
PERK/Sestrin2 Signaling Pathway Mediated Autophagy Regulates Human Cardiomyocytes Apoptosis Induced by Traffic-Related PM2.5 and Diverse Constituents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
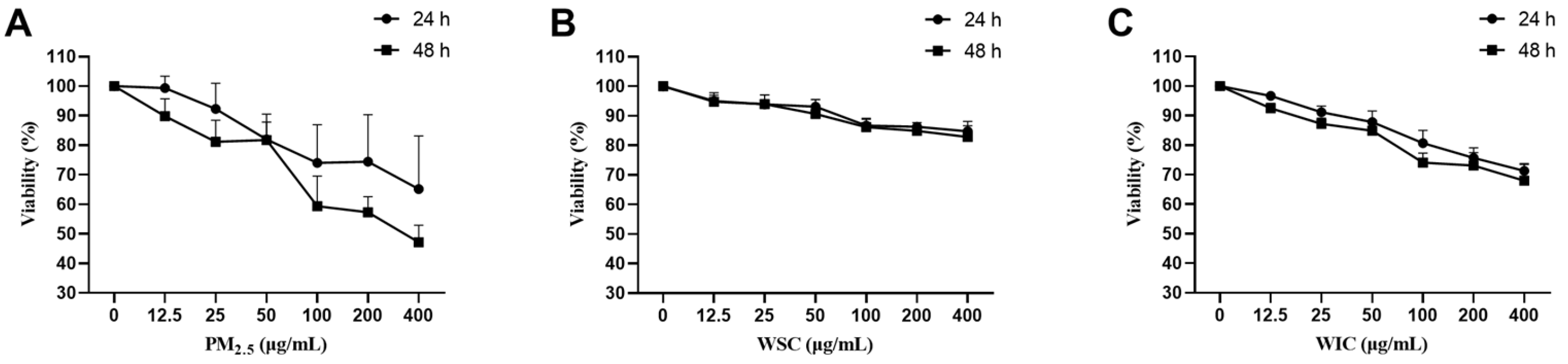
2.1. TRPM2.5, WSC, and WIC Descended the Cell Viability Rates of AC16
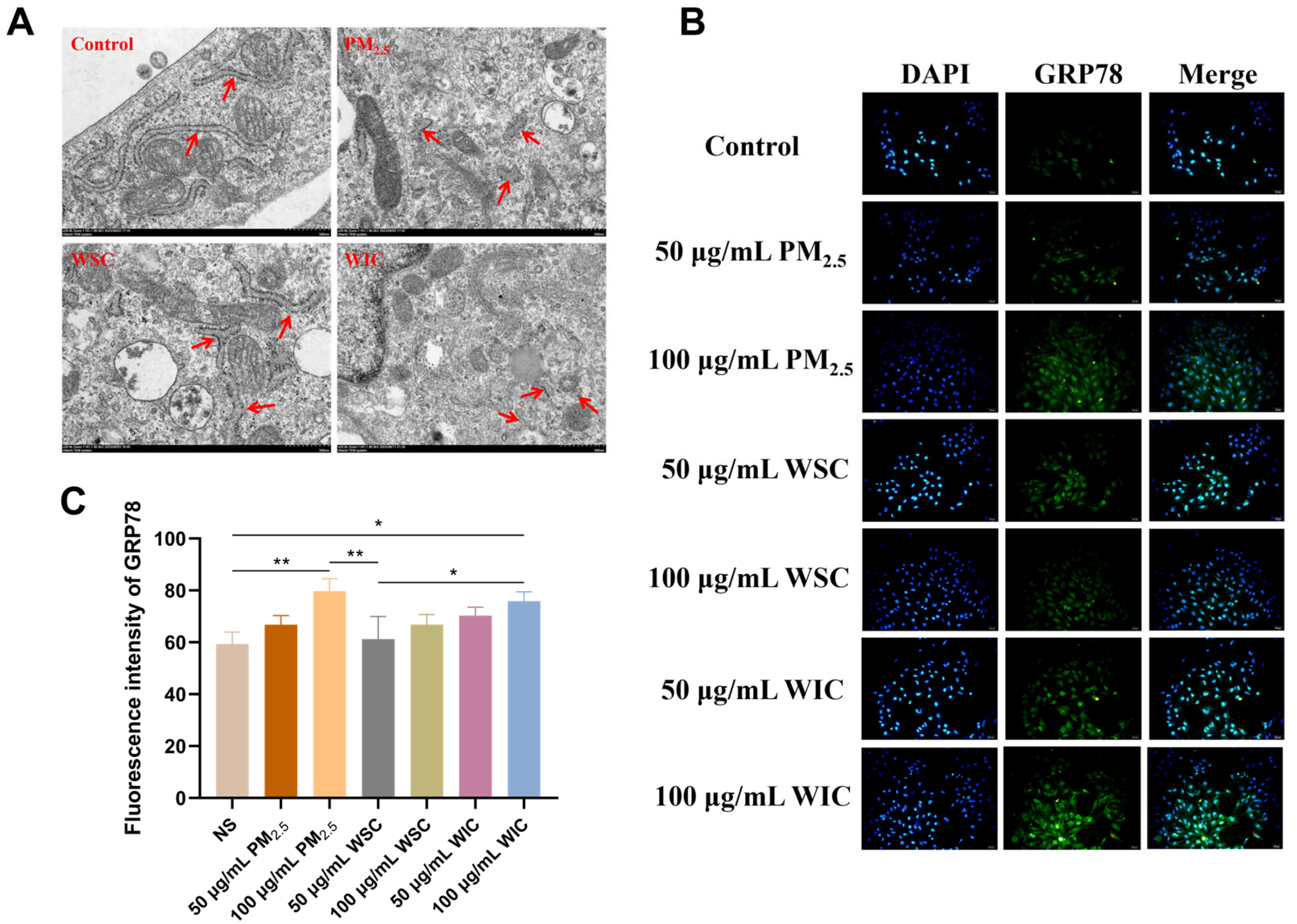
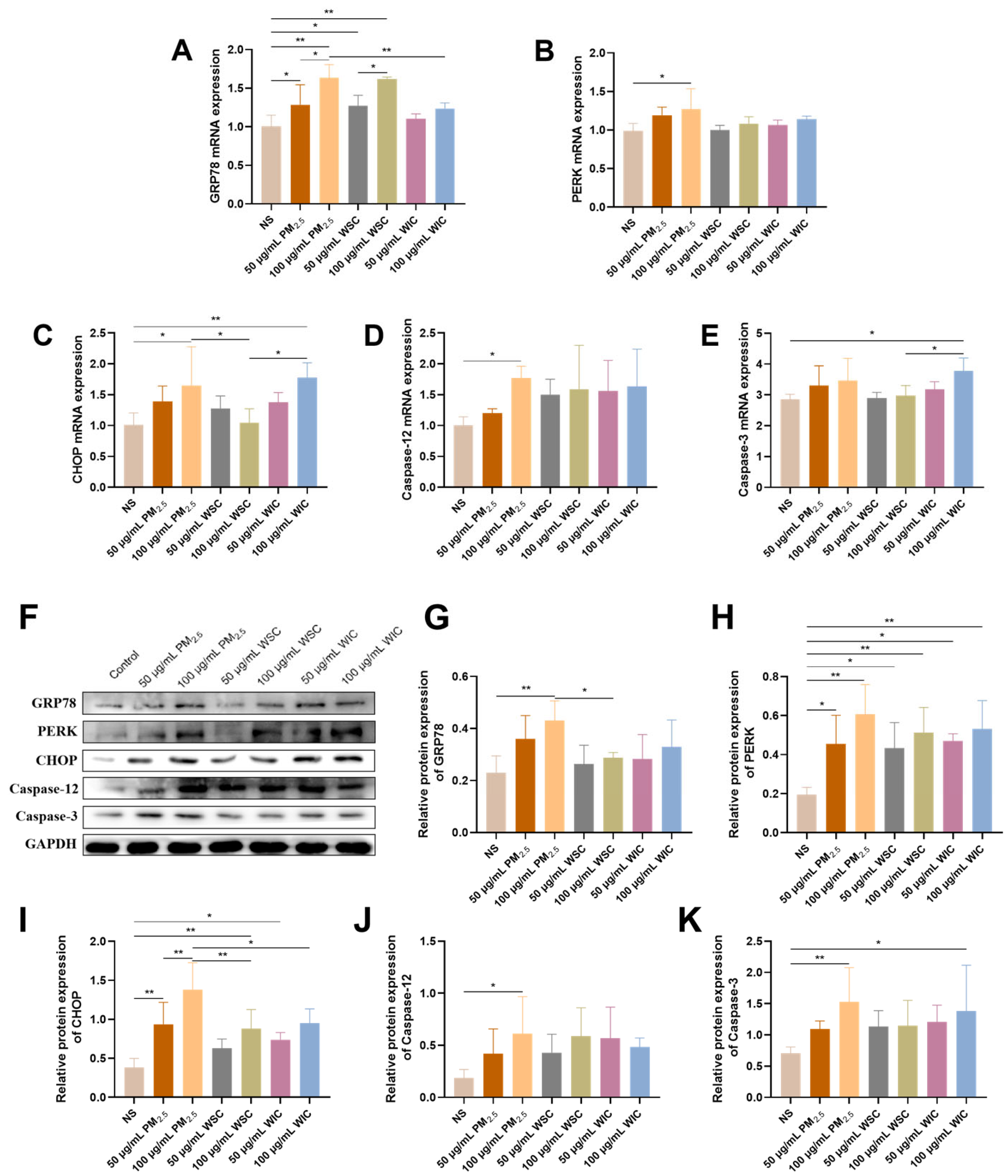
2.2. TRPM2.5 and Components Induced AC16 Cells Apoptosis Through ERS
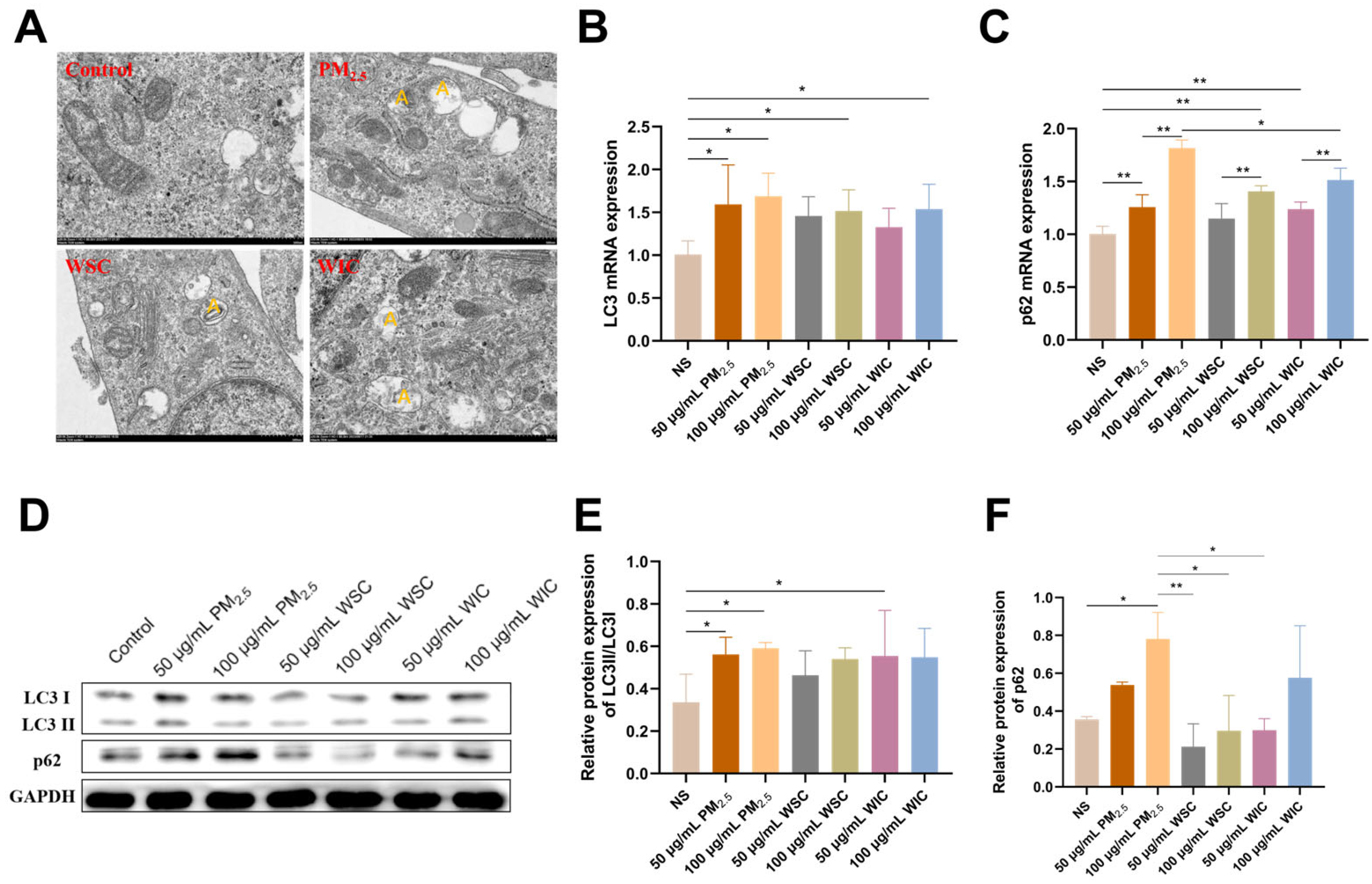
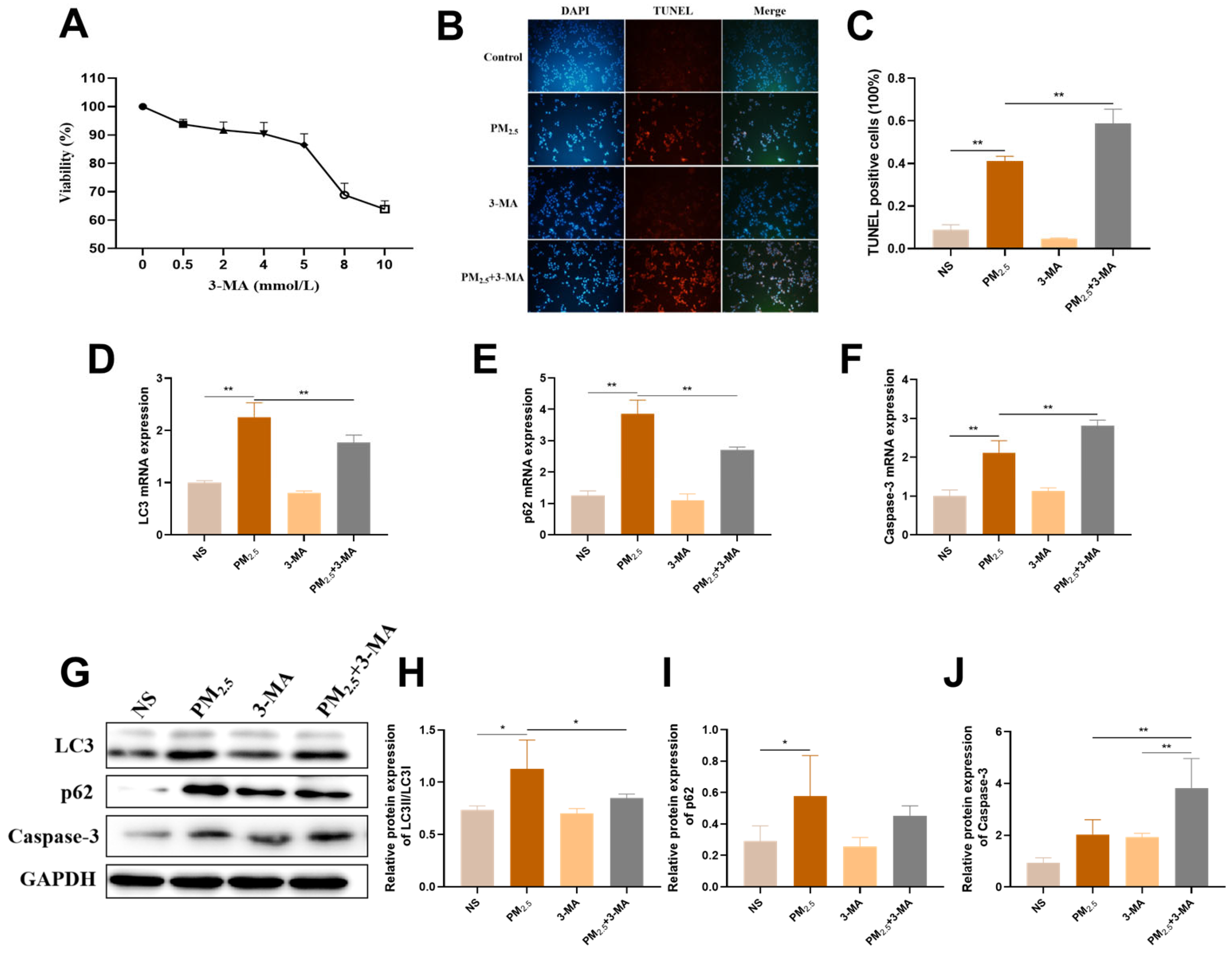
2.3. TRPM2.5 and Components Triggered AC16 Cells Autophagy
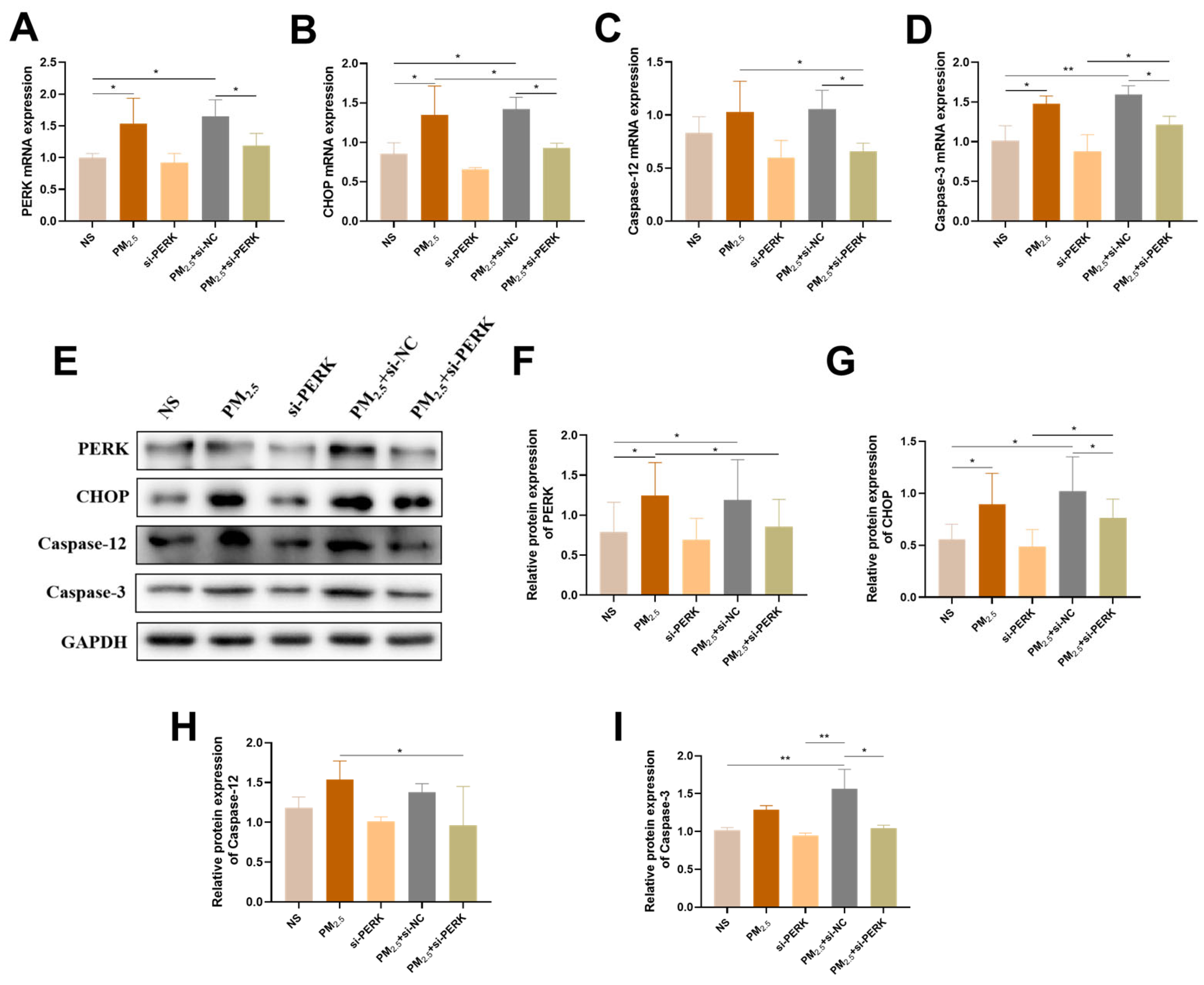
2.4. PERK Gene Knockdown Alleviated Sensitivity of AC16 Cells to TRPM2.5
2.5. PERK/Sestrin2 Signaling Pathway Participated in the Initiation of Autophagy
2.6. Activation of Protective Autophagy Resists TRPM2.5-Induced Cell Damage
3. Discussion
4. Materials and Methods
4.1. TRPM2.5 Collection and Extraction
4.2. Cell Culture and TRPM2.5 Exposure
4.3. Assessment of Cell Viability
4.4. Ultrastructure Observation
4.5. Immunofluorescence (If)
4.6. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Staining
4.7. Cell Transfection
4.8. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.9. Western Blotting (WB) Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Health Effects Institute. State of Global Air 2024. In Special Report; Health Effects Institute: Boston, MA, USA, 2024. [Google Scholar]
- IQAir. 2023 World Air Quality Report; IQAir: Goldach, Switzerland, 2023. [Google Scholar]
- Li, Z.; Liang, D.; Ebelt, S.; Gearing, M.; Kobor, M.S.; Konwar, C.; Maclsaac, J.L.; Dever, K.; Wingo, A.P.; Levey, A.I.; et al. Differential DNA methylation in the brain as potential mediator of the association between traffic-related PM2.5 and neuropathology markers of Alzheimer’s disease. Alzheimer’s Dement. 2024, 20, 2538–2551. [Google Scholar] [CrossRef]
- Wang, C.; Wang, D.; Zhao, H.; Wang, J.; Liu, N.; Shi, H.; Tian, J.; Wang, X.; Zhang, Z. Traffic-related PM2.5 and diverse constituents disturb the balance of Th17/Treg cells by STAT3/RORγt-STAT5/Foxp3 signaling pathway in a rat model of asthma. Int. Immunopharmacol. 2021, 96, 107788. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.L.S.; Andersson, N.; Stockfelt, L.; Andersson, E.M.; Nilsson Sommar, J.; Eneroth, K.; Gidhagen, L.; Johansson, C.; Lager, A.; Leander, K.; et al. Long-Term Exposure to Particulate Air Pollution, Black Carbon, and Their Source Components in Relation to Ischemic Heart Disease and Stroke. Environ. Health Perspect. 2019, 127, 107012. [Google Scholar] [CrossRef]
- Institute for Health Metrics and Evaluation (IHME). Global Burden of Disease 2021: Findings from the GBD 2021 Study; IHME: Seattle, WA, USA, 2024. [Google Scholar]
- Alexeeff, S.E.; Deosaransingh, K.; Van Den Eeden, S.; Schwartz, J.; Liao, N.S.; Sidney, S. Association of Long-term Exposure to Particulate Air Pollution with Cardiovascular Events in California. JAMA Netw. Open 2023, 6, e230561. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, W.; Li, S.; He, C.; Dai, Y.; Feng, S.; Zeng, C.; Yang, T.; Meng, Q.; Meng, J.; et al. Ambient PM2.5 and its components associated with 10-year atherosclerotic cardiovascular disease risk in Chinese adults. Ecotoxicol. Environ. Saf. 2023, 263, 115371. [Google Scholar] [CrossRef] [PubMed]
- Huynh, Q.; Marwick, T.H.; Venkataraman, P.; Knibbs, L.D.; Johnston, F.H.; Negishi, K. Long-term exposure to ambient air pollution is associated with coronary artery calcification among asymptomatic adults. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 922–929. [Google Scholar] [CrossRef]
- Gao, L.; Qin, J.X.; Shi, J.Q.; Jiang, T.; Wang, F.; Xie, C.; Gao, Q.; Zhi, N.; Dong, Q.; Guan, Y.T. Fine particulate matter exposure aggravates ischemic injury via NLRP3 inflammasome activation and pyroptosis. CNS Neurosci. Ther. 2022, 28, 1045–1058. [Google Scholar] [CrossRef]
- Ren, J.; Yin, B.; Guo, Z.; Sun, X.; Pei, H.; Wen, R.; Wang, Z.; Zhu, S.; Zuo, J.; Zhang, Y.; et al. Astaxanthin alleviates PM2.5-induced cardiomyocyte injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2023, 28, 95. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, M.; Sun, X.; Yang, J.; Guo, M. Microplastics induced endoplasmic reticulum stress to format an inflammation and cell death in hepatocytes of carp (Cyprinus carpio). Aquat. Toxicol. 2024, 269, 106870. [Google Scholar] [CrossRef]
- Liu, D.; Qin, H.; Gao, Y.; Sun, M.; Wang, M. Cardiovascular disease: Mitochondrial dynamics and mitophagy crosstalk mechanisms with novel programmed cell death and macrophage polarisation. Pharmacol. Res. 2024, 206, 107258. [Google Scholar] [CrossRef]
- Qi, J.; Li, Q.; Xin, T.; Lu, Q.; Lin, J.; Zhang, Y.; Luo, H.; Zhang, F.; Xing, Y.; Wang, W.; et al. MCOLN1/TRPML1 in the lysosome: A promising target for autophagy modulation in diverse diseases. Autophagy 2024, 20, 1712–1722. [Google Scholar] [CrossRef]
- Wang, J.; Lu, W.; Zhang, J.; Du, Y.; Fang, M.; Zhang, A.; Sungcad, G.; Chon, S.; Xing, J. Loss of TRIM29 mitigates viral myocarditis by attenuating PERK-driven ER stress response in male mice. Nat. Commun. 2024, 15, 3481. [Google Scholar] [CrossRef]
- He, W.; Sun, Z.; Tong, G.; Zeng, L.; He, W.; Chen, X.; Zhen, C.; Chen, P.; Tan, N.; He, P. FUNDC1 alleviates doxorubicin-induced cardiotoxicity by restoring mitochondrial-endoplasmic reticulum contacts and blocked autophagic flux. Theranostics 2024, 14, 3719–3738. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.C.; Xu, Z.M.; Lyu, Y.; Li, X.H.; Li, Z.F.; He, H.; Tian, F.J.; Zheng, J.P. PM2.5 induces autophagy-dependent ferroptosis by endoplasmic reticulum stress in SH-SY5Y cells. J. Appl. Toxicol. 2023, 43, 1013–1025. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Kavinda, M.H.D.; Choi, Y.H.; Lee, H.; Kang, C.H.; Lee, M.H.; Lee, C.M.; Kim, G.Y. Fisetin Protects HaCaT Human Keratinocytes from Fine Particulate Matter (PM2.5)-Induced Oxidative Stress and Apoptosis by Inhibiting the Endoplasmic Reticulum Stress Response. Antioxidants 2021, 10, 1492. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jiang, Y.; Xu, W.; Bao, X. Sestrin2: Multifaceted functions, molecular basis, and its implications in liver diseases. Cell Death Dis. 2023, 14, 160. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.A.; Abdelsalam, S.S.; Raïq, H.; Parray, A.; Korashy, H.M.; Zeidan, A.; Elrayess, M.A.; Agouni, A. Sestrin2 as a Protective Shield against Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 4880. [Google Scholar] [CrossRef]
- Dai, G.; Li, M.; Xu, H.; Quan, N. Status of Research on Sestrin2 and Prospects for its Application in Therapeutic Strategies Targeting Myocardial Aging. Curr. Probl. Cardiol. 2023, 48, 101910. [Google Scholar] [CrossRef]
- Bi, D.; Zheng, D.; Shi, M.; Hu, Q.; Wang, H.; Zhi, H.; Lou, D.; Zhang, A.; Hu, Y. Role of SESTRIN2/AMPK/ULK1 pathway activation and lysosomes dysfunction in NaAsO(2)-induced liver injury under oxidative stress. Ecotoxicol. Environ. Saf. 2023, 254, 114751. [Google Scholar] [CrossRef]
- Gong, H.; Lyu, X.; Liu, Y.; Peng, N.; Tan, S.; Dong, L.; Zhang, X. Eupatilin inhibits pulmonary fibrosis by activating Sestrin2/PI3K/Akt/mTOR dependent autophagy pathway. Life Sci. 2023, 334, 122218. [Google Scholar] [CrossRef]
- Liu, H.; Lai, W.; Nie, H.; Shi, Y.; Zhu, L.; Yang, L.; Tian, L.; Li, K.; Bian, L.; Xi, Z.; et al. PM2.5 triggers autophagic degradation of Caveolin-1 via endoplasmic reticulum stress (ERS) to enhance the TGF-β1/Smad3 axis promoting pulmonary fibrosis. Environ. Int. 2023, 181, 108290. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Huang, Y.; Yu, F.; Feng, X.; Zheng, Y.Y.; Li, Q.; Niu, Q.; Jiang, Y.H.; Zhao, L.Q.; Wang, M.; et al. BMAL1/p53 mediating bronchial epithelial cell autophagy contributes to PM2.5-aggravated asthma. Cell Commun. Signal. 2023, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shao, T.; Qin, M.; Miao, X.; Chang, Y.; Sheng, W.; Wu, F.; Yu, Y. The effects of autophagy on vascular endothelial cells induced by airborne PM2.5. J. Environ. Sci. 2018, 66, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, W.; Yue, H.; Zhao, P.; Li, J. Effective-components combination alleviates PM2.5-induced inflammation by evoking macrophage autophagy in COPD. J. Ethnopharmacol. 2024, 321, 117537. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, T.; Feng, L.; Shi, Y.; Jiang, J.; Liang, S.; Sun, B.; Xu, Q.; Duan, J.; Sun, Z. PM2.5-induced ADRB2 hypermethylation contributed to cardiac dysfunction through cardiomyocytes apoptosis via PI3K/Akt pathway. Environ. Int. 2019, 127, 601–614. [Google Scholar] [CrossRef]
- Yang, X.; Feng, L.; Zhang, Y.; Hu, H.; Shi, Y.; Liang, S.; Zhao, T.; Fu, Y.; Duan, J.; Sun, Z. Cytotoxicity induced by fine particulate matter (PM2.5) via mitochondria-mediated apoptosis pathway in human cardiomyocytes. Ecotoxicol. Environ. Saf. 2018, 161, 198–207. [Google Scholar] [CrossRef]
- Qi, Z.; Song, Y.; Ding, Q.; Liao, X.; Li, R.; Liu, G.; Tsang, S.; Cai, Z. Water soluble and insoluble components of PM2.5 and their functional cardiotoxicities on neonatal rat cardiomyocytes in vitro. Ecotoxicol. Environ. Saf. 2019, 168, 378–387. [Google Scholar] [CrossRef]
- Shi, M.; Liu, K.; Li, X.; Zeng, X.L.; Liu, X.J. Melatonin ameliorates PM2.5-induced airway inflammation and apoptosis by PERK/eIF2α/ATF4/CHOP in chronic obstructive pulmonary disease mice. Toxicol. Appl. Pharmacol. 2025, 499, 117314. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, M. PM2.5 induces autophagy and apoptosis through endoplasmic reticulum stress in human endothelial cells. Sci. Total Environ. 2020, 710, 136397. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Zhou, K.; Xie, L.; Xiang, G.; Fang, M.; Han, W.; Wang, X.; Xiao, J. Elevating sestrin2 attenuates endoplasmic reticulum stress and improves functional recovery through autophagy activation after spinal cord injury. Cell Biol. Toxicol. 2021, 37, 401–419. [Google Scholar] [CrossRef]
- Park, H.J.; Yang, S.G.; Koo, D.B. SESN2/NRF2 signaling activates as a direct downstream regulator of the PERK pathway against endoplasmic reticulum stress to improve the in vitro maturation of porcine oocytes. Free Radic. Biol. Med. 2022, 178, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, S.; Li, J.; Han, L.; He, Q.; Wang, R.; Wang, X.; Liu, K. Developmental toxicity induced by PM2.5 through endoplasmic reticulum stress and autophagy pathway in zebrafish embryos. Chemosphere 2018, 197, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Shi, Z.; Wu, J.; Yang, X.; Feng, L.; Ren, L.; Duan, J.; Sun, Z. Metabolic impact induced by total, water soluble and insoluble components of PM2.5 acute exposure in mice. Chemosphere 2018, 207, 337–346. [Google Scholar] [CrossRef]
- Yan, Z.; Ge, P.; Lu, Z.; Liu, X.; Cao, M.; Chen, W.; Chen, M. The Cytotoxic Effects of Fine Particulate Matter (PM2.5) from Different Sources at the Air-Liquid Interface Exposure on A549 Cells. Toxics 2023, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, D.; Shi, H.; Liu, N.; Wang, C.; Tian, J.; Wang, X.; Zhang, Z. PM2.5 and water-soluble components induce airway fibrosis through TGF-β1/Smad3 signaling pathway in asthmatic rats. Mol. Immunol. 2021, 137, 1–10. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Niu, Z.; Yang, H.; Wang, C.; Guan, L.; Zhao, L.; Shi, D.; Zhang, Z. PERK/Sestrin2 Signaling Pathway Mediated Autophagy Regulates Human Cardiomyocytes Apoptosis Induced by Traffic-Related PM2.5 and Diverse Constituents. Int. J. Mol. Sci. 2025, 26, 3784. https://doi.org/10.3390/ijms26083784
Tian J, Niu Z, Yang H, Wang C, Guan L, Zhao L, Shi D, Zhang Z. PERK/Sestrin2 Signaling Pathway Mediated Autophagy Regulates Human Cardiomyocytes Apoptosis Induced by Traffic-Related PM2.5 and Diverse Constituents. International Journal of Molecular Sciences. 2025; 26(8):3784. https://doi.org/10.3390/ijms26083784
Chicago/Turabian StyleTian, Jiayu, Zeyu Niu, Huan Yang, Caihong Wang, Linlin Guan, Lifang Zhao, Dongxing Shi, and Zhihong Zhang. 2025. "PERK/Sestrin2 Signaling Pathway Mediated Autophagy Regulates Human Cardiomyocytes Apoptosis Induced by Traffic-Related PM2.5 and Diverse Constituents" International Journal of Molecular Sciences 26, no. 8: 3784. https://doi.org/10.3390/ijms26083784
APA StyleTian, J., Niu, Z., Yang, H., Wang, C., Guan, L., Zhao, L., Shi, D., & Zhang, Z. (2025). PERK/Sestrin2 Signaling Pathway Mediated Autophagy Regulates Human Cardiomyocytes Apoptosis Induced by Traffic-Related PM2.5 and Diverse Constituents. International Journal of Molecular Sciences, 26(8), 3784. https://doi.org/10.3390/ijms26083784