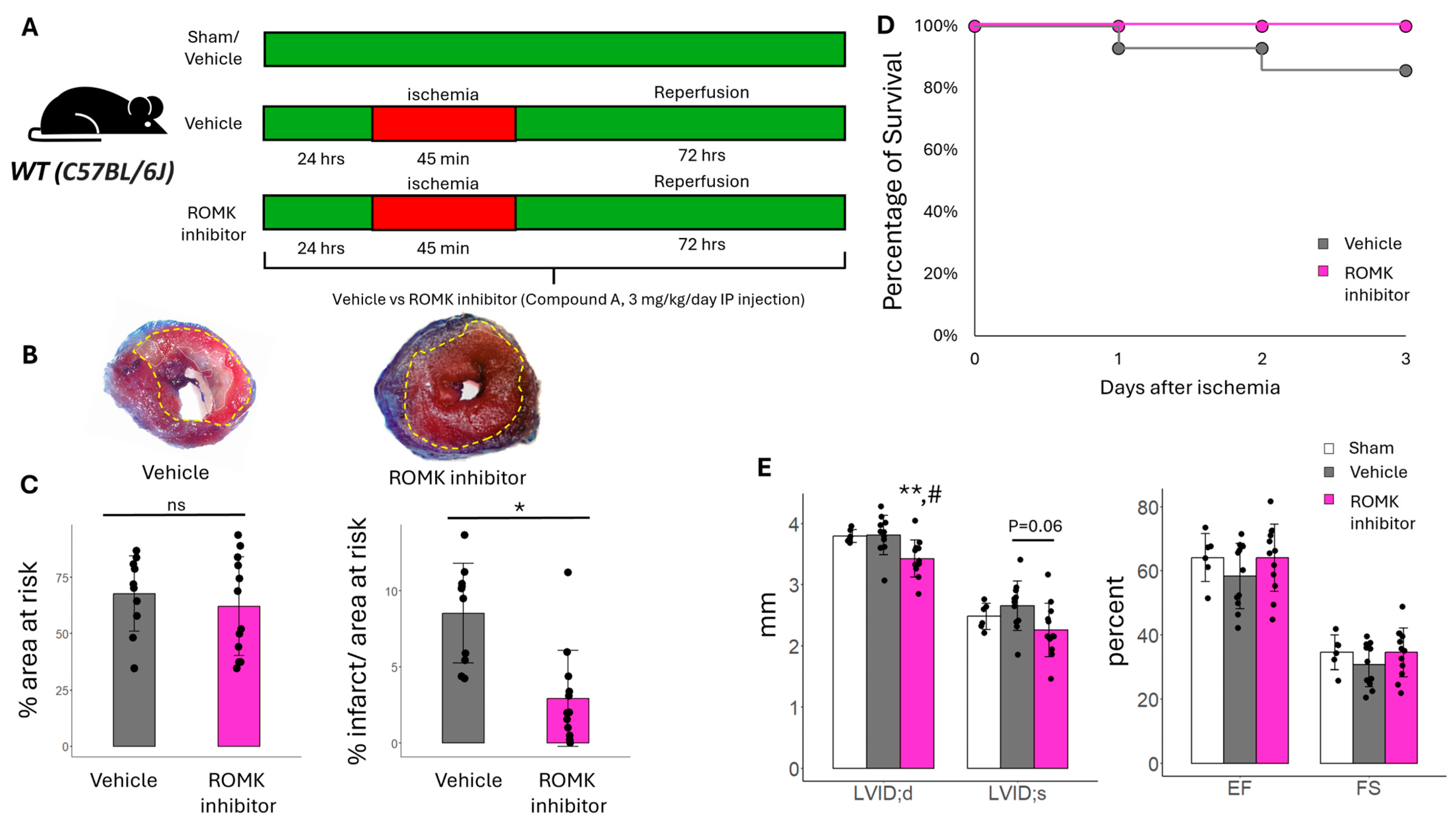
2.1. Pharmacologic ROMK Block Improves Infarct Size in Mice After In Vivo IR Injury
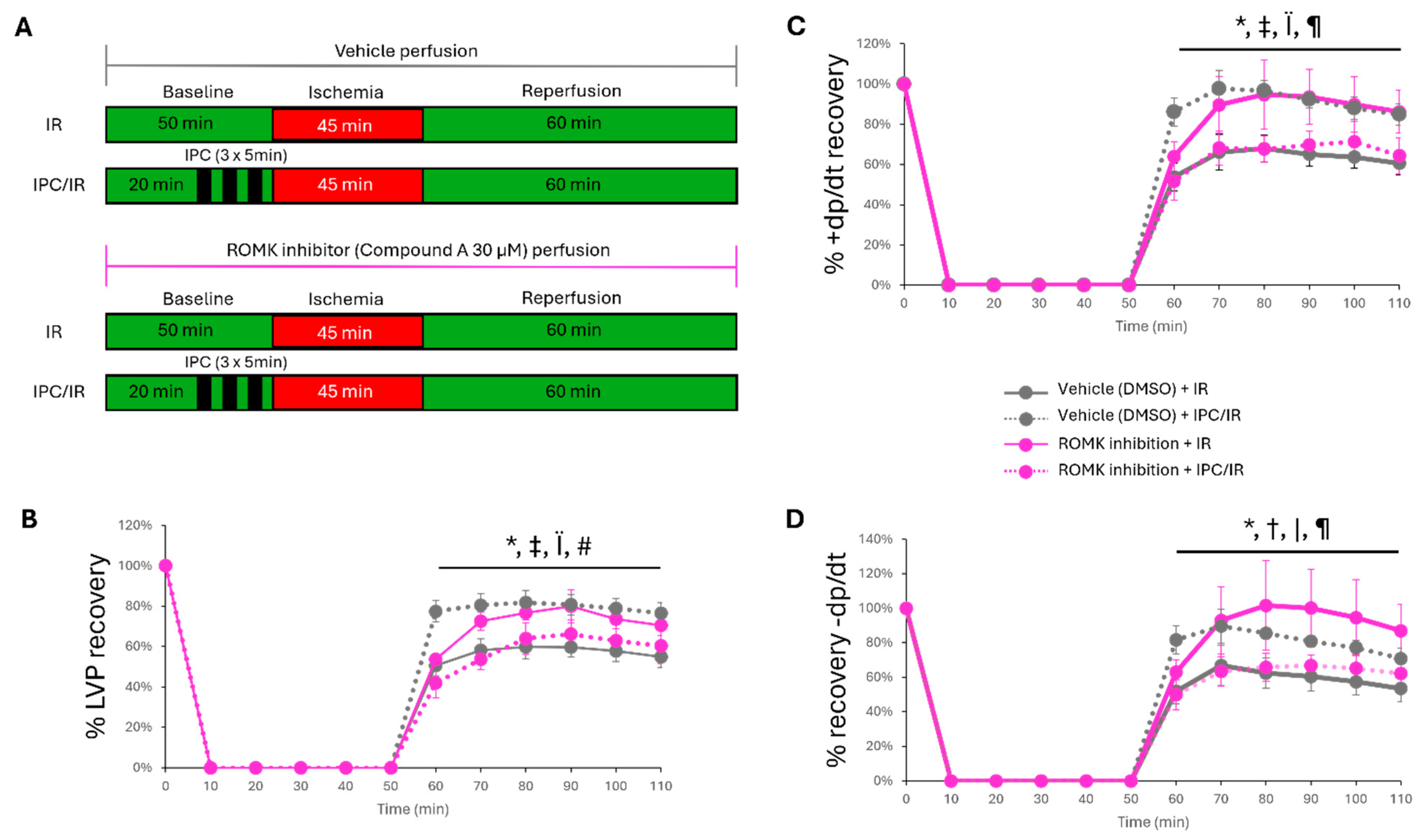
2.2. Pharmacologic ROMK2 Block Protects the Heart During Ex Vivo Myocardial IR Injury
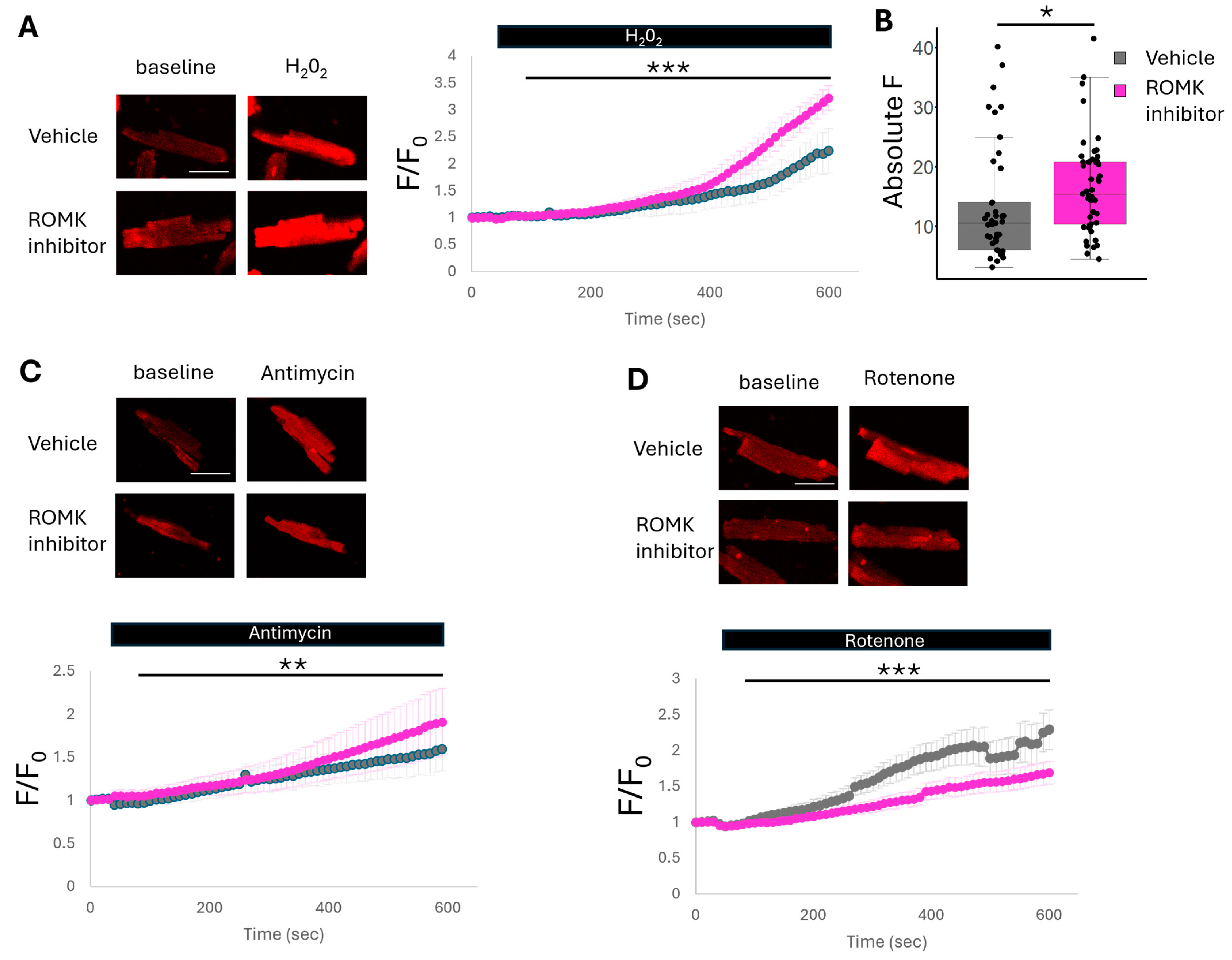
2.3. ROMK2 Inhibition Enhances Flavoprotein Oxidation in Cardiomyocytes
2.4. ROMK2 Inhibition Increases Mitochondrial ROS Production at ETC Complex III and Reduces ROS Production at Complex I
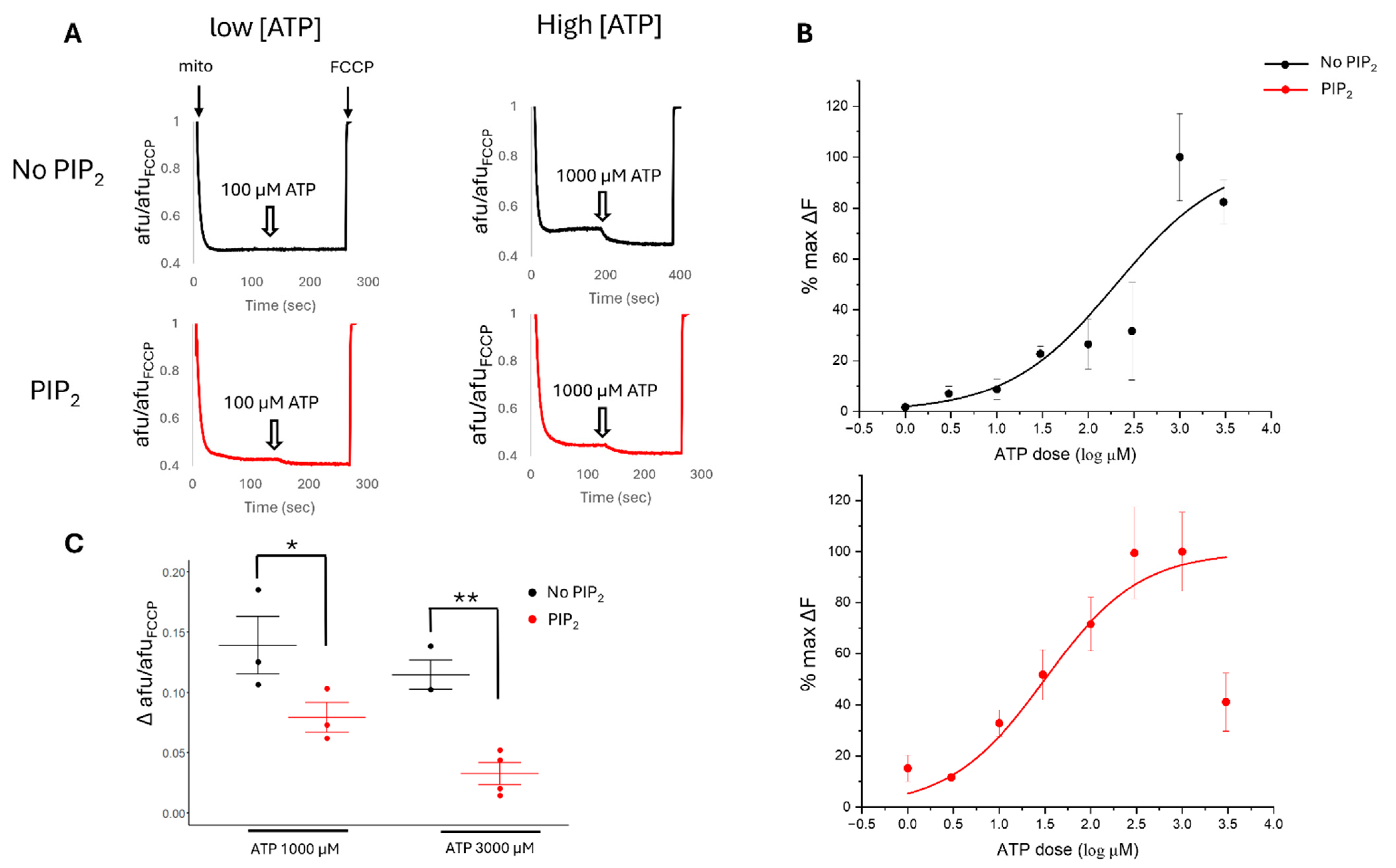
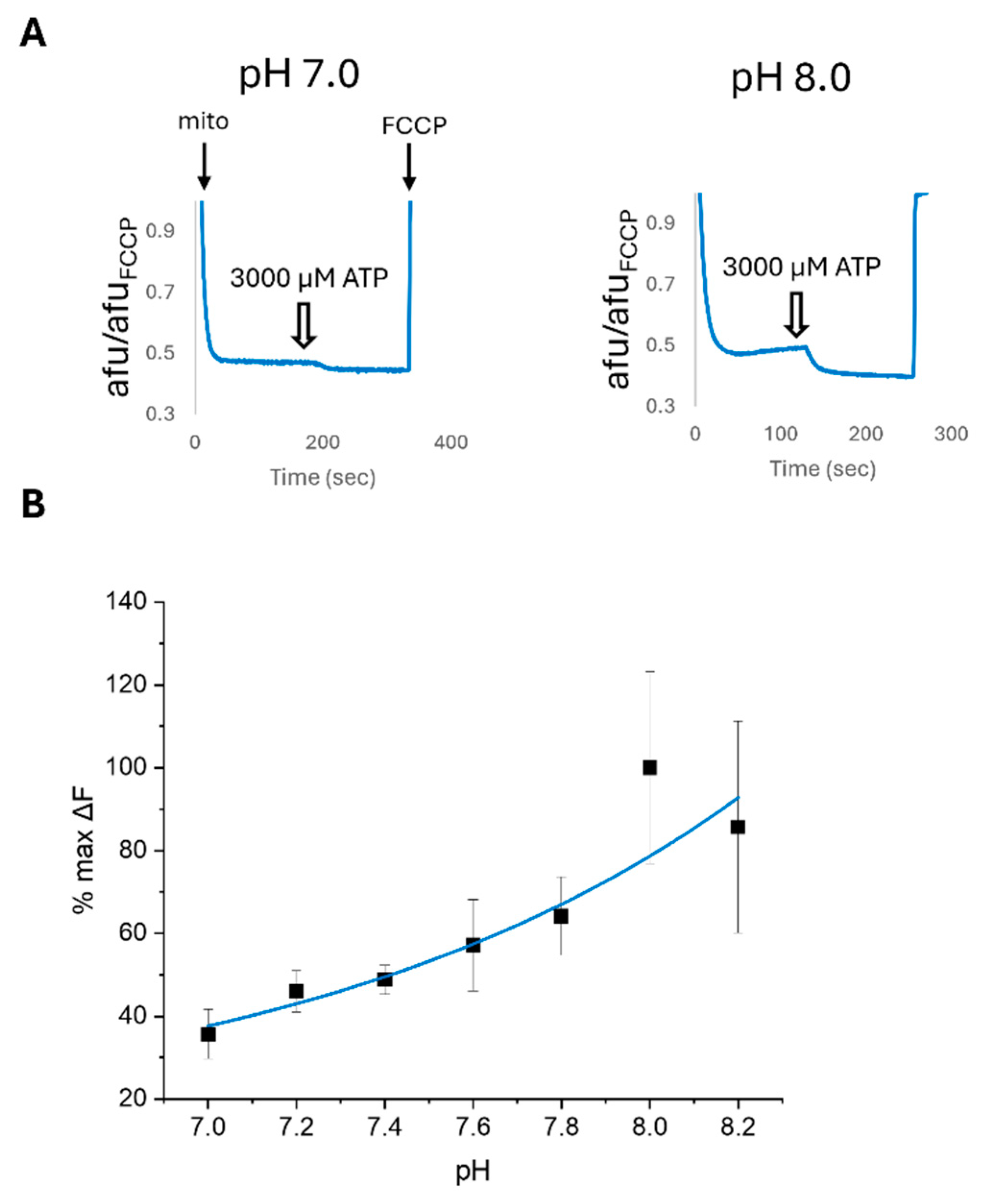
2.5. PIP2 and pH Affect the ATP Sensitivity of Cardiac Mitochondrial Membrane Potential
2.6. ATP Sensitivity of Cardiac Mitochondria Is Altered by ROMK2 Inhibition Only in the Setting of PIP2
2.7. Pharmacologic ROMK2 Blockade Alters Swelling in Isolated Cardiac Mitochondria
3. Discussion
4. Materials and Methods
4.1. Animal Usage
4.2. In Vivo Murine Myocardial IR Injury
4.3. Echocardiography
4.4. Isolated Murine Heart IR Injury Experiments
4.5. Isolation of Cardiomyocytes
4.6. FAD/FADH2 Autofluorescence Measurements in Isolated Cardiomyocytes
4.7. Measurement of Mitochondrial ROS Production in Isolated Cardiomyocytes
4.8. Isolation of Mitochondria
4.9. Mitochondrial Membrane Potential Assessment in Isolated Cardiac Mitochondria
4.10. Isolated Cardiac Mitochondria mitoKATP Swelling Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mitoKATP | Mitochondrial ATP-sensitive potassium channel |
| IR | Ischemia reperfusion |
| KATP | ATP-regulated potassium channel |
| ROMK | Renal outer medullary potassium channel |
| ROMK2 | Renal outer medullary K+ channel isoform 2 |
| Ψm | Mitochondrial membrane potential |
References
- Lascano, E.C.; Negroni, J.A.; del Valle, H.F. Ischemic shortening of action potential duration as a result of KATP channel opening attenuates myocardial stunning by reducing calcium influx. Mol. Cell. Biochem. 2002, 236, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Flagg, T.P.; Nichols, C.G. Cardiac sarcolemmal K(ATP) channels: Latest twists in a questing tale! J. Mol. Cell. Cardiol. 2010, 48, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D. Cation transport in mitochondria—The potassium cycle. Biochim. Biophys. Acta 1996, 1275, 123–126. [Google Scholar] [CrossRef]
- Facundo, H.T.; Fornazari, M.; Kowaltowski, A.J. Tissue protection mediated by mitochondrial K+ channels. Biochim. Biophys. Acta 2006, 1762, 202–212. [Google Scholar] [CrossRef]
- Costa, A.D.; Quinlan, C.L.; Andrukhiv, A.; West, I.C.; Jaburek, M.; Garlid, K.D. The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H406–H415. [Google Scholar] [CrossRef]
- Dos Santos, P.; Kowaltowski, A.J.; Laclau, M.N.; Seetharaman, S.; Paucek, P.; Boudina, S.; Thambo, J.B.; Tariosse, L.; Garlid, K.D. Mechanisms by which opening the mitochondrial ATP- sensitive K+ channel protects the ischemic heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H284–H295. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Seetharaman, S.; Paucek, P.; Garlid, K.D. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H649–H657. [Google Scholar] [CrossRef]
- Munch-Ellingsen, J.; Lokebo, J.E.; Bugge, E.; Jonassen, A.K.; Ravingerova, T.; Ytrehus, K. 5-HD abolishes ischemic preconditioning independently of monophasic action potential duration in the heart. Basic. Res. Cardiol. 2000, 95, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sasaki, N.; Seharaseyon, J.; O’Rourke, B.; Marban, E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation 2000, 101, 2418–2423. [Google Scholar] [CrossRef]
- Liu, Y.; Sato, T.; O’Rourke, B.; Marban, E. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation 1998, 97, 2463–2469. [Google Scholar] [CrossRef]
- Pain, T.; Yang, X.M.; Critz, S.D.; Yue, Y.; Nakano, A.; Liu, G.S.; Heusch, G.; Cohen, M.V.; Downey, J.M. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ. Res. 2000, 87, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabo, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Foster, D.B.; Ho, A.S.; Rucker, J.; Garlid, A.O.; Chen, L.; Sidor, A.; Garlid, K.D.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef]
- Welling, P.A.; Ho, K. A comprehensive guide to the ROMK potassium channel: Form and function in health and disease. Am. J. Physiol. Ren. Physiol. 2009, 297, F849–F863. [Google Scholar] [CrossRef]
- Laskowski, M.; Augustynek, B.; Bednarczyk, P.; Zochowska, M.; Kalisz, J.; O’Rourke, B.; Szewczyk, A.; Kulawiak, B. Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. Int. J. Mol. Sci. 2019, 20, 5323. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Kicinska, A.; Laskowski, M.; Kulawiak, B.; Kampa, R.; Walewska, A.; Krajewska, M.; Jarmuszkiewicz, W.; Szewczyk, A. Evidence for a mitochondrial ATP-regulated potassium channel in human dermal fibroblasts. Biochim. Biophys. Acta 2018, 1859, 309–318. [Google Scholar] [CrossRef]
- El-Meanawy, S.K.; Dooge, H.; Wexler, A.C.; Kosmach, A.C.; Serban, L.; Santos, E.A.; Alvarado, F.J.; Hacker, T.A.; Ramratnam, M. Overexpression of a Short Sulfonylurea Splice Variant Increases Cardiac Glucose Uptake and Uncouples Mitochondria by Regulating ROMK Activity. Life 2023, 13, 1015. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Ashok, D.; Liu, T.; Bauer, T.M.; Sun, J.; Li, Z.; da Costa, E.; D’Orleans, C.C.; Nathan, S.; Lefer, D.J.; et al. Global knockout of ROMK potassium channel worsens cardiac ischemia-reperfusion injury but cardiomyocyte-specific knockout does not: Implications for the identity of mitoKATP. J. Mol. Cell. Cardiol. 2020, 139, 176–189. [Google Scholar] [CrossRef]
- Wagner, C.A.; Loffing-Cueni, D.; Yan, Q.; Schulz, N.; Fakitsas, P.; Carrel, M.; Wang, T.; Verrey, F.; Geibel, J.P.; Giebisch, G.; et al. Mouse model of type II Bartter’s syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am. J. Physiol. Ren. Physiol. 2008, 294, F1373–F1380. [Google Scholar] [CrossRef]
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Li, D.; Welling, P.A. Hypertension resistance polymorphisms in ROMK (Kir1.1) alter channel function by different mechanisms. Am. J. Physiol. Ren. Physiol. 2010, 299, F1359–F1364. [Google Scholar] [CrossRef]
- Garcia, M.L.; Priest, B.T.; Alonso-Galicia, M.; Zhou, X.; Felix, J.P.; Brochu, R.M.; Bailey, T.; Thomas-Fowlkes, B.; Liu, J.; Swensen, A.; et al. Pharmacologic inhibition of the renal outer medullary potassium channel causes diuresis and natriuresis in the absence of kaliuresis. J. Pharmacol. Exp. Ther. 2014, 348, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Forrest, M.J.; Sharif-Rodriguez, W.; Forrest, G.; Szeto, D.; Urosevic-Price, O.; Zhu, Y.; Stevenson, A.S.; Zhou, Y.; Stribling, S.; et al. Chronic Inhibition of Renal Outer Medullary Potassium Channel Not Only Prevented but Also Reversed Development of Hypertension and End-Organ Damage in Dahl Salt-Sensitive Rats. Hypertension 2017, 69, 332–338. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ. Res. 2004, 94, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Bezerra Palacio, P.; Brito Lucas, A.M.; Varlla de Lacerda Alexandre, J.; Oliveira Cunha, P.L.; Ponte Viana, Y.I.; Albuquerque, A.C.; Nunes Varela, A.L.; Facundo, H.T. Pharmacological and molecular docking studies reveal that glibenclamide competitively inhibits diazoxide-induced mitochondrial ATP-sensitive potassium channel activation and pharmacological preconditioning. Eur. J. Pharmacol. 2021, 908, 174379. [Google Scholar] [CrossRef]
- Walewska, A.; Krajewska, M.; Stefanowska, A.; Buta, A.; Bilewicz, R.; Krysinski, P.; Bednarczyk, P.; Koprowski, P.; Szewczyk, A. Methods of Measuring Mitochondrial Potassium Channels: A Critical Assessment. Int. J. Mol. Sci. 2022, 23, 1210. [Google Scholar] [CrossRef]
- Lawrence, C.L.; Billups, B.; Rodrigo, G.C.; Standen, N.B. The KATP channel opener diazoxide protects cardiac myocytes during metabolic inhibition without causing mitochondrial depolarization or flavoprotein oxidation. Br. J. Pharmacol. 2001, 134, 535–542. [Google Scholar] [CrossRef]
- Forbes, R.A.; Steenbergen, C.; Murphy, E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ. Res. 2001, 88, 802–809. [Google Scholar] [CrossRef]
- Vander Heide, R.S.; Steenbergen, C. Cardioprotection and myocardial reperfusion: Pitfalls to clinical application. Circ. Res. 2013, 113, 464–477. [Google Scholar] [CrossRef]
- Vanden Hoek, T.; Becker, L.B.; Shao, Z.H.; Li, C.Q.; Schumacker, P.T. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circ. Res. 2000, 86, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, C.M.; MacGregor, G.G.; Islas, L.D.; Yang, Y.; Hebert, S.C.; Giebisch, G. pH-dependent modulation of the cloned renal K+ channel, ROMK. Am. J. Physiol. 1998, 275, F972–F981. [Google Scholar] [CrossRef]
- Baukrowitz, T.; Schulte, U.; Oliver, D.; Herlitze, S.; Krauter, T.; Tucker, S.J.; Ruppersberg, J.P.; Fakler, B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 1998, 282, 1141–1144. [Google Scholar] [CrossRef]
- Leung, Y.M.; Zeng, W.Z.; Liou, H.H.; Solaro, C.R.; Huang, C.L. Phosphatidylinositol 4,5-bisphosphate and intracellular pH regulate the ROMK1 potassium channel via separate but interrelated mechanisms. J. Biol. Chem. 2000, 275, 10182–10189. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Williams, D.M.; Karcz, M.K.; Lopes, C.M.; Gray, D.A.; Nehrke, K.W.; Brookes, P.S. A novel mitochondrial K(ATP) channel assay. Circ. Res. 2010, 106, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.T.; Pravdic, D.; McNally, E.M.; Bosnjak, Z.J.; Shi, N.Q.; Makielski, J.C. The mitochondrial bioenergetic phenotype for protection from cardiac ischemia in SUR2 mutant mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1884–H1890. [Google Scholar] [CrossRef]
- Keung, W.; Ren, L.; Sen, L.; Wong, A.O.; Chopra, A.; Kong, C.W.; Tomaselli, G.F.; Chen, C.S.; Li, R.A. Non-cell autonomous cues for enhanced functionality of human embryonic stem cell-derived cardiomyocytes via maturation of sarcolemmal and mitochondrial K(ATP) channels. Sci. Rep. 2016, 6, 34154. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- DiResta, D.J.; Kutschke, K.P.; Hottois, M.D.; Garlid, K.D. K+-H+ exchange and volume homeostasis in brown adipose tissue mitochondria. Am. J. Physiol. 1986, 251, R787–R793. [Google Scholar] [CrossRef] [PubMed]
- Paucek, P.; Yarov-Yarovoy, V.; Sun, X.; Garlid, K.D. Inhibition of the mitochondrial KATP channel by long-chain acyl-CoA esters and activation by guanine nucleotides. J. Biol. Chem. 1996, 271, 32084–32088. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- O’Rourke, B. Myocardial K(ATP) channels in preconditioning. Circ. Res. 2000, 87, 845–855. [Google Scholar] [CrossRef]
- Oldenburg, O.; Cohen, M.V.; Downey, J.M. Mitochondrial K(ATP) channels in preconditioning. J. Mol. Cell. Cardiol. 2003, 35, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Shivkumar, K.; Deutsch, N.A.; Lamp, S.T.; Khuu, K.; Goldhaber, J.I.; Weiss, J.N. Mechanism of hypoxic K loss in rabbit ventricle. J. Clin. Investig. 1997, 100, 1782–1788. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Maack, C. Calcium release microdomains and mitochondria. Cardiovasc. Res. 2013, 98, 259–268. [Google Scholar] [CrossRef]
- Tang, H.; de Jesus, R.K.; Walsh, S.P.; Zhu, Y.; Yan, Y.; Priest, B.T.; Swensen, A.M.; Alonso-Galicia, M.; Felix, J.P.; Brochu, R.M.; et al. Discovery of a novel sub-class of ROMK channel inhibitors typified by 5-(2-(4-(2-(4-(1H-Tetrazol-1-yl)phenyl)acetyl)piperazin-1-yl)ethyl)isobenzofuran- 1(3H)-one. Bioorg. Med. Chem. Lett. 2013, 23, 5829–5832. [Google Scholar] [CrossRef]
- Bischof, H.; Rehberg, M.; Stryeck, S.; Artinger, K.; Eroglu, E.; Waldeck-Weiermair, M.; Gottschalk, B.; Rost, R.; Deak, A.T.; Niedrist, T.; et al. Novel genetically encoded fluorescent probes enable real-time detection of potassium in vitro and in vivo. Nat. Commun. 2017, 8, 1422. [Google Scholar] [CrossRef]
- Ramratnam, M.; Kenny, B.; Kyle, J.W.; Wiedmeyer, B.; Hacker, T.A.; Barefield, D.Y.; McNally, E.M.; Makielski, J.C. Transgenic overexpression of the SUR2A-55 splice variant in mouse heart reduces infract size and promotes protective mitochondrial function. Heliyon 2018, 4, e00677. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.E.; Bell, J.R.; Pepe, S.; Delbridge, L.M. Benchmarking ventricular arrhythmias in the mouse--revisiting the ’Lambeth Conventions’ 20 years on. Heart Lung Circ. 2008, 17, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Abramov, A.Y. Measurement of mitochondrial NADH and FAD autofluorescence in live cells. Methods Mol. Biol. 2015, 1264, 263–270. [Google Scholar] [PubMed]
- Emaus, R.K.; Grunwald, R.; Lemasters, J.J. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: Spectral and metabolic properties. Biochim. Biophys. Acta 1986, 850, 436–448. [Google Scholar] [CrossRef]
- Baracca, A.; Sgarbi, G.; Solaini, G.; Lenaz, G. Rhodamine 123 as a probe of mitochondrial membrane potential: Evaluation of proton flux through F(0) during ATP synthesis. Biochim. Biophys. Acta 2003, 1606, 137–146. [Google Scholar] [CrossRef]
- Blass, B. Inhibitors of Renal Outer Medullary Potassium Channel. ACS Med. Chem. Lett. 2016, 7, 447–448. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT Vehicle (N = 7) | WT Compound A (N = 7) | p Value | |
|---|---|---|---|
| LVDP, mmHg | 61 ± 5.9 | 54 ± 7.6 | 0.23 |
| +dp/dt, mmHg/s | 1947 ± 198 | 1590 ± 214 | 0.12 |
| −dp/dt, mmHg/s | −1339 ± 174 | −1024 ± 183 | 0.11 |
| HR, bpm | 310 ± 16 | 247 ± 44 | 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wexler, A.C.; Dooge, H.; Serban, L.; Tewari, A.; Tehrani, B.M.; Alvarado, F.J.; Ramratnam, M. Pharmacologic ROMK Inhibition Protects Against Myocardial Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2025, 26, 3795. https://doi.org/10.3390/ijms26083795
Wexler AC, Dooge H, Serban L, Tewari A, Tehrani BM, Alvarado FJ, Ramratnam M. Pharmacologic ROMK Inhibition Protects Against Myocardial Ischemia Reperfusion Injury. International Journal of Molecular Sciences. 2025; 26(8):3795. https://doi.org/10.3390/ijms26083795
Chicago/Turabian StyleWexler, Allison C., Holly Dooge, Lara Serban, Aditya Tewari, Babak M. Tehrani, Francisco J. Alvarado, and Mohun Ramratnam. 2025. "Pharmacologic ROMK Inhibition Protects Against Myocardial Ischemia Reperfusion Injury" International Journal of Molecular Sciences 26, no. 8: 3795. https://doi.org/10.3390/ijms26083795
APA StyleWexler, A. C., Dooge, H., Serban, L., Tewari, A., Tehrani, B. M., Alvarado, F. J., & Ramratnam, M. (2025). Pharmacologic ROMK Inhibition Protects Against Myocardial Ischemia Reperfusion Injury. International Journal of Molecular Sciences, 26(8), 3795. https://doi.org/10.3390/ijms26083795