

Synthesis and In Vitro Evaluation of the Anticancer Effect of Novel Phosphonium Vindoline Derivatives

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
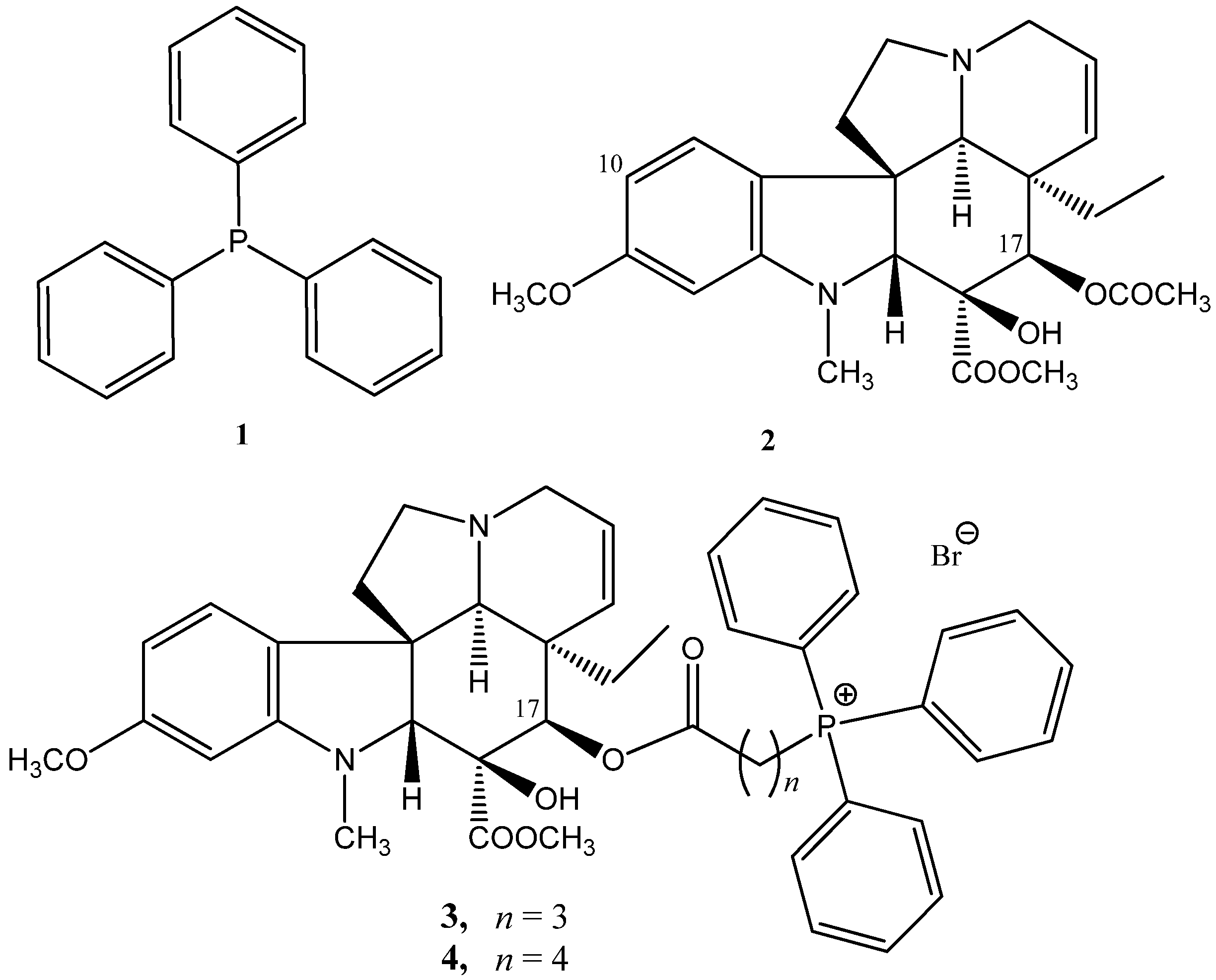
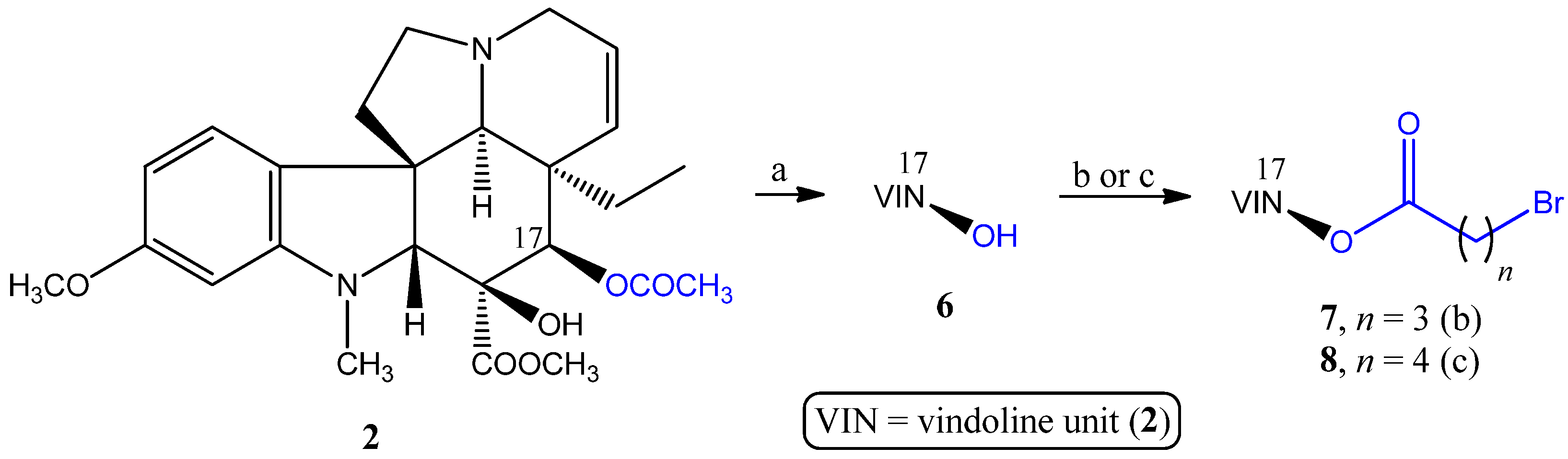
2.1.1. Coupling Trisubstituted Phosphines at Position 17 of Vindoline
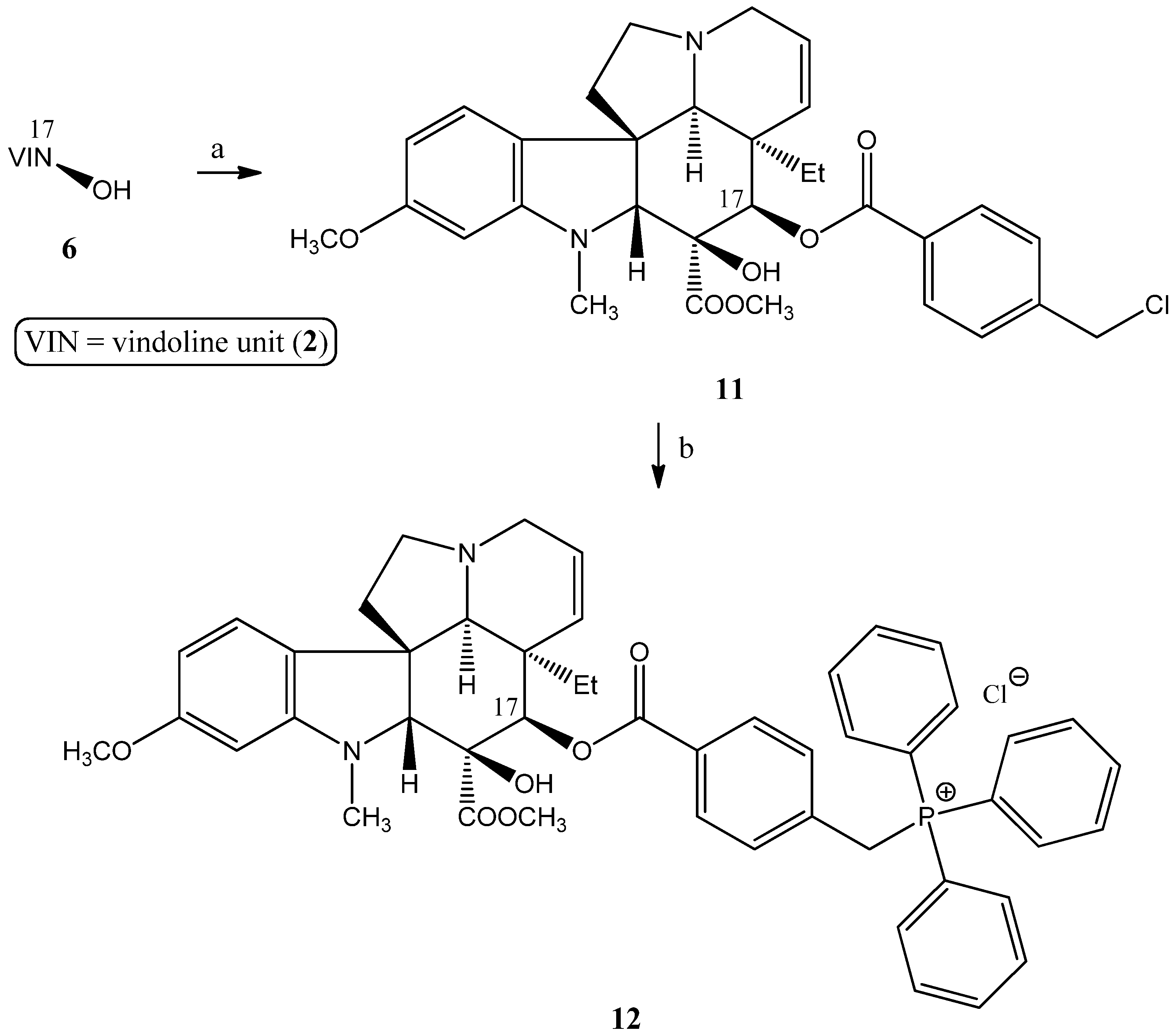
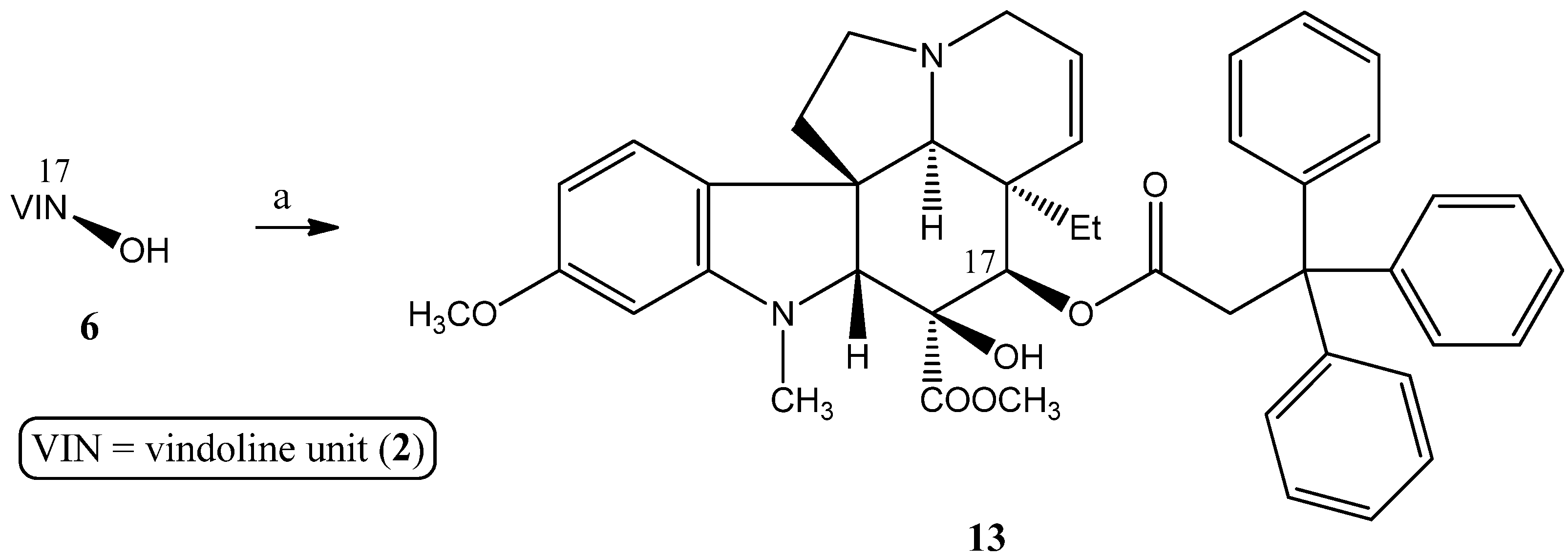
2.1.2. Synthesis of a Conjugate via a More Rigid Linker
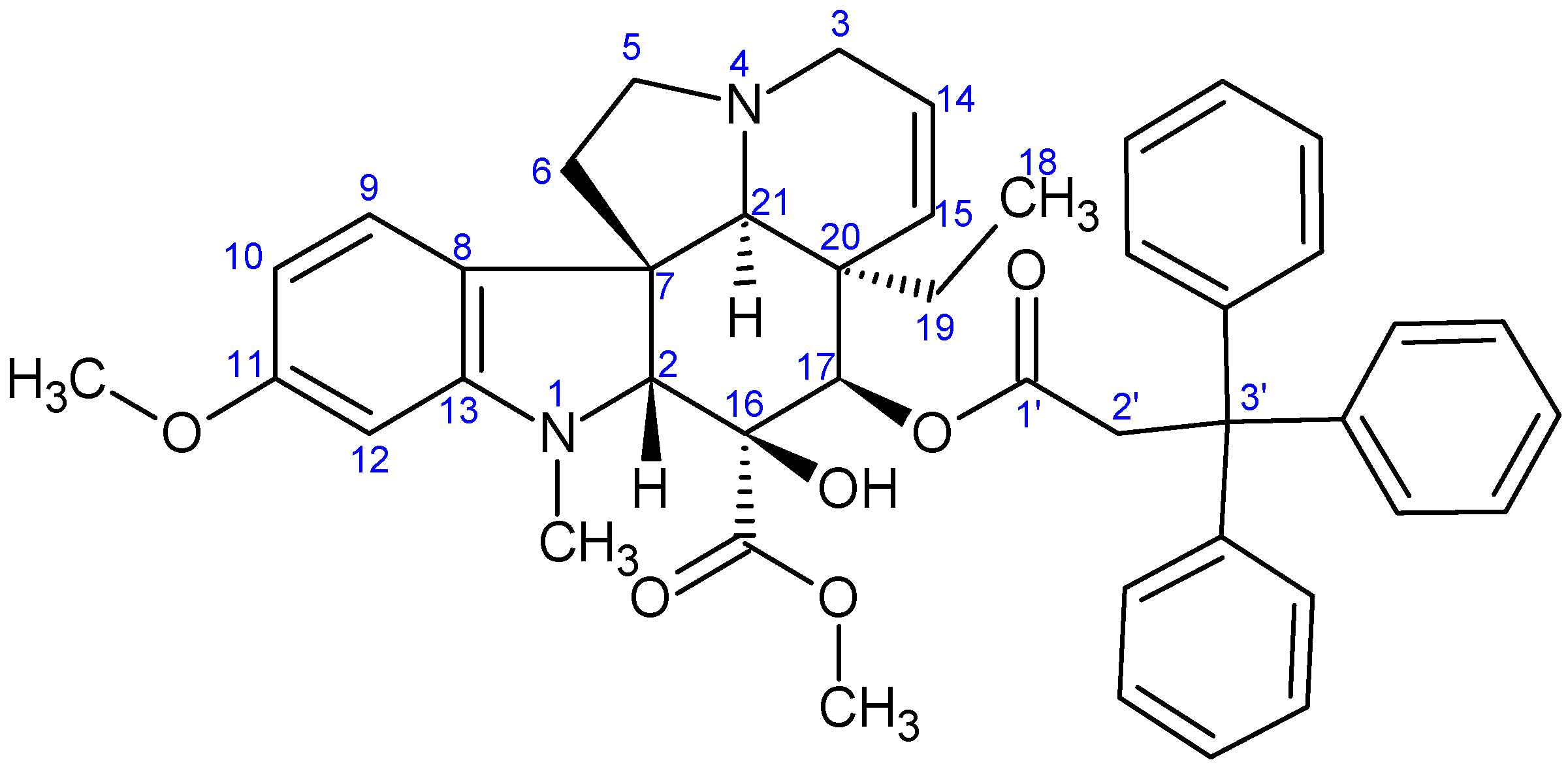
2.1.3. Formation of Triphenylmethyl Group at Position 17 of Vindoline
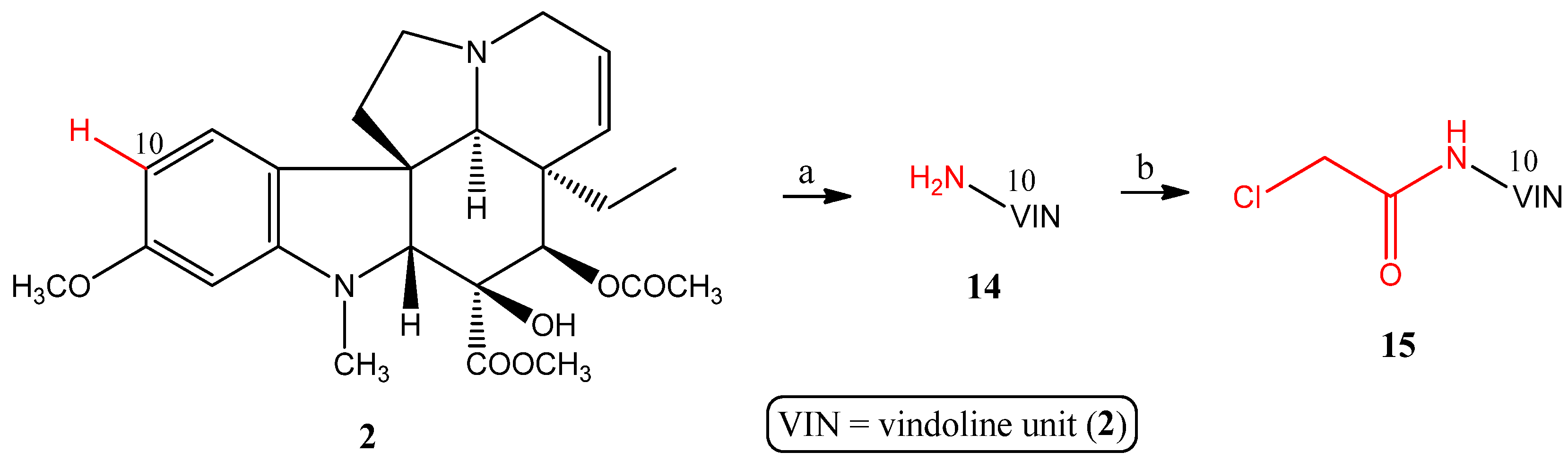
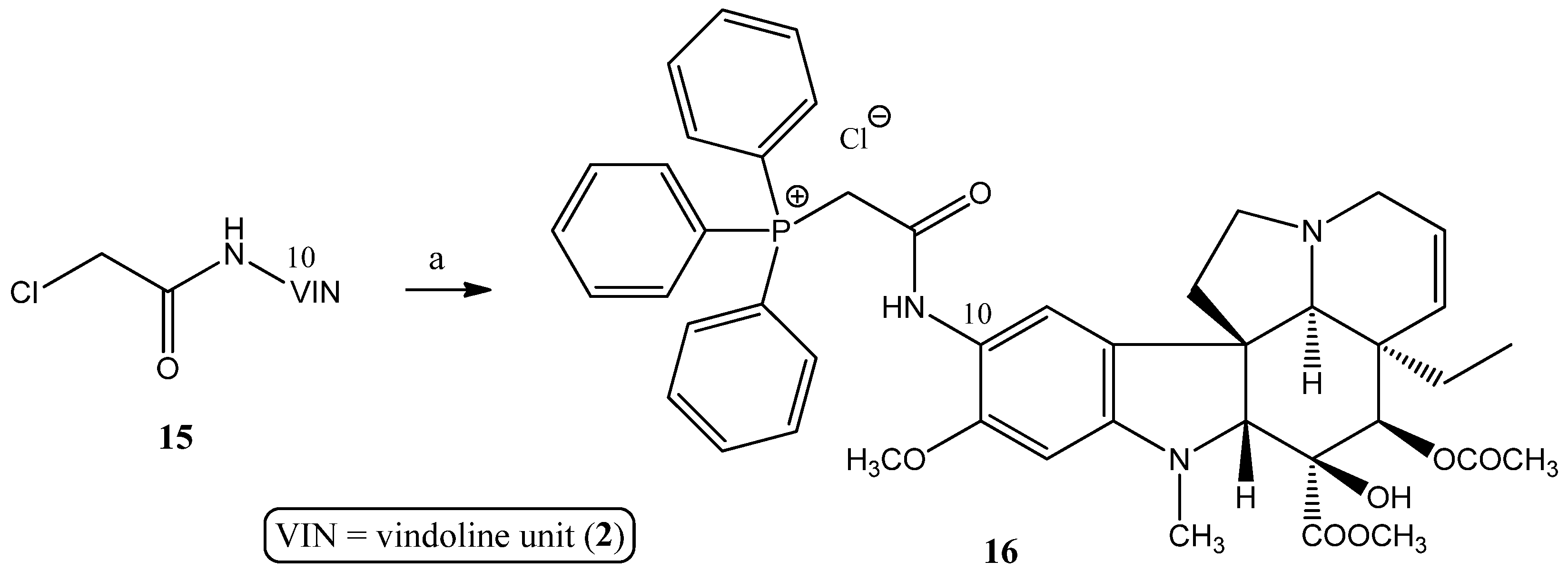
2.1.4. Coupling Triphenylphosphine at Position 10 of Vindoline
2.2. Evaluation of the Biological Activities
2.2.1. NCI60 Screening
2.2.2. MTT Assay of Compounds 9g, 13, and 16
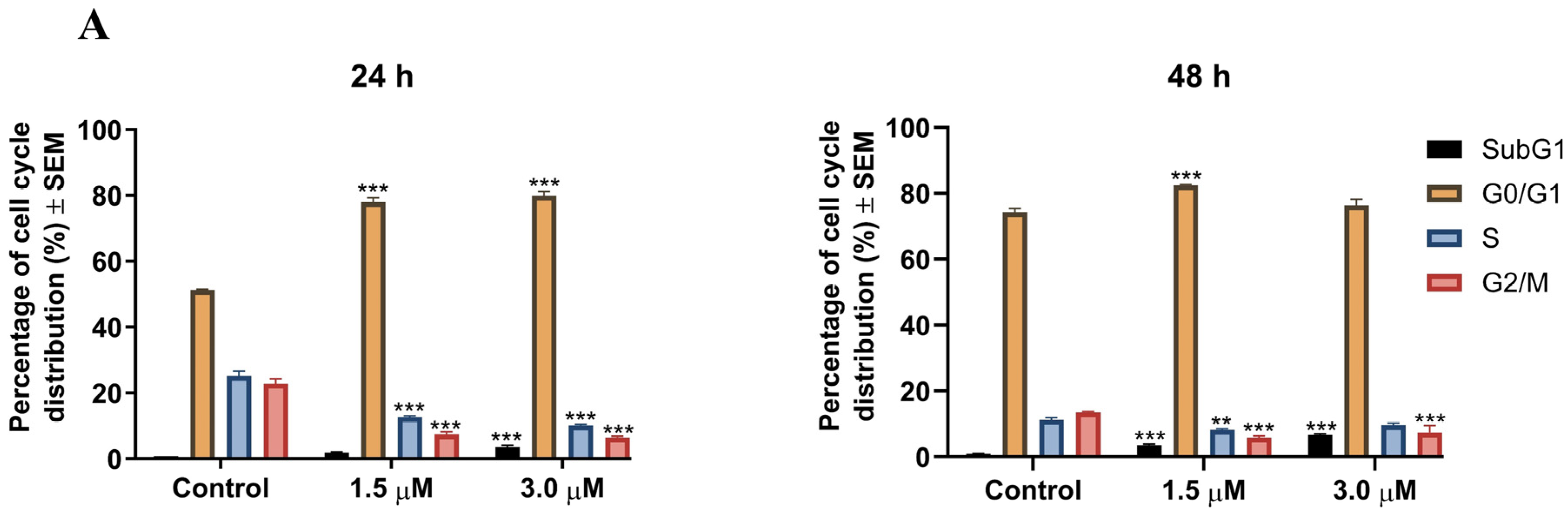
2.2.3. Cell Cycle Analysis
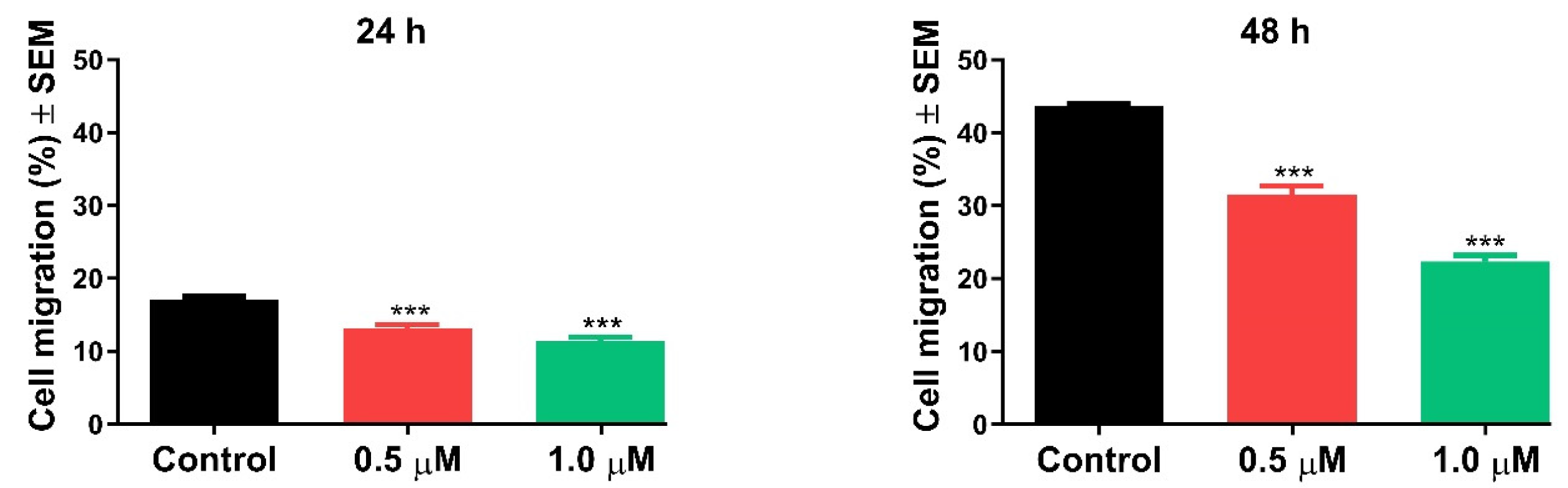
2.2.4. Antimigratory Activity
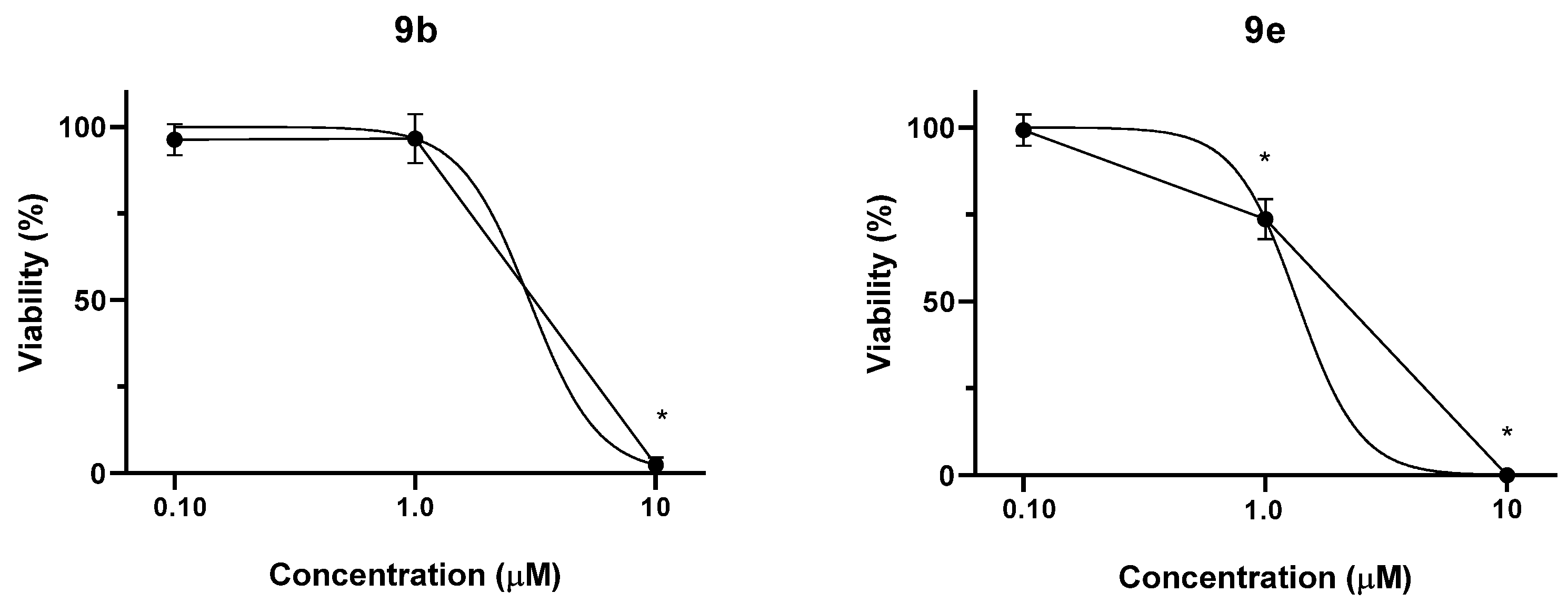
2.2.5. Effect of Selected Conjugates on the Viability of Non-Tumor Chinese Hamster Ovary (CHO) Cells
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. Synthesis of the Linker-Containing Vindoline Derivatives (7, 8, 11, and 15) and Their Precursors (6 and 14)
- Preparation of 17-desacetylvindoline (6)
- Preparation of 17-(O-4-bromobutanoyl)vindoline (7)
- Preparation of 17-(O-5-bromopentanoyl)vindoline (8)
- Preparation of 17-(O-4-(chloromethyl)benzoyl)vindoline (11)

- Preparation of 10-aminovindoline (14)
- Preparation of 10-chloroacetamidovindoline (15)
3.2.2. Synthesis of Vindoline–TPP Phosphonium Salts (9a–g, 10a–d, 12, and 16)
- Preparation of product 9a

- Preparation of product 9b
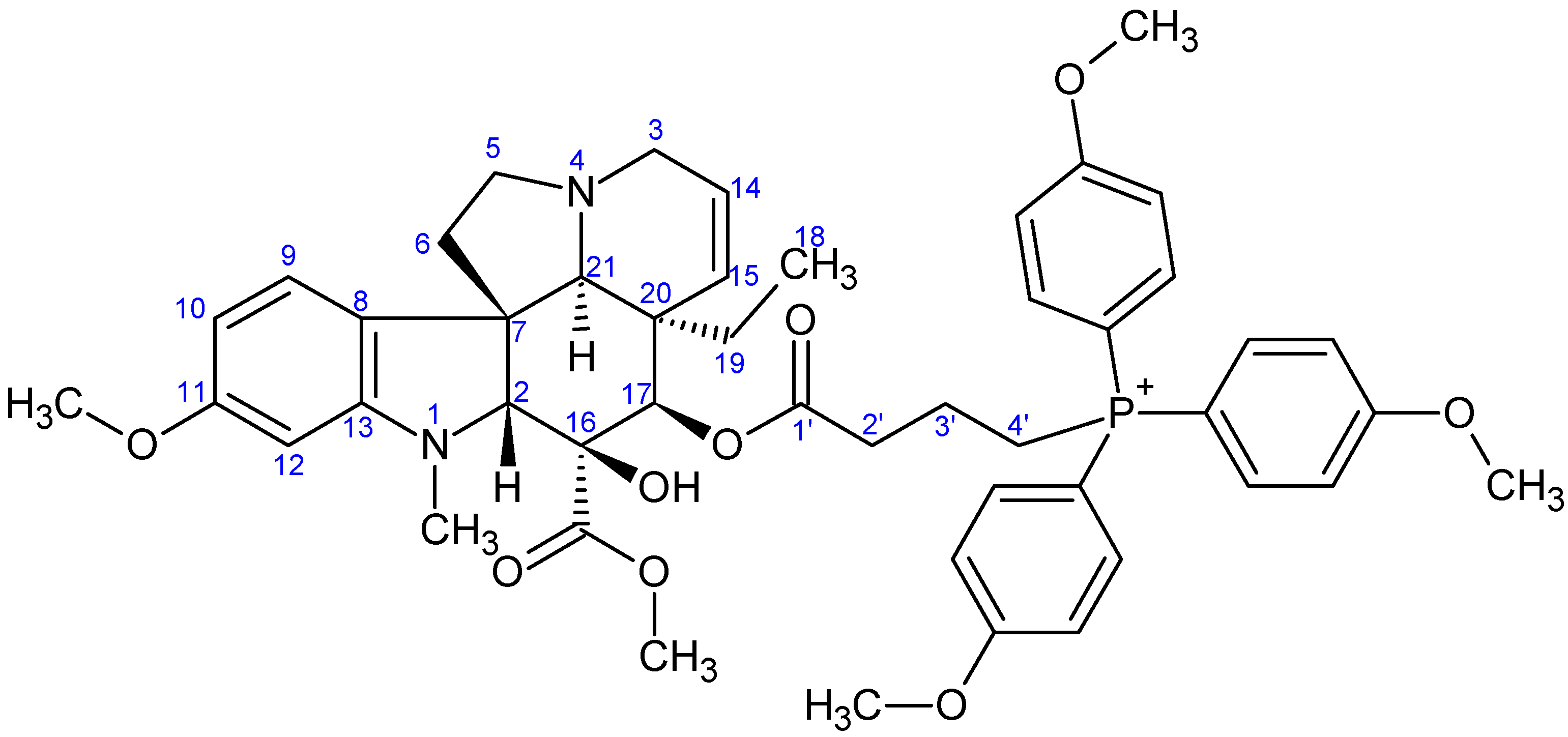
- Preparation of product 9c

- Preparation of product 9d
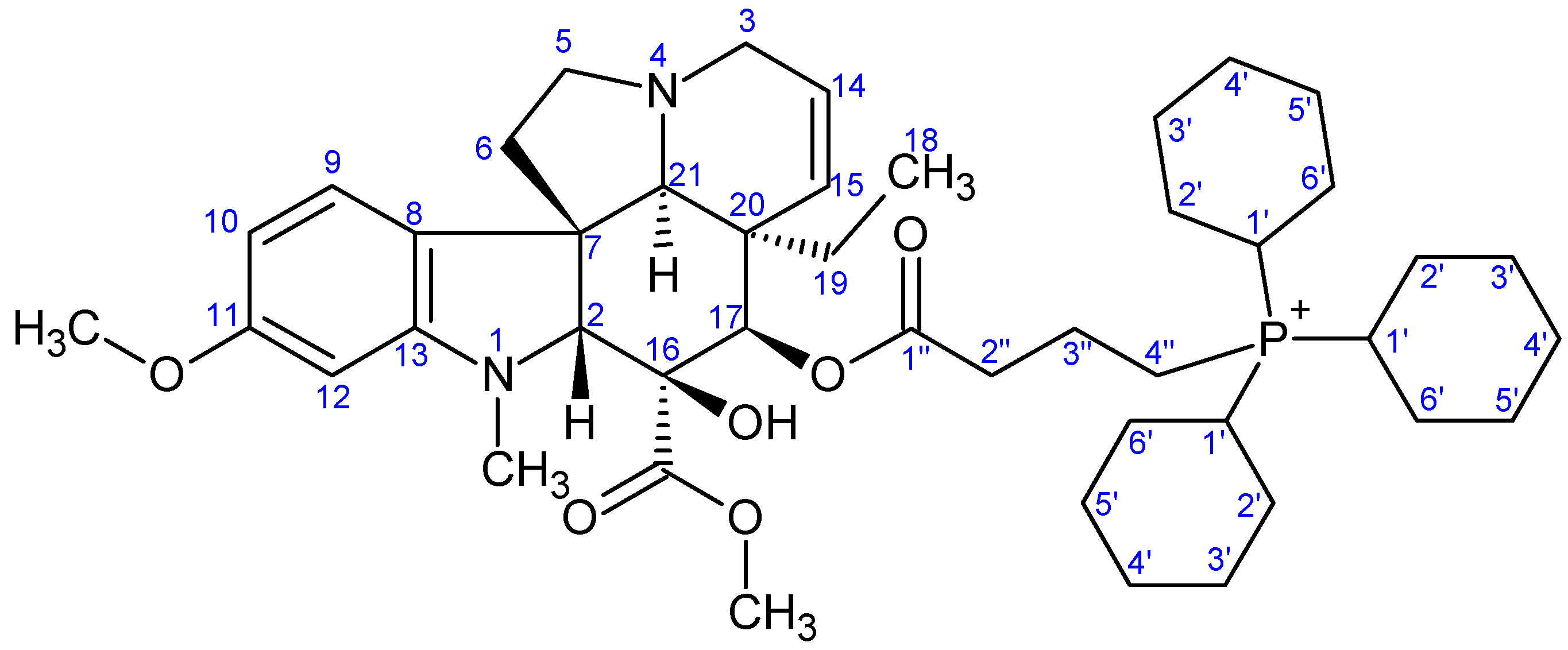
- Preparation of product 9e

- Preparation of product 9f
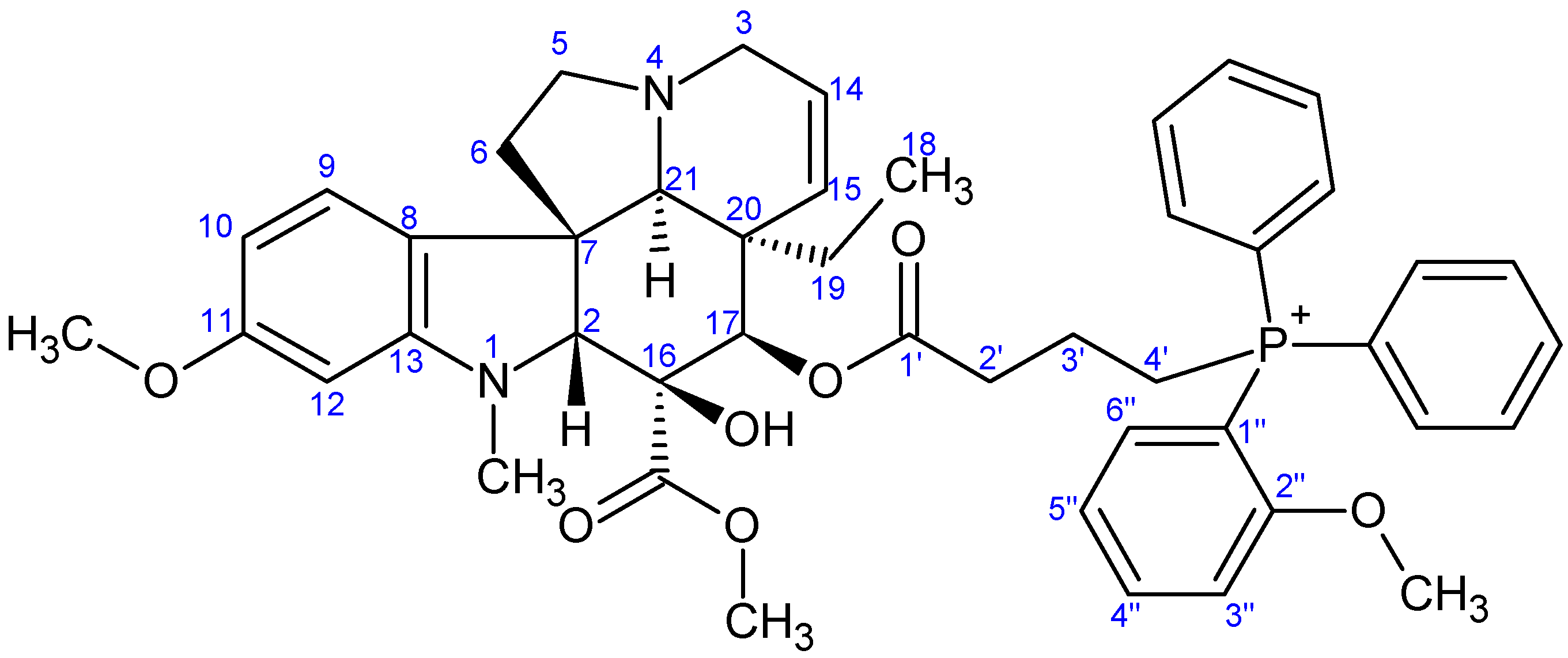
- Preparation of product 9g

- Preparation of product 10a

- Preparation of product 10b
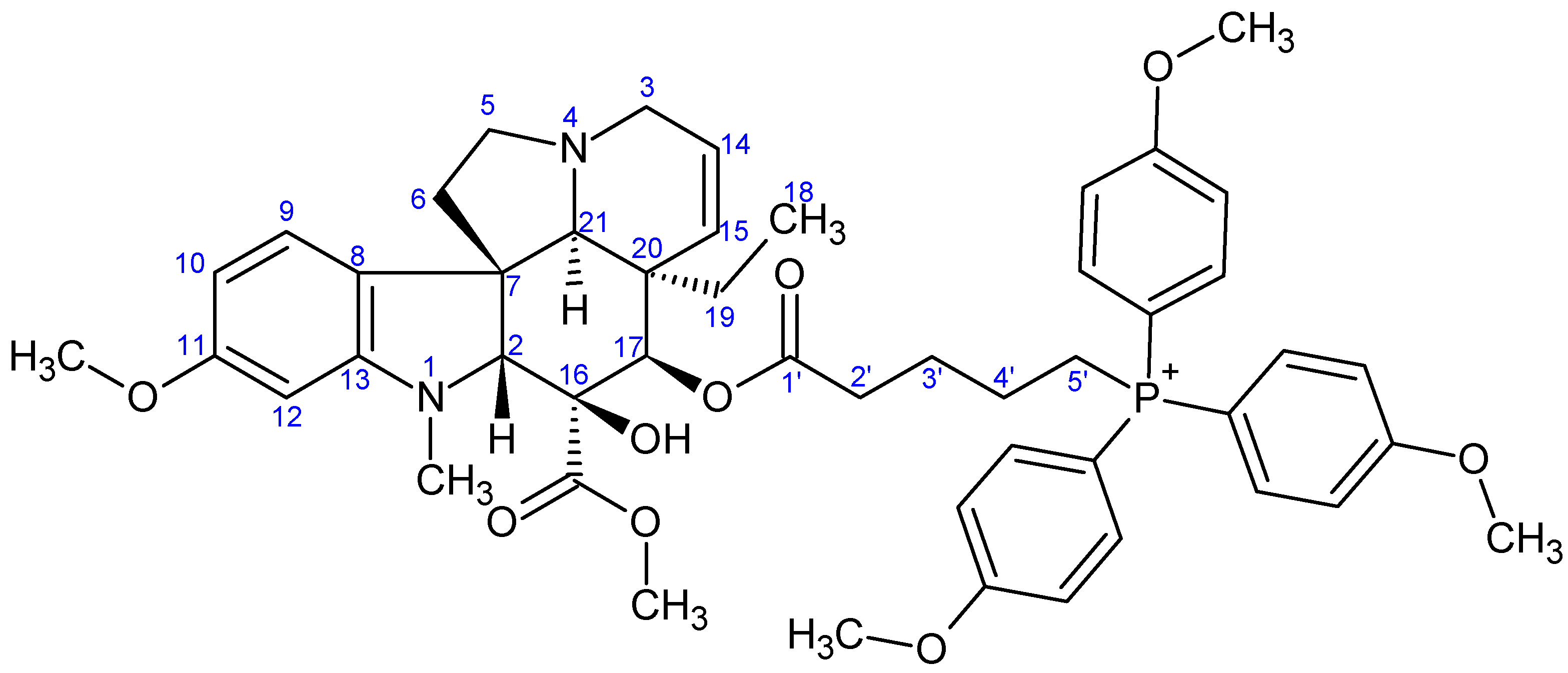
- Preparation of product 10c
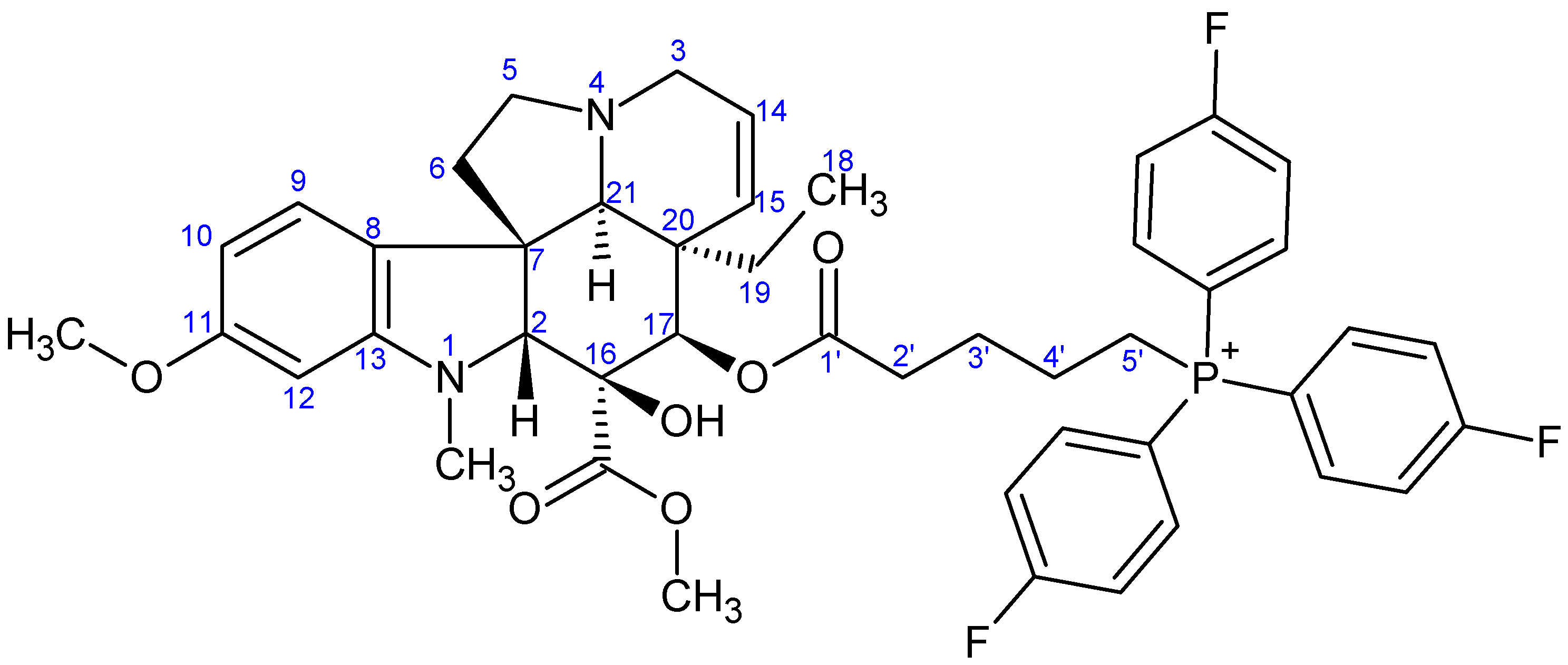
- Preparation of product 10d
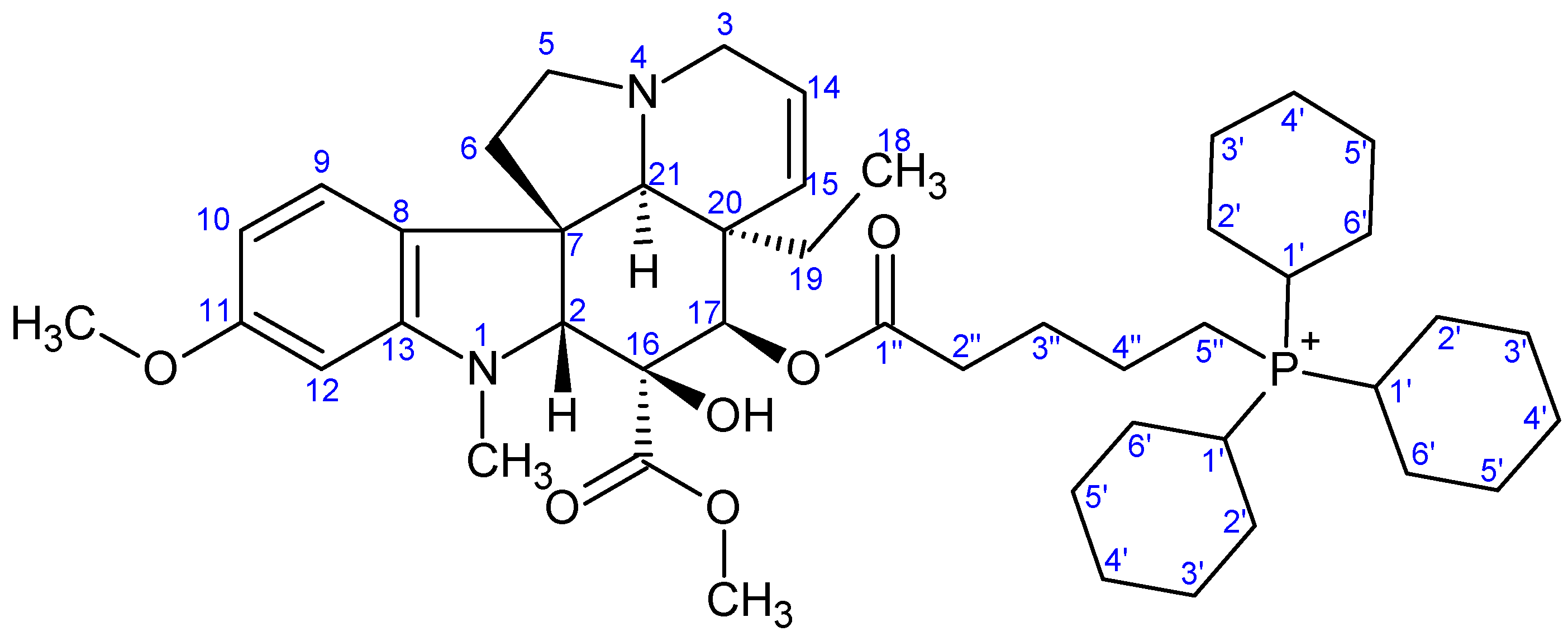
- Preparation of product 12
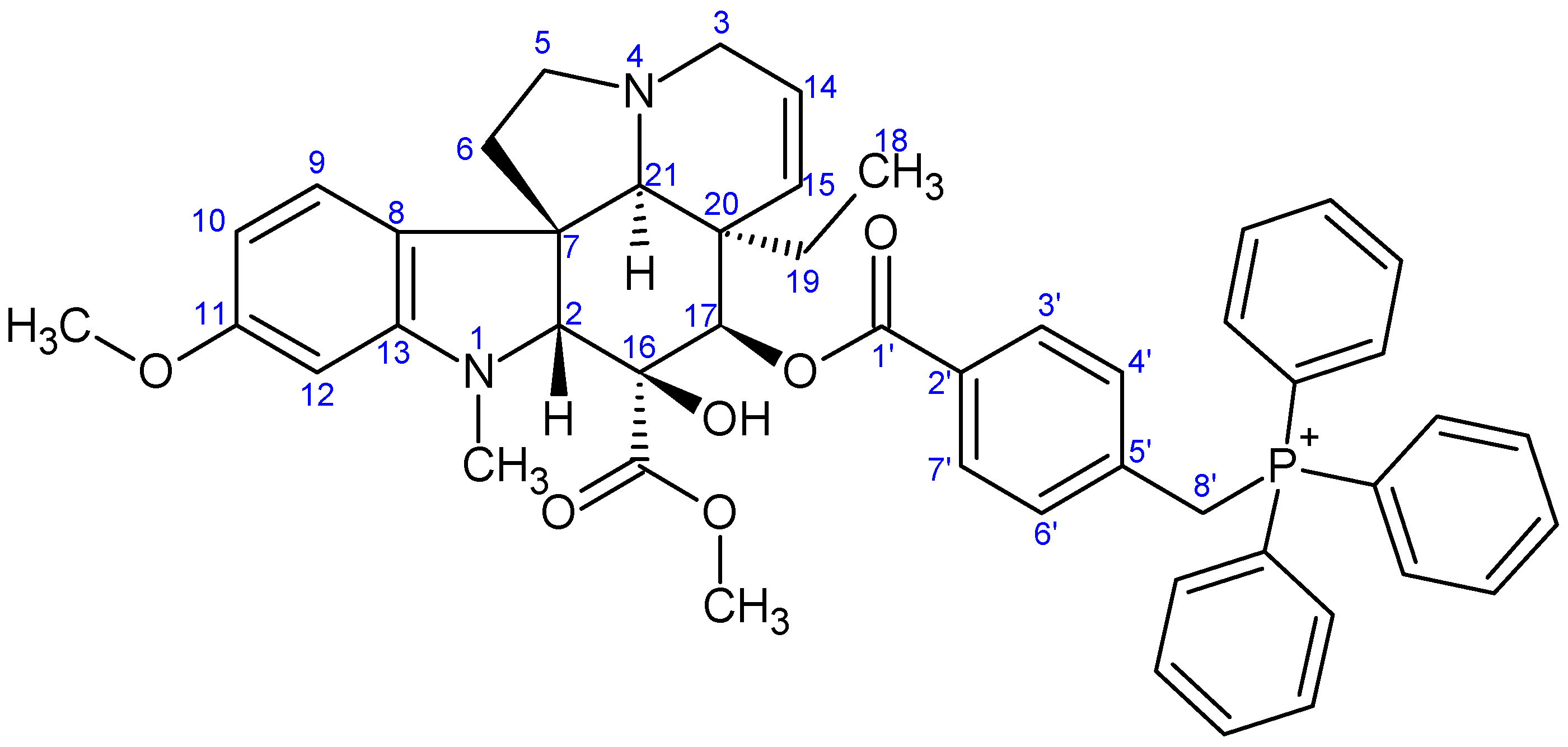
- Preparation of product 16
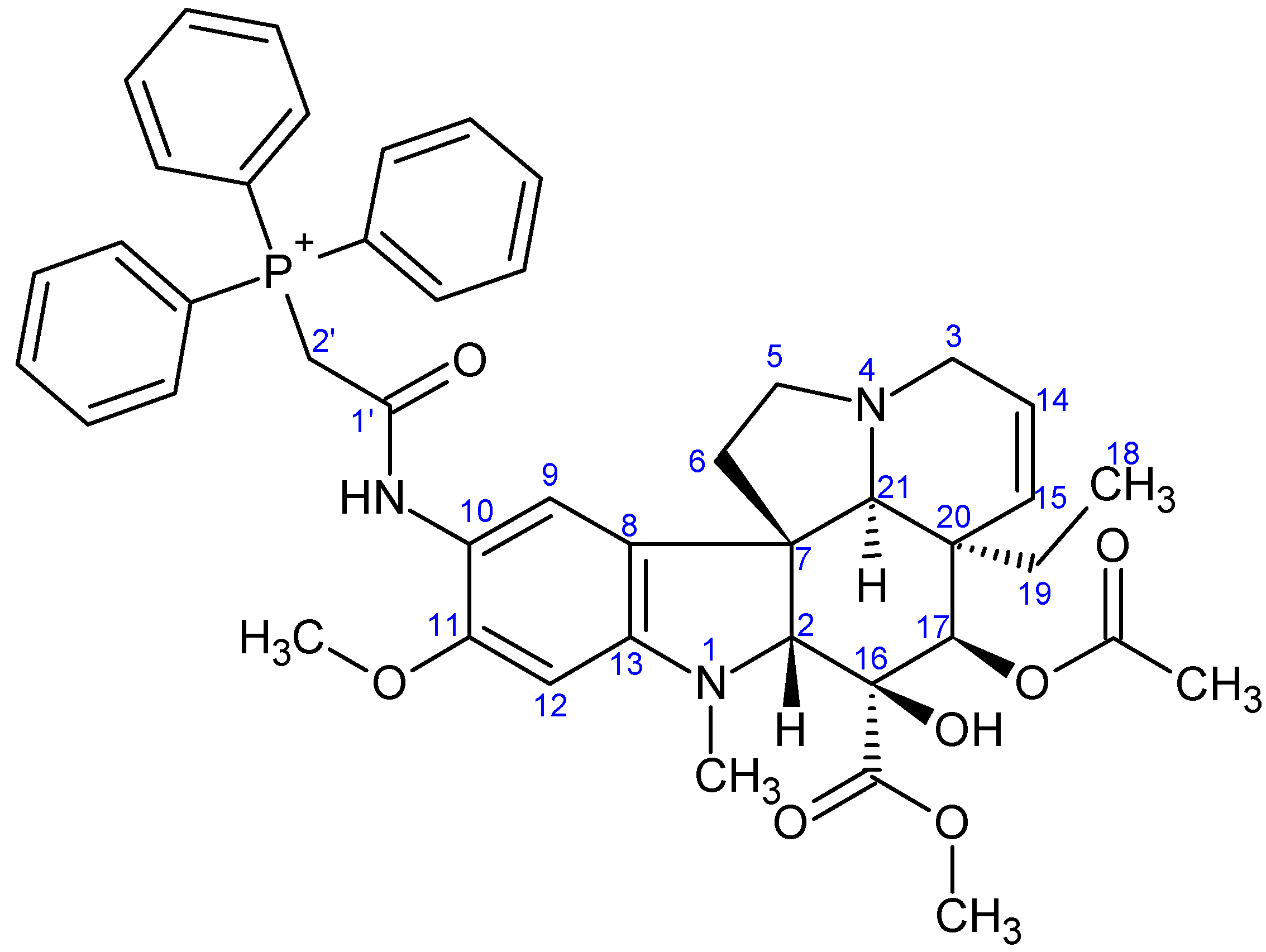
3.2.3. Synthesis of 17-(O-3,3,3-Triphenylpropanoyl)vindoline (13)
3.3. Biological Evaluation
3.3.1. NCI60 Screening
3.3.2. Determination of Antiproliferative Activities on Adherent Cell Lines
3.3.3. Cell Cycle Analysis
3.3.4. Migration Assay
3.3.5. CellTiter-Glo Luminescent Cell Viability Assay on Non-Tumor CHO Cells
3.3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miyajima, Y.; Ochiai, K.; Fujii, S. Design, Synthesis, and Evaluation of B-(Trifluoromethyl)phenyl Phosphine–Borane Derivatives as Novel Progesterone Receptor Antagonists. Molecules 2024, 29, 1587. [Google Scholar] [CrossRef] [PubMed]
- Demkowicz, S.; Rachon, J.; Dasko, M.; Kozak, W. Selected organophosphorus compounds with biological activity. Appl. Med. RSC Adv. 2016, 6, 7101–7112. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Chiral Organophosphorus Pharmaceuticals: Properties and Application. Symmetry 2023, 15, 1550. [Google Scholar] [CrossRef]
- Aroniadou-Anderjaska, V.; Figueiredo, T.H.; de Araujo Furtado, M.; Pidoplichko, V.I.; Braga, M.F.M. Mechanisms of Organophosphate Toxicity and the Role of Acetylcholinesterase Inhibition. Toxics 2023, 11, 866. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Advances in the Synthesis and Analysis of Biologically Active Phosphometabolites. Int. J. Mol. Sci. 2023, 24, 3150. [Google Scholar] [CrossRef]
- Voráčová, M.; Zore, M.; Yli-Kauhaluoma, J.; Kiuru, P. Harvesting phosphorus-containing moieties for their antibacterial effects. Bioorg. Med. Chem. 2023, 96, 117512. [Google Scholar] [CrossRef]
- Mdeni, N.L.; Adeniji, A.O.; Okoh, A.I.; Okoh, O.O. Analytical Evaluation of Carbamate and Organophosphate Pesticides in Human and Environmental Matrices: A Review. Molecules 2022, 27, 618. [Google Scholar] [CrossRef]
- Pundir, C.S.; Malik, A. Preety Bio-sensing of organophosphorus pesticides: A review. Biosens. Bioelectron. 2019, 140, 111348. [Google Scholar] [CrossRef]
- Camacho-Pérez, M.R.; Covantes-Rosales, C.E.; Toledo-Ibarra, G.A.; Mercado-Salgado, U.; Ponce-Regalado, M.D.; Díaz-Resendiz, K.J.G.; Girón-Pérez, M.I. Organophosphorus Pesticides as Modulating Substances of Inflammation through the Cholinergic Pathway. Int. J. Mol. Sci. 2022, 23, 4523. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry 2021, 13, 889. [Google Scholar] [CrossRef]
- Elahmer, N.R.; Wong, S.K.; Mohamed, N.; Alias, E.; Chin, K.-Y.; Muhammad, N. Mechanistic Insights and Therapeutic Strategies in Osteoporosis: A Comprehensive Review. Biomedicines 2024, 12, 1635. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. Oral bisphosphonates in the treatment of osteoporosis: A review. Curr. Ther. Res. 1995, 56, 831–851. [Google Scholar] [CrossRef]
- Mbese, Z.; Aderibigbe, B.A. Bisphosphonate-Based Conjugates and Derivatives as Potential Therapeutic Agents in Osteoporosis, Bone Cancer and Metastatic Bone Cancer. Int. J. Mol. Sci. 2021, 22, 6869. [Google Scholar] [CrossRef] [PubMed]
- Hudson, H.R.; Keglevich, G. The Preparation and Anticancer Activity of Some Phosphorus Heterocycles. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 2256–2261. [Google Scholar] [CrossRef]
- Grymel, M.; Lalik, A.; Kazek-Kęsik, A.; Szewczyk, M.; Grabiec, P.; Erfurt, K. Design, Synthesis and Preliminary Evaluation of the Cytotoxicity and Antibacterial Activity of Novel Triphenylphosphonium Derivatives of Betulin. Molecules 2022, 27, 5156. [Google Scholar] [CrossRef]
- Baren, M.H.; Ibrahim, S.A.; Al-Rooqi, M.M.; Ahmed, S.A.; El-Gamil, M.M.; Hekal, H.A. A new class of anticancer activity with computational studies for a novel bioactive aminophosphonates based on pyrazole moiety. Sci. Rep. 2023, 13, 14680. [Google Scholar] [CrossRef]
- Tripolszky, A.; Tóth, E.; Szabó, P.T.; Hackler, L., Jr.; Kari, B.; Puskás, L.G.; Bálint, E. Synthesis and In Vitro Cytotoxicity and Antibacterial Activity of Novel 1,2,3-Triazol-5-yl-Phosphonates. Molecules 2020, 25, 2643. [Google Scholar] [CrossRef]
- Caminade, A.-M. Phosphorus Dendrimers as Nanotools against Cancers. Molecules 2020, 25, 3333. [Google Scholar] [CrossRef]
- Mai, N.N.H.; Yamaguchi, Y.; Choijookhuu, N.; Matsumoto, J.; Nanashima, A.; Takagi, H.; Sato, K.; Tuan, L.Q.; Hishikawa, Y. Photodynamic Therapy Using a Novel Phosphorus Tetraphenylporphyrin Induces an Anticancer Effect via Bax/Bcl-xL-related Mitochondrial Apoptosis in Biliary Cancer Cells. Acta Histochem. Cytochem. 2020, 53, 61–72. [Google Scholar] [CrossRef]
- Li, S.; Zhao, J.; Guo, Y.; Mei, Y.; Yuan, B.; Gan, N.; Zhang, J.; Hu, J.; Hou, H. Influence of the Introduction of a Triphenylphosphine Group on the Anticancer Activity of a Copper Complex. J. Inorg. Biochem. 2020, 210, 111102. [Google Scholar] [CrossRef]
- Cheng, X.; Feng, D.; Lv, J.; Cui, X.; Wang, Y.; Wang, Q.; Zhang, L. Application Prospects of Triphenylphosphine-Based Mitochondria-Targeted Cancer Therapy. Cancers 2023, 15, 666. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Vakili, M.R.; Abyaneh, H.S.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial Delivery of Doxorubicin via Triphenylphosphine Modification for Overcoming Drug Resistance in MDA-MB-435/DOX Cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Leite, C.M.; Araujo-Neto, J.H.; Guedes, A.P.M.; Costa, A.R.; Demidoff, F.C.; Netto, C.D.; Castellano, E.E.; Nascimento, O.R.; Batista, A.A. Copper(I)/Triphenylphosphine Complexes Containing Naphthoquinone Ligands as Potential Anticancer Agents. Inorganics 2023, 11, 367. [Google Scholar] [CrossRef]
- Ye, L.; Yao, Q.; Xu, F.; He, L.; Ding, J.; Xiao, R.; Ding, L.; Luo, B. Preparation and antitumor activity of triphenylphosphine-based mitochondrial targeting polylactic acid nanoparticles loaded with 7-hydroxyl coumarin. J. Biomater. Appl. 2022, 36, 1064–1075. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Honig, B.H.; Hubbell, W.L.; Flewelling, R.F. Electrostatic interactions in membranes and proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 163–193. [Google Scholar] [CrossRef]
- Rideout, D.C.; Calogeropoulou, T.; Jaworski, J.S.; Dagnino, R.J.; McCarthy, M.R. Phosphonium Salts Exhibiting Selective Anti-Carcinoma Activity in Vitro. Anti-Cancer Drug Des. 1989, 4, 265–280. [Google Scholar]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective Targeting of an Antioxidant to Mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, M.I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Dubinin, M.V.; Belosludtsev, K.N. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Dis. 2013, 8, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef]
- Keglevich, A.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Keglevich, P.; Hazai, L. Synthesis of Vinca Alkaloid–Triphenylphosphine Derivatives Having Potential Antitumor Effect. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 606–609. [Google Scholar] [CrossRef]
- Keglevich, A.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Keglevich, P.; Hazai, L. Synthesis and in vitro Antitumor Effect of New Vindoline Derivatives Coupled with Triphenylphosphine. Curr. Org. Chem. 2019, 23, 852–858. [Google Scholar] [CrossRef]
- Passarella, D.; Giardini, A.; Peretto, B.; Fontana, G.; Sacchetti, A.; Silvani, A.; Ronchi, C.; Cappelletti, G.; Cartelli, D.; Borlak, J.; et al. Inhibitors of tubulin polymerization: Synthesis and biological evaluation of hybrids of vindoline, anhydrovinblastine and vinorelbine with thiocolchicine, podophyllotoxin and baccatin III. Bioorg. Med. Chem. 2008, 16, 6269–6285. [Google Scholar] [CrossRef]
- Keglevich, A.; Zsiros, V.; Keglevich, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Mernyák, E.; Wölfling, J.; Hazai, L. Synthesis and In Vitro Antitumor Effect of New Vindoline-steroid Hybrids. Curr. Org. Chem. 2019, 23, 959–967. [Google Scholar] [CrossRef]
- Mayer, S.; Keglevich, A.; Sepsey Für, C.; Bölcskei, H.; Ilkei, V.; Keglevich, P.; Hazai, L. Results in Chemistry of Natural Organic Compounds. Synthesis of New Anticancer Vinca Alkaloids and Flavone Alkaloids. Chemistry 2020, 2, 714–726. [Google Scholar] [CrossRef]
- Mayer, S.; Nagy, N.; Keglevich, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Hazai, L. Synthesis of Novel Vindoline-Chrysin Hybrids. Chem. Biodivers. 2022, 19, e2100725. [Google Scholar] [CrossRef]
- Mayer, S.; Keglevich, P.; Keglevich, A.; Hazai, L. New Anticancer Vinca Alkaloids in the Last Decade—A Mini-Review. Curr. Org. Chem. 2021, 25, 1224–1234. [Google Scholar] [CrossRef]
- Zsoldos, B.; Nagy, N.; Donkó-Tóth, V.; Keglevich, P.; Weber, M.; Dékány, M.; Nehr-Majoros, A.; Szőke, É.; Helyes, Z.; Hazai, L. Novel Piperazine Derivatives of Vindoline as Anticancer Agents. Int. J. Mol. Sci. 2024, 25, 7929. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.H.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.M.; Hursey, L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Shoemaker, R.H.; Monks, A.; Alley, M.C.; Scudiero, D.A.; Fine, D.L.; McLemore, T.L.; Abbott, B.J.; Paull, K.D.; Mayo, J.G.; Boyd, M.R. Development of Human Tumor Cell Line Panels for Use in Disease-Oriented Drug Screening. Prog. Clin. Biol. Res. 1988, 276, 265–286. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesh, H.; Kennedy, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- NCI-60 Screening Methodology. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 1 April 2025).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Reutelingsperger, C. Flow cytometry of apoptotic cell death. J. Immunol. Methods 2000, 243, 167–190. [Google Scholar] [CrossRef]
- Wang, D.; Shen, M.; Kitamura, N.; Sennari, Y.; Morita, K.; Tsukada, J.; Kanazawa, T.; Yoshida, Y. Mitogen-activated protein kinases are involved in cucurbitacin D-induced antitumor effects on adult T-cell leukemia cells. Investig. New Drugs 2021, 39, 122–130. [Google Scholar] [CrossRef]
- Nehr-Majoros, A.K.; Erostyák, J.; Fenyvesi, É.; Szabó-Meleg, E.; Szőcs, L.; Sétáló, G., Jr.; Helyes, Z.; Szőke, É. Cyclodextrin derivatives decrease Transient Receptor Potential vanilloid 1 and Ankyrin 1 ion channel activation via altering the surrounding membrane microenvironment by cholesterol depletion. Front. Cell Dev. Biol. 2024, 12, 1334130. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 9a | 9b | 9c | 9d | 9e | 9f | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Panel | Cell Line | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean |
| Leukemia | CCRF-CEM | 0.487 | 0.36 | 0.359 | 0.26 | 0.887 | 0.55 | 12.70 | 7.25 | 1.690 | 1.20 | 0.114 | 0.11 | 0.256 | 0.22 |
| HL-60(TB) | 0.124 | 0.162 | 0.256 | 1.770 | 0.322 | 0.039 | 0.170 | ||||||||
| K-562 | 0.360 | 0.291 | 0.556 | 9.830 | 1.320 | 0.195 | 0.125 | ||||||||
| MOLT-4 | 0.614 | 0.294 | 0.933 | 9.690 | 2.160 | 0.164 | 0.419 | ||||||||
| RPMI-8226 | 0.219 | 0.186 | 0.275 | 2.050 | 0.383 | 0.020 | 0.106 | ||||||||
| SR | 0.346 | 0.281 | 0.389 | 7.450 | 1.350 | 0.144 | 0.221 | ||||||||
| Non-small Cell Lung Cancer | A549/ATCC | 1.350 | 1.11 | 0.334 | 0.41 | 0.731 | 1.09 | 21.50 | 19.58 | 3.460 | 3.67 | 0.275 | 0.23 | 0.945 | 1.05 |
| EKVX | 0.470 | 0.534 | 1.060 | 19.40 | 3.090 | n.d. | 1.220 | ||||||||
| HOP-62 | 1.370 | 0.470 | 1.660 | 20.50 | 3.680 | 0.173 | 2.160 | ||||||||
| HOP-92 | 0.066 | 0.187 | 0.344 | 5.440 | 1.010 | 0.201 | 0.172 | ||||||||
| NCI-H226 | 2.040 | 0.521 | 1.430 | 24.20 | 4.060 | 0.230 | 1.210 | ||||||||
| NCI-H23 | 0.971 | 0.303 | 0.671 | 15.80 | 2.310 | 0.183 | 0.413 | ||||||||
| NCI-H322M | 2.330 | 0.785 | 2.860 | >48.5 | 5.790 | 0.342 | 2.230 | ||||||||
| NCI-H460 | 1.220 | 0.327 | 0.666 | 17.00 | 2.770 | 0.229 | 0.774 | ||||||||
| NCI-H522 | 0.216 | 0.258 | 0.391 | 3.880 | 6.880 | 0.198 | 0.339 | ||||||||
| Colon Cancer | COLO 205 | 0.161 | 1.69 | 0.176 | 0.51 | 0.194 | 0.98 | 4.980 | 16.80 | 0.430 | 3.82 | 0.091 | 0.24 | 0.179 | 1.61 |
| HCC-2998 | 0.436 | 0.415 | 0.498 | 9.530 | 1.700 | 0.195 | 0.325 | ||||||||
| HCT-116 | 0.965 | 0.307 | 0.712 | 14.80 | 2.850 | 0.223 | 0.383 | ||||||||
| HCT-15 | 8.500 | 1.620 | 3.640 | >48.5 | 15.30 | 0.760 | 9.070 | ||||||||
| HT29 | 0.266 | 0.219 | 0.371 | 12.50 | 1.880 | 0.151 | 0.356 | ||||||||
| KM12 | 0.297 | 0.376 | 0.628 | 8.960 | 1.630 | 0.047 | 0.331 | ||||||||
| SW-620 | 1.200 | 0.434 | 0.844 | 18.30 | 2.950 | 0.245 | 0.637 | ||||||||
| CNS Cancer | SF-268 | 0.975 | 0.76 | 0.343 | 0.36 | 1.210 | 1.02 | 15.00 | 20.21 | 3.230 | 2.71 | 0.231 | 0.18 | 0.372 | 0.57 |
| SF-295 | 1.450 | 0.521 | 1.920 | >48.5 | 4.410 | 0.213 | 1.830 | ||||||||
| SF-539 | 0.662 | 0.338 | 1.230 | 16.80 | 2.940 | 0.307 | 0.434 | ||||||||
| SNB-19 | 0.334 | 0.306 | 0.391 | 11.40 | 2.220 | 0.059 | 0.313 | ||||||||
| SNB-75 | 0.750 | n.d. | n.d. | n.d. | n.d. | 0.195 | 0.197 | ||||||||
| U251 | 0.371 | 0.291 | 0.360 | 9.360 | 0.746 | 0.054 | 0.248 | ||||||||
| Melanoma | LOX IMVI | 0.858 | 0.45 | 0.333 | 0.35 | 0.614 | 0.76 | 16.60 | 8.13 | 2.880 | 1.70 | 0.145 | 0.18 | 0.429 | 0.49 |
| MALME-3M | 0.283 | 0.220 | 0.624 | 6.850 | 1.460 | 0.288 | 0.295 | ||||||||
| M14 | 0.790 | 0.456 | 1.120 | 9.100 | 1.920 | 0.299 | 1.060 | ||||||||
| MDA-MB-435 | 0.278 | 0.331 | 0.480 | 8.140 | 1.830 | 0.231 | 0.299 | ||||||||
| SK-MEL-2 | 0.322 | n.d. | n.d. | n.d. | n.d. | 0.060 | 0.322 | ||||||||
| SK-MEL-28 | 0.261 | 0.361 | 0.453 | 5.340 | 1.080 | 0.272 | 0.506 | ||||||||
| SK-MEL-5 | 0.766 | 0.532 | 1.550 | 8.260 | 1.760 | 0.138 | 0.777 | ||||||||
| UACC-257 | 0.240 | 0.272 | 0.646 | 4.670 | 1.260 | 0.038 | 0.406 | ||||||||
| UACC-62 | 0.222 | 0.275 | 0.603 | 6.050 | 1.410 | 0.160 | 0.286 | ||||||||
| Ovarian Cancer | IGROV1 | 0.237 | 7.78 | 0.383 | 1.57 | 0.533 | 3.46 | 7.090 | 19.04 | 1.390 | 7.12 | 0.193 | 0.94 | 0.510 | 4.20 |
| OVCAR-3 | 0.369 | 0.374 | 0.624 | 6.470 | 1.740 | 0.147 | 0.297 | ||||||||
| OVCAR-4 | 0.469 | 0.370 | 0.760 | 16.20 | 2.390 | 0.271 | 0.540 | ||||||||
| OVCAR-5 | 1.200 | 0.331 | 1.770 | 20.80 | 3.190 | 0.339 | 0.735 | ||||||||
| OVCAR-8 | 1.270 | 0.372 | 1.413 | 18.40 | 2.970 | 0.300 | 0.790 | ||||||||
| NCI/ADR-RES | >50.0 | 8.870 | 18.10 | >48.5 | 35.60 | 5.110 | 26.10 | ||||||||
| SK-OV-3 | 0.894 | 0.290 | 1.023 | 15.80 | 2.590 | 0.232 | 0.411 | ||||||||
| Renal Cancer | 786-0 | 2.740 | 3.52 | 1.060 | 1.05 | 1.930 | 3.01 | >48.5 | 34.21 | 4.450 | 7.93 | 0.239 | 0.44 | 1.980 | 3.08 |
| A498 | 1.710 | 1.630 | 2.344 | >48.5 | 12.40 | 0.314 | 2.650 | ||||||||
| ACHN | 4.530 | 1.610 | 0.458 | >48.5 | 15.20 | 0.319 | 4.580 | ||||||||
| CAKI-1 | 5.570 | 1.870 | 13.70 | >48.5 | 16.40 | 0.313 | 1.900 | ||||||||
| RXF 393 | 0.705 | 0.279 | 0.482 | 8.460 | 1.470 | n.d. | 0.247 | ||||||||
| SN12C | 0.590 | 0.291 | 0.672 | 14.80 | 2.890 | 0.159 | 0.325 | ||||||||
| TK-10 | 1.480 | 0.600 | 1.514 | 22.20 | 2.730 | 0.277 | 1.940 | ||||||||
| UO-31 | 10.80 | n.d. | n.d. | n.d. | n.d. | 1.480 | 11.00 | ||||||||
| Prostate Cancer | PC-3 | 0.204 | 0.64 | 0.310 | 0.38 | 0.470 | 0.71 | 12.60 | 18.15 | 1.710 | 2.73 | 0.192 | 0.25 | 0.268 | 0.34 |
| DU-145 | 1.070 | 0.457 | 0.955 | 23.70 | 3.750 | 0.300 | 0.418 | ||||||||
| Breast Cancer | MCF7 | 0.306 | 0.45 | 0.281 | 0.27 | 0.383 | 0.64 | 8.260 | 8.04 | 1.190 | 1.54 | 0.044 | 0.14 | 0.288 | 0.31 |
| MDA-MB231ATCC | 0.406 | n.d. | n.d. | n.d. | n.d. | 0.293 | 0.397 | ||||||||
| HS 578T | 1.260 | 0.344 | 1.180 | 11.80 | 2.450 | 0.185 | 0.473 | ||||||||
| BT-549 | 0.380 | 0.247 | 1.050 | 8.130 | 2.440 | 0.170 | 0.262 | ||||||||
| T-47D | 0.192 | 0.314 | 0.353 | 10.500 | 1.320 | 0.105 | 0.302 | ||||||||
| MDA-MB-468 | 0.126 | 0.165 | 0.241 | 1.500 | 0.313 | 0.027 | 0.129 | ||||||||
| Mean | 1.985 | 0.605 | 1.449 | 17.010 | 3.799 | 0.309 | 1.439 | ||||||||
| 4 | 10a | 10b | 10c | 10d | 12 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Panel | Cell Line | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean | GI50 | Mean |
| Leukemia | CCRF-CEM | 0.697 | 0.60 | 0.768 | 0.48 | n.d. | 0.56 | 13.60 | 9.86 | 2.550 | 1.22 | 1.300 | 0.77 |
| HL-60(TB) | n.d. | 0.221 | 0.234 | 2.110 | 0.293 | 0.297 | |||||||
| K-562 | n.d. | 0.654 | 0.675 | 11.30 | 0.983 | 0.448 | |||||||
| MOLT-4 | n.d. | 0.611 | 1.180 | 14.70 | 1.720 | 1.470 | |||||||
| RPMI-8226 | 0.497 | 0.283 | 0.299 | 2.970 | 0.412 | 0.325 | |||||||
| SR | n.d. | 0.360 | 0.428 | 14.50 | 1.390 | n.d. | |||||||
| Non-small Cell Lung Cancer | A549/ATCC | 1.020 | 1.44 | 0.819 | 1.07 | 1.650 | 1.36 | 33.80 | 24.32 | 3.130 | 2.95 | 2.090 | 2.05 |
| EKVX | 0.586 | 1.050 | 0.185 | 21.00 | 3.130 | 2.030 | |||||||
| HOP-62 | 2.600 | 1.370 | 2.420 | 25.50 | 3.900 | 2.670 | |||||||
| HOP-92 | 0.268 | 0.490 | 0.520 | 6.320 | 1.020 | 0.404 | |||||||
| NCI-H226 | 1.930 | 1.430 | 2.030 | 27.30 | 4.410 | 1.540 | |||||||
| NCI-H23 | 1.220 | 0.855 | 1.190 | 20.10 | 2.340 | 2.810 | |||||||
| NCI-H322M | 3.000 | 2.670 | 2.790 | 50.00 | 5.300 | 4.760 | |||||||
| NCI-H460 | 2.110 | 0.600 | 1.150 | 29.40 | 2.710 | 1.660 | |||||||
| NCI-H522 | 0.229 | 0.306 | 0.341 | 5.440 | 0.642 | 0.491 | |||||||
| Colon Cancer | COLO 205 | 0.242 | 3.19 | 0.200 | 0.72 | 0.214 | 1.88 | 4.630 | 26.90 | 0.377 | 3.80 | 0.313 | 4.69 |
| HCC-2998 | 0.367 | 0.520 | 0.617 | 12.50 | 1.310 | 0.991 | |||||||
| HCT-116 | 1.060 | 0.452 | 1.300 | 25.90 | 2.940 | 1.590 | |||||||
| HCT-15 | 18.60 | 2.170 | 8.790 | >100 | 16.10 | 27.40 | |||||||
| HT29 | 0.404 | 0.358 | 0.511 | 15.40 | 1.430 | 0.541 | |||||||
| KM12 | 0.442 | 0.596 | 0.675 | 13.40 | 1.720 | 0.514 | |||||||
| SW-620 | 1.180 | 0.761 | 1.060 | 16.50 | 2.750 | 1.500 | |||||||
| CNS Cancer | SF-268 | 0.634 | 0.94 | 1.170 | 1.09 | 1.470 | 1.17 | 17.60 | 18.76 | 3.170 | 2.60 | 1.280 | 1.32 |
| SF-295 | 2.750 | 1.620 | 1.970 | 30.60 | 4.400 | 2.920 | |||||||
| SF-539 | 1.300 | 1.220 | 1.520 | 21.90 | 2.850 | 1.260 | |||||||
| SNB-19 | 0.397 | 0.960 | 0.484 | 13.00 | 1.920 | 0.725 | |||||||
| SNB-75 | 0.221 | n.d. | n.d. | n.d. | n.d. | 1.110 | |||||||
| U251 | 0.312 | 0.470 | 0.392 | 10.70 | 0.640 | 0.644 | |||||||
| Melanoma | LOX IMVI | 1.170 | 0.75 | 0.797 | 0.92 | 1.110 | 0.98 | 16.80 | 12.52 | 2.900 | 1.96 | 1.820 | 1.03 |
| MALME-3M | 0.273 | 1.170 | 1.130 | 13.50 | 1.630 | 0.652 | |||||||
| M14 | 1.790 | 1.110 | 1.200 | 14.70 | 2.200 | 1.350 | |||||||
| MDA-MB-435 | 0.357 | 0.678 | 0.521 | 12.90 | 2.620 | 0.483 | |||||||
| SK-MEL-2 | 0.279 | n.d. | n.d. | n.d. | n.d. | 1.270 | |||||||
| SK-MEL-28 | 0.532 | 0.485 | 0.486 | 8.280 | 1.380 | 0.520 | |||||||
| SK-MEL-5 | 1.710 | 1.570 | 1.600 | 17.10 | 1.880 | 1.210 | |||||||
| UACC-257 | 0.215 | 0.817 | 0.792 | 5.590 | 1.570 | 0.918 | |||||||
| UACC-62 | 0.403 | 0.705 | 0.963 | 11.30 | 1.530 | 1.030 | |||||||
| Ovarian Cancer | IGROV1 | 0.553 | 15.08 | 0.479 | 2.54 | 0.548 | 4.11 | 10.80 | 32.46 | 1.360 | 11.42 | 0.885 | 15.79 |
| OVCAR-3 | 0.398 | 0.604 | 0.634 | 9.300 | 1.930 | 0.704 | |||||||
| OVCAR-4 | 0.326 | 1.090 | 1.570 | 25.10 | 2.720 | 1.050 | |||||||
| OVCAR-5 | 1.820 | 1.440 | 2.130 | 29.10 | 3.190 | 1.990 | |||||||
| OVCAR-8 | 1.740 | 1.130 | 1.990 | 29.40 | 2.930 | 3.180 | |||||||
| NCI/ADR-RES | >100 | 12.00 | 20.50 | >100 | 65.50 | >100 | |||||||
| SK-OV-3 | 0.725 | 1.040 | 1.400 | 23.50 | 2.280 | 2.700 | |||||||
| Renal Cancer | 786-0 | 2.900 | 4.87 | 1.640 | 2.34 | 2.590 | 4.34 | 34.10 | 56.74 | 5.430 | 8.36 | 3.580 | 8.30 |
| A498 | 2.840 | 2.170 | 2.930 | >100 | 13.30 | 16.40 | |||||||
| ACHN | 14.60 | 2.540 | 6.280 | >100 | 16.00 | 18.50 | |||||||
| CAKI-1 | 2.820 | 7.460 | 15.20 | >100 | 16.70 | 3.260 | |||||||
| RXF 393 | 0.308 | 0.468 | 0.524 | 13.30 | 1.240 | 1.140 | |||||||
| SN12C | 0.467 | 0.619 | 1.180 | 28.60 | 2.770 | 0.708 | |||||||
| TK-10 | 2.820 | 1.490 | 1.670 | 21.20 | 3.080 | 2.840 | |||||||
| UO-31 | 12.20 | n.d. | n.d. | n.d. | n.d. | 20.00 | |||||||
| Prostate Cancer | PC-3 | 0.369 | 0.57 | 0.450 | 0.80 | 0.562 | 8.28 | 16.40 | 25.90 | 1.460 | 2.56 | 0.657 | 1.13 |
| DU-145 | 0.763 | 1.150 | 16.00 | 35.40 | 3.650 | 1.610 | |||||||
| Breast Cancer | MCF7 | 0.322 | 0.50 | 0.325 | 0.58 | 0.105 | 0.70 | 11.50 | 11.08 | 1.050 | 1.48 | 0.442 | 0.61 |
| MDA-MB231ATCC | 0.370 | n.d. | n.d. | n.d. | n.d. | 0.686 | |||||||
| HS 578T | 1.230 | 1.200 | 1.510 | 16.90 | 2.610 | 1.270 | |||||||
| BT-549 | 0.583 | 0.763 | 1.250 | 12.90 | 2.450 | 0.538 | |||||||
| T-47D | 0.303 | 0.370 | 0.383 | 11.50 | 0.920 | 0.525 | |||||||
| MDA-MB-468 | 0.170 | 0.264 | 0.264 | 2.610 | 0.378 | 0.186 | |||||||
| Mean | 3.525 | 1.232 | 2.202 | 24.856 | 4.289 | 4.325 | |||||||
| Cell Line | Conc. (μM) | Growth Inhibition (%) ± SEM | ||
|---|---|---|---|---|
| 9g | 13 | 16 | ||
| HeLa | 10 30 | 61.45 ± 2.35 68.50 ± 2.36 | 39.86 ± 2.57 40.15 ± 1.12 | 21.67 ± 0.84 29.96 ± 0.45 |
| SiHa | 10 30 | 62.46 ± 0.87 83.76 ± 3.07 | >10 * >10 | 14.96 ± 0.34 36.78 ± 3.30 |
| MCF-7 | 10 30 | 81.94 ± 0.84 89.92 ± 0.72 | - 20.82 ± 1.54 | 31.15 ± 3.10 47.13 ± 0.81 |
| MDA-MB-231 | 10 30 | 72.04 ± 0.35 87.71 ± 1.74 | >10 >10 | 12.64 ± 0.03 13.46 ± 0.38 |
| A2780 | 10 30 | 79.37 ± 2.63 85.47 ± 2.25 | >10 11.90 ± 0.85 | 19.42 ± 0.64 29.48 ± 0.56 |
| NIH/3T3 (fibroblast) | 10 30 | 34.63 ± 1.11 77.24 ± 2.35 | 26.12 ± 1.55 22.14 ± 0.67 | 24.95 ± 0.78 27.48 ± 0.90 |
| Cell Line | IC50 Value (μM) ± SEM | |
|---|---|---|
| Selectivity Ratio | ||
| 9g | Cisplatin | |
| HeLa | 6.48 ± 0.12 0.45 | 13.60 ± 0.44 3.23 |
| SiHa | 2.42 ± 0.05 0.17 | 15.50 ± 0.93 3.68 |
| MCF-7 | 2.56 ± 0.10 0.18 | 2.60 ± 0.17 0.62 |
| MDA-MB-231 | 3.35 ± 0.12 0.23 | 19.13 ± 0.12 4.54 |
| A2780 | 1.50 ± 0.20 0.10 | 1.40 ± 0.11 0.33 |
| NIH/3T3 (fibroblast) | 14.37 ± 0.02 | 2.70 ± 0.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halmai, M.; Donkó-Tóth, V.; Keglevich, P.; Kánai, K.; Weber, M.; Dékány, M.; Abdallah, E.A.; Bózsity, N.; Zupkó, I.; Nehr-Majoros, A.; et al. Synthesis and In Vitro Evaluation of the Anticancer Effect of Novel Phosphonium Vindoline Derivatives. Int. J. Mol. Sci. 2025, 26, 3775. https://doi.org/10.3390/ijms26083775
Halmai M, Donkó-Tóth V, Keglevich P, Kánai K, Weber M, Dékány M, Abdallah EA, Bózsity N, Zupkó I, Nehr-Majoros A, et al. Synthesis and In Vitro Evaluation of the Anticancer Effect of Novel Phosphonium Vindoline Derivatives. International Journal of Molecular Sciences. 2025; 26(8):3775. https://doi.org/10.3390/ijms26083775
Chicago/Turabian StyleHalmai, Mónika, Viktória Donkó-Tóth, Péter Keglevich, Károly Kánai, Márton Weber, Miklós Dékány, Ejlal A. Abdallah, Noémi Bózsity, István Zupkó, Andrea Nehr-Majoros, and et al. 2025. "Synthesis and In Vitro Evaluation of the Anticancer Effect of Novel Phosphonium Vindoline Derivatives" International Journal of Molecular Sciences 26, no. 8: 3775. https://doi.org/10.3390/ijms26083775
APA StyleHalmai, M., Donkó-Tóth, V., Keglevich, P., Kánai, K., Weber, M., Dékány, M., Abdallah, E. A., Bózsity, N., Zupkó, I., Nehr-Majoros, A., Szőke, É., Helyes, Z., & Hazai, L. (2025). Synthesis and In Vitro Evaluation of the Anticancer Effect of Novel Phosphonium Vindoline Derivatives. International Journal of Molecular Sciences, 26(8), 3775. https://doi.org/10.3390/ijms26083775