
Current Development of iPSC-Based Modeling in Neurodegenerative Diseases

Abstract
1. Introduction
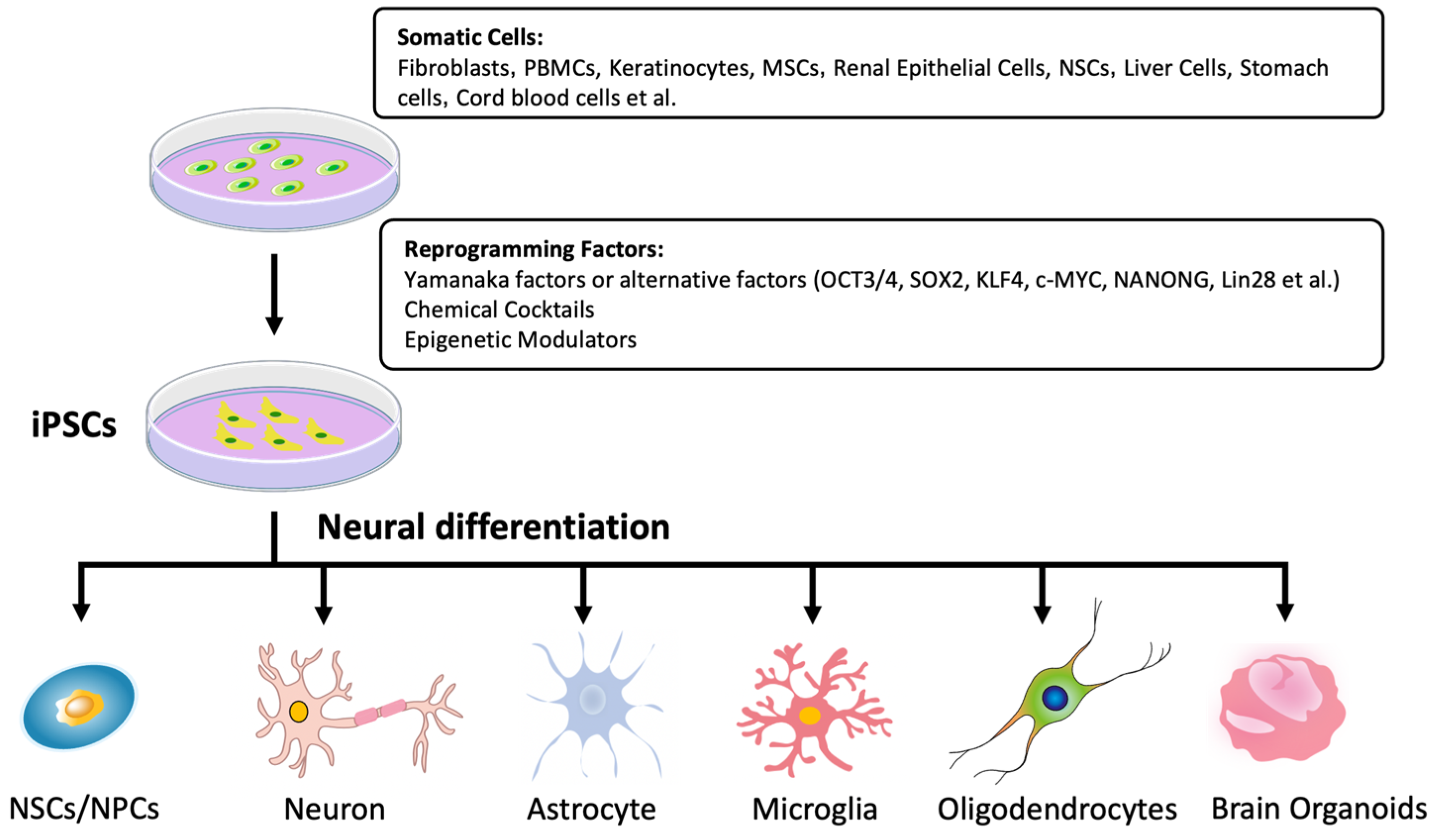
2. Generation of iPSCs
2.1. Delivery Methods
2.2. Reprogramming Factors
2.3. Somatic Cell Sources
3. Neural Differentiation of iPSCs
3.1. Neural Stem Cells
3.2. Neurons
3.3. Astrocytes
3.4. Microglia
3.5. Oligodendrocytes
3.6. Brain Organoids
3.7. Assembloids
4. Neurodegenerative Disease Modeling with Patient-Derived iPSCs
4.1. Alzheimer’s Disease
4.2. Parkinson’s Disease
4.3. Huntington’s Disease
4.4. Rare Neurodegenerative Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef] [PubMed]
- Tabar, V.; Studer, L. Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nat. Rev. Genet. 2014, 15, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, A.E.; Siegert, S.; Tsai, L.-H. Modeling Alzheimer’s Disease with Human Induced Pluripotent Stem (iPS) Cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef]
- Chang, C.Y.; Ting, H.C.; Liu, C.A.; Su, H.L.; Chiou, T.W.; Lin, S.Z.; Harn, H.J.; Ho, T.J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef]
- Akter, M.; Ding, B. Modeling Movement Disorders via Generation of hiPSC-Derived Motor Neurons. Cells 2022, 11, 3796. [Google Scholar] [CrossRef]
- Soubannier, V.; Maussion, G.; Chaineau, M.; Sigutova, V.; Rouleau, G.; Durcan, T.M.; Stifani, S. Characterization of human iPSC-derived astrocytes with potential for disease modeling and drug discovery. Neurosci. Lett. 2020, 731, 135028. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent stem cell-based cell therapy-Promise and challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Prabhu, A.V.; Fernandopulle, M.S.; Patel, R.; Abshari, M.; et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 2019, 104, 239–255.e12. [Google Scholar] [CrossRef]
- Hendriks, D.; Clevers, H.; Artegiani, B. CRISPR-Cas Tools and Their Application in Genetic Engineering of Human Stem Cells and Organoids. Cell Stem Cell 2020, 27, 705–731. [Google Scholar] [CrossRef]
- Haridhasapavalan, K.K.; Borgohain, M.P.; Dey, C.; Saha, B.; Narayan, G.; Kumar, S.; Thummer, R.P. An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 2019, 686, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hamalainen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. PiggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Tsukiyama, T.; Kimura, K.; Matsuyama, S.; Minami, N.; Yamada, M.; Imai, H. Generation of Naïve Bovine Induced Pluripotent Stem Cells Using PiggyBac Transposition of Doxycycline- Inducible Transcription Factors. PLoS ONE 2015, 10, e0135403. [Google Scholar] [CrossRef]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc frome mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Tsubooka, N.; Ichisaka, T.; Okita, K.; Takahashi, K.; Nakagawa, M.; Yamanaka, S. Roles of Sall4 in the generation of pluripotent stem cells from blastocysts and fibroblasts. Genes Cells 2009, 14, 683–694. [Google Scholar] [CrossRef]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef]
- Heng, J.C.; Feng, B.; Han, J.; Jiang, J.; Kraus, P.; Ng, J.H.; Orlov, Y.L.; Huss, M.; Yang, L.; Lufkin, T.; et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 2010, 6, 167–174. [Google Scholar] [CrossRef]
- Yu, J.; Chau, K.F.; Vodyanik, M.A.; Jiang, J.; Jiang, Y. Efficient feeder-free episomal reprogramming with small molecules. PLoS ONE 2011, 6, e17557. [Google Scholar] [CrossRef]
- Li, D.; Wang, L.; Hou, J.; Shen, Q.; Chen, Q.; Wang, X.; Du, J.; Cai, X.; Shan, Y.; Zhang, T.; et al. Optimized Approaches for Generation of Integration-free iPSCs from Human Urine-Derived Cells with Small Molecules and Autologous Feeder. Stem Cell Rep. 2016, 6, 717–728. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yamaguchi, T.; Hamanaka, S.; Kato-Itoh, M.; Yamazaki, Y.; Ibata, M.; Sato, H.; Lee, Y.S.; Usui, J.; Knisely, A.S.; et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010, 142, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.A.; Conrad, J.V.; Rusteika, M.; Chu, L.F. The progress of induced pluripotent stem cells derived from pigs: A mini review of recent advances. Front. Cell Dev. Biol. 2024, 12, 1371240. [Google Scholar] [CrossRef]
- Harding, J.; Roberts, R.M.; Mirochnitchenko, O. Large animal models for stem cell therapy. Stem Cell Res. Ther. 2013, 4, 23. [Google Scholar] [CrossRef]
- Honda, A.; Hirose, M.; Hatori, M.; Matoba, S.; Miyoshi, H.; Inoue, K.; Ogura, A. Generation of induced pluripotent stem cells in rabbits: Potential experimental models for human regenerative medicine. J. Biol. Chem. 2010, 285, 31362–31369. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, F.; Yong, J.; Zhang, P.; Hou, P.; Li, H.; Jiang, W.; Cai, J.; Liu, M.; Cui, K.; et al. Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell 2008, 3, 587–590. [Google Scholar] [CrossRef]
- Nagy, K.; Sung, H.K.; Zhang, P.; Laflamme, S.; Vincent, P.; Agha-Mohammadi, S.; Woltjen, K.; Monetti, C.; Michael, I.P.; Smith, L.C.; et al. Induced pluripotent stem cell lines derived from equine fibroblasts. Stem Cell Rev. 2011, 7, 693–702. [Google Scholar] [CrossRef]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014, 768391. [Google Scholar] [CrossRef]
- Ray, A.; Joshi, J.M.; Sundaravadivelu, P.K.; Raina, K.; Lenka, N.; Kaveeshwar, V.; Thummer, R.P. An Overview on Promising Somatic Cell Sources Utilized for the Efficient Generation of Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2021, 17, 1954–1974. [Google Scholar] [CrossRef]
- Cerneckis, J.; Cai, H.; Shi, Y. Induced pluripotent stem cells (iPSCs): Molecular mechanisms of induction and applications. Signal Transduct. Target. Ther. 2024, 9, 112. [Google Scholar] [CrossRef]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef]
- Merling, R.K.; Sweeney, C.L.; Choi, U.; De Ravin, S.S.; Myers, T.G.; Otaizo-Carrasquero, F.; Pan, J.; Linton, G.; Chen, L.; Koontz, S.; et al. Transgene-free iPSCs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood 2013, 121, e98–e107. [Google Scholar] [CrossRef]
- Mack, A.A.; Kroboth, S.; Rajesh, D.; Wang, W.B. Generation of Induced Pluripotent Stem Cells from CD34+ Cells across Blood Drawn from Multiple Donors with Non-Integrating Episomal Vectors. PLoS ONE 2011, 6, e27956. [Google Scholar] [CrossRef]
- de Leeuw, V.C.; van Oostrom, C.T.M.; Imholz, S.; Piersma, A.H.; Hessel, E.V.S.; Dollé, M.E.T. Going Back and Forth: Episomal Vector Reprogramming of Peripheral Blood Mononuclear Cells to Induced Pluripotent Stem Cells and Subsequent Differentiation into Cardiomyocytes and Neuron-Astrocyte Co-cultures. Cell. Reprogramming 2020, 22, 300–310. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, W.; Ma, K.; Zhang, C.; Fu, X. Reprogramming of Keratinocytes as Donor or Target Cells Holds Great Promise for Cell Therapy and Regenerative Medicine. Stem Cell Rev. Rep. 2019, 15, 680–689. [Google Scholar] [CrossRef]
- Sun, N.; Panetta, N.J.; Gupta, D.M.; Wilson, K.D.; Lee, A.; Jia, F.; Hu, S.; Cherry, A.M.; Robbins, R.C.; Longaker, M.T.; et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15720–15725. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of Induced Pluripotent Stem Cells from Urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef]
- Eminli, S.; Utikal, J.; Arnold, K.; Jaenisch, R.; Hochedlinger, K. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells 2008, 26, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Aoi, T.; Yae, K.; Nakagawa, M.; Ichisaka, T.; Okita, K.; Takahashi, K.; Chiba, T.; Yamanaka, S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 2008, 321, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kumar, N.; Kalsan, M.; Saini, A.; Chandra, R. Mechanism of Induction: Induced Pluripotent Stem Cells (iPSCs). J. Stem Cells 2015, 10, 43–62. [Google Scholar] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Barak, M.; Fedorova, V.; Pospisilova, V.; Raska, J.; Vochyanova, S.; Sedmik, J.; Hribkova, H.; Klimova, H.; Vanova, T.; Bohaciakova, D. Human iPSC-Derived Neural Models for Studying Alzheimer’s Disease: From Neural Stem Cells to Cerebral Organoids. Stem Cell Rev. Rep. 2022, 18, 792–820. [Google Scholar] [CrossRef]
- Elkabetz, Y.; Panagiotakos, G.; Al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef]
- Koch, P.; Opitz, T.; Steinbeck, J.A.; Ladewig, J.; Brüstle, O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. USA 2009, 106, 3225–3230. [Google Scholar] [CrossRef]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brüstle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Flitsch, L.J.; Laupman, K.E.; Brüstle, O. Transcription Factor-Based Fate Specification and Forward Programming for Neural Regeneration. Front. Cell. Neurosci. 2020, 14, 121. [Google Scholar] [CrossRef]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2001, 5, e18648. [Google Scholar] [CrossRef]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- Cintrón-Colón, A.F.; Almeida-Alves, G.; Boynton, A.M.; Spitsbergen, J.M. GDNF synthesis, signaling, and retrograde transport in motor neurons. Cell Tissue Res. 2020, 382, 47–56. [Google Scholar] [CrossRef]
- Lepski, G.P.; Jannes, C.E.; Nikkhah, G.; Bischofberger, J. cAMP promotes the differentiation of neural progenitor cells in vitro via modulation of voltage-gated calcium channels. Front. Cell. Neurosci. 2013, 7, 155. [Google Scholar] [CrossRef]
- Jang, S.; Cho, H.H.; Cho, Y.B.; Park, J.S.; Jeong, H.S. Functional neural differentiation of human adipose tissue-derived stem cells using bFGF and forskolin. BMC Cell Biol. 2010, 11, 25. [Google Scholar] [CrossRef]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signaling and neuronal differentiation. Cell. Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef]
- Busskamp, V.; Lewis, N.E.; Guye, P.; Ng, A.H.; Shipman, S.L.; Byrne, S.M.; Sanjana, N.E.; Murn, J.; Li, Y.; Li, S.; et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Caiazzo, M.; Giannelli, S.; Valente, P.; Lignani, G.; Carissimo, A.; Sessa, A.; Colasante, G.; Bartolomeo, R.; Massimino, L.; Ferroni, S.; et al. Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Rep. 2015, 4, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Pang, Z.P.; Yang, N.; Tsai, M.C.; Qu, K.; Chang, H.Y.; Südhof, T.C.; Wernig, M. Direct Lineage Conversion of Terminally Differentiated Hepatocytes to Functional Neurons. Cell Stem Cell 2011, 9, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Ambasudhan, R.; Talantova, M.; Coleman, R.; Yuan, X.; Zhu, S.; Lipton, S.A.; Ding, S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 2011, 9, 113–118. [Google Scholar] [CrossRef]
- Zhang, W.; Jiao, B.; Zhou, M.; Zhou, T.; Shen, L. Modeling Alzheimer’s Disease with Induced Pluripotent Stem Cells: Current Challenges and Future Concerns. Stem Cells Int. 2016, 2016, 7828049. [Google Scholar] [CrossRef]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; Slesinger, P.A.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Byun, J.S.; Lee, C.O.; Oh, M.; Cha, D.; Kim, W.K.; Oh, K.J.; Bae, K.H.; Lee, S.C.; Han, B.S. Rapid differentiation of astrocytes from human embryonic stem cells. Neurosci. Lett. 2020, 716, 134681. [Google Scholar] [CrossRef]
- Ren, B.; Dunaevsky, A. Modeling Neurodevelopmental and Neuropsychiatric Diseases with Astrocytes Derived from Human-Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2021, 22, 1692. [Google Scholar] [CrossRef]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 2019, 37, 267–275. [Google Scholar] [CrossRef]
- Janssen, K.; Bahnassawy, L.; Kiefer, C.; Korffmann, J.; Terstappen, G.C.; Lakics, V.; Cik, M.; Reinhardt, P. Generating Human iPSC-Derived Astrocytes with Chemically Defined Medium for In vitro Disease Modeling. In Cell-Based Assays Using iPSCs for Drug Development and Testing; Mandenius, C.F., Ross, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1994, pp. 31–39. [Google Scholar]
- Raman, S.; Srinivasan, G.; Brookhouser, N.; Nguyen, T.; Henson, T.; Morgan, D.; Cutts, J.; Brafman, D.A. A Defined and Scalable Peptide-Based Platform for the Generation of Human Pluripotent Stem Cell-Derived Astrocytes. ACS Biomater. Sci. Eng. 2020, 6, 3477–3490. [Google Scholar] [CrossRef] [PubMed]
- Foo, L.C.; Allen, N.J.; Bushong, E.A.; Ventura, P.B.; Chung, W.S.; Zhou, L.; Cahoy, J.D.; Daneman, R.; Zong, H.; Ellisman, M.H.; et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 2011, 71, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; McConnell, E.; Pare, J.F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 399, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef]
- Ijaz, L.; Nijsure, M.; Fossati, V. Human Pluripotent Stem Cell Differentiation to Microglia. In Induced Pluripotent Stem (iPS) Cells; Nagy, A., Turksen, K., Eds.; Methods in Molecular Biology; Humana: New York, NY, USA, 2021; Volume 2454. [Google Scholar]
- McQuade, A.; Coburn, M.; Tu, C.H.; Hasselmann, J.; Davtyan, H.; Blurton-Jones, M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 2018, 13, 67. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia- like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating microglia from human pluripotent stem cells: Novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 2019, 14, 46. [Google Scholar] [CrossRef]
- Cakir, B.; Tanaka, Y.; Kiral, F.R.; Xiang, Y.; Dagliyan, O.; Wang, J.; Lee, M.; Greaney, A.M.; Yang, W.S.; duBoulay, C.; et al. Expression of the transcription factor, P.U.1 induces the generation of microglia-like cells in human cortical organoids. Nat. Commun. 2022, 13, 430. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.A.; Kuypers, N.J. How to make an oligodendrocyte. Development 2015, 142, 3983–3995. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.-M.; Sun, S.; Wang, C.; Huang, H.; Hu, X.; Zhang, Z.; Lu, Q.R.; Qiu, M. Stage-Specific Regulation of Oligodendrocyte Development by Wnt/β-Catenin Signaling. J. Neurosci. 2014, 34, 8467–8473. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Fossati, V. Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 1143–1154. [Google Scholar] [CrossRef]
- Shi, B.; Ding, J.; Liu, Y.; Zhuang, X.; Zhuang, X.; Chen, X.; Fu, C. ERK1/2 pathway-mediated differentiation of IGF- 1-transfected spinal cord-derived neural stem Cells into oligodendrocytes. PLoS ONE 2014, 9, e106038. [Google Scholar] [CrossRef]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.L.; Cui, Q.L.; Schambach, A.; Kim, K.P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef]
- Chanoumidou, K.; Hernández-Rodríguez, B.; Windener, F.; Thomas, C.; Stehling, M.; Mozafari, S.; Albrecht, S.; Ottoboni, L.; Antel, J.; Kim, K.P.; et al. One-step Reprogramming of Human Fibroblasts into Oligodendrocyte-like Cells by SOX10, OLIG2, and NKX6.2. Stem Cell Rep. 2021, 16, 771–783. [Google Scholar] [CrossRef]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef]
- Hubler, Z.; Allimuthu, D.; Bederman, I.; Elitt, M.S.; Madhavan, M.; Allan, K.C.; Shick, H.E.; Garrison, E.; T Karl, M.; Factor, D.C.; et al. Accumulation of 8, 9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 2018, 560, 372–376. [Google Scholar] [CrossRef]
- Swire, M.; Ffrench-Constant, C. Oligodendrocyte-Neuron Myelinating Coculture. In Oligodendrocytes; Lyons, D., Kegel, L., Eds.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1936. [Google Scholar]
- Sim, F.J.; McClain, C.R.; Schanz, S.J.; Protack, T.L.; Windrem, M.S.; Goldman, S.A. CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nat. Biotechnol. 2011, 29, 934–941. [Google Scholar] [CrossRef]
- Abe-Fukasawa, N.; Otsuka, K.; Aihara, A.; Itasaki, N.; Nishino, T. Novel 3D Liquid Cell Culture Method for Anchorage-independent Cell Growth, Cell Imaging and Automated Drug Screening. Sci. Rep. 2018, 8, 3627. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, M.; Mazzoldi, E.L.; Ferraro, R.M.; Pinelli, M.; Parigi, M.; Aghel, S.A.M.; Bugatti, M.; Collo, G.; Stocco, G.; Vermi, W.; et al. Progesterone receptor is constitutively expressed in induced Pluripotent Stem Cells (iPSCs). Stem Cell Rev. Rep. 2024, 20, 2303–2317. [Google Scholar] [CrossRef]
- Corsini, N.S.; Knoblich, J.A. Human organoids: New strategies and methods for analyzing human development and disease. Cell 2022, 185, 2756–2769. [Google Scholar] [CrossRef]
- Koo, B.; Choi, B.; Park, H.; Yoon, K.J. Past, Present, and Future of Brain Organoid Technology. Mol. Cells 2019, 42, 617–627. [Google Scholar]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human organoids: Tools for understanding biology and treating diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Kyrousi, C.; Cappello, S. Using brain organoids to study human neurodevelopment, evolution and disease. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e347. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Cederquist, G.Y.; Asciolla, J.J.; Tchieu, J.; Walsh, R.M.; Cornacchia, D.; Resh, M.D.; Studer, L. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 2019, 37, 436–444. [Google Scholar] [CrossRef]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 2020, 26, 766–781.e769. [Google Scholar] [CrossRef]
- Abbott, J.; Tambe, M.; Pavlinov, I.; Farkhondeh, A.; Nguyen, H.N.; Xu, M.; Pradhan, M.; York, T.; Might, M.; Baumgärtel, K.; et al. Generation and characterization of NGLY1 patient-derived midbrain organoids. Front. Cell Dev. Biol. 2023, 11, 1039182. [Google Scholar] [CrossRef]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef]
- Ciarpella, F.; Zamfir, R.G.; Campanelli, A.; Pedrotti, G.; Di Chio, M.; Bottani, E.; Decimo, I. Generation of mouse hippocampal brain organoids from primary embryonic neural stem cells. STAR Protoc. 2023, 4, 102413. [Google Scholar] [CrossRef]
- Sarkar, A.; Mei, A.; Paquola, A.C.M.; Stern, S.; Bardy, C.; Klug, J.R.; Kim, S.; Neshat, N.; Kim, H.J.; Ku, M.; et al. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell 2018, 22, 684–697.e9. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Onesto, M.M.; Kim, J.I.; Pasca, S.P. Assembloid models of cell-cell interaction to study tissue and disease biology. Cell Stem Cell 2024, 31, 1563–1573. [Google Scholar] [CrossRef]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119, Erratum in Nat. Biotechnol. 2012, 30, 112. [Google Scholar] [CrossRef]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schröter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E.; et al. Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genom. 2015, 16, 84. [Google Scholar]
- Young, J.E.; Boulanger-Weill, J.; Williams, D.A.; Woodruff, G.; Buen, F.; Revilla, A.C.; Herrera, C.; Israel, M.A.; Yuan, S.H.; Edland, S.D.; et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 2015, 16, 373–385. [Google Scholar] [CrossRef]
- Knupp, A.; Mishra, S.; Martinez, R.; Braggin, J.E.; Szabo, M.; Kinoshita, C.; Hailey, D.W.; Small, S.A.; Jayadev, S.; Young, J.E. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020, 31, 107719. [Google Scholar] [CrossRef]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stüber, K.; Esselmann, H.; Wiltfang, J.; Brustle, O.; Walter, J. Presenilin-1 L166P mutant human pluripotent stem cell–derived neurons exhibit partial loss of γ-secretase activity in endogenous Amyloid-β generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Moore, S.; Evans, L.D.; Andersson, T.; Portelius, E.; Smith, J.; Dias, T.B.; Saurat, N.; McGlade, A.; Kirwan, P.; Blennow, K.; et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015, 11, 689–696. [Google Scholar] [CrossRef]
- Hu, N.W.; Corbett, G.T.; Moore, S.; Klyubin, I.; O’Malley, T.T.; Walsh, D.M.; Livesey, F.J.; Rowan, M.J. Extracellular Forms of Aβ and Tau from iPSC Models of Alzheimer’s Disease Disrupt Synaptic Plasticity. Cell Rep. 2018, 23, 1932–1938. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020, 11, 5540. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Elsworthy, R.J.; King, M.C.; Grainger, A.; Fisher, E.; Crowe, J.A.; Alqattan, S.; Ludlam, A.; Hill, D.E.J.; Aldred, S. Amyloid-β Precursor Protein Processing and Oxidative Stress are Altered in Human iPSC-Derived Neuron and Astrocyte Co-Cultures Carrying Pre- senillin-1 Gene Mutations Following Spontaneous Differentiation. Mol. Cell. Neurosci. 2021, 114, 103631. [Google Scholar] [CrossRef]
- Armijo, E.; Gonzalez, C.; Shahnawaz, M.; Flores, A.; Davis, B.; Soto, C. Increased susceptibility to Aβ toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 2017, 639, 74–81. [Google Scholar] [CrossRef]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Giudice, M.L.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyper-phosphorylation, increased amyloid levels, and GSK3B activation. Alzheimer’s Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.T.; et al. REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer’s Disease. Cell Rep. 2019, 26, 1112–1127.e9. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, H.; Ma, Y.; Shi, G.; Song, J.; Tang, Y.; Li, S.; Li, T.; Liu, N.; Tang, F.; et al. Early pathogenic event of Alzheimer’s disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 2017, 8, 7900–7913. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef]
- Claes, C.; Daele, J.V.D.; Boon, R.; Schouteden, S.; Colombo, A.; Monasor, L.S.; Fiers, M.; Ordovás, L.; Nami, F.; Bohrmann, B.; et al. Human stem cell–derived monocytes and microglia-like cells reveal impaired amyloid plaque clearance upon heterozygous or homozygous loss of TREM2. Alzheimer’s Dement. 2019, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Olivé, M.G.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.K.; Yang, M.M.; Dos Santos Chaves, R.; Reyna, S.M.; Langness, V.F.; Woodruff, G.; Roberts, E.A.; Young, J.E.; Goldstein, L.S.B. Full-length amyloid precursor protein regulates lipoprotein metabolism and amyloid-β clearance in human astrocytes. J. Biol. Chem. 2018, 293, 11341–11357. [Google Scholar] [CrossRef]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 2021, 13, eaaz4564. [Google Scholar] [CrossRef]
- Zou, P.; Wu, C.; Liu, T.C.; Duan, R.; Yang, L. Oligodendrocyte progenitor cells in Alzheimer’s disease: From physiology to pathology. Transl. Neurodegener. 2003, 12, 52. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Alić, I.; Goh, P.A.; Murray, A.; Portelius, E.; Gkanatsiou, E.; Gough, G.; Mok, K.Y.; Koschut, D.; Brunmeir, R.; Yeap, Y.J.; et al. Patient-specific Alzheimer-like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dose-sensitive AD suppressor in human brain. Mol. Psychiatry 2020, 26, 5766–5788. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Pavoni, S.; Jarray, R.; Nassor, F.; Guyot, A.C.; Cottin, S.; Rontard, J.; Mikol, J.; Mabondzo, A.; Deslys, J.P.; Yates, F. Small-molecule induction of Aβ-42 peptide production in human cerebral organoids to model Alzheimer’s disease associated phenotypes. PLoS ONE 2018, 13, e0209150. [Google Scholar] [CrossRef]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Jang, S.Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.H.; Seo, J.; Choi, M.; et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Duan, Y.; Tsang, H.W.S.; Xu, H.; Chen, Y.; Cao, H.; Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Efficient manipulation of gene dosage in human iPSCs using CRISPR/Cas9 nickases. Commun. Biol. 2021, 4, 195. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a smallmolecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Claes, C.; Danhash, E.P.; Hasselmann, J.; Chadarevian, J.P.; Shabestari, S.K.; England, W.E.; Lim, T.E.; Hidalgo, J.L.S.; Spitale, R.C.; Davtyan, H.; et al. Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 50. [Google Scholar] [CrossRef]
- Bose, A.; Petsko, G.A.; Studer, L. Induced pluripotent stem cells: A tool for modeling Parkinson’s disease. Trends Neurosci. 2022, 45, 608–620. [Google Scholar] [CrossRef]
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012, 3, e350. [Google Scholar] [CrossRef]
- Torrent, R.; De Angelis Rigotti, F.; Dell’Era, P.; Memo, M.; Raya, A.; Consiglio, A. Using iPS Cells toward the Understanding of Parkinson’s Disease. J. Clin. Med. 2015, 4, 548–566. [Google Scholar] [CrossRef]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.L.; et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.-W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. Alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.R.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.E.; et al. Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1 transcription. Cell 2013, 155, 1351–1364, Erratum in Cell 2013, 155, 1652–1653. [Google Scholar] [CrossRef]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef]
- Ho, G.P.H.; Ramalingam, N.; Imberdis, T.; Wilkie, E.C.; Dettmer, U.; Selkoe, D.J. Upregulation of Cellular Palmitoylation Mitigates alpha-Synuclein Accumulation and Neurotoxicity. Mov. Disord. 2021, 36, 348–359. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Danés, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jiménez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef]
- Bono, F.; Mutti, V.; Devoto, P.; Bolognin, S.; Schwamborn, J.C.; Missale, C.; Fiorentini, C. Impaired dopamine D3 and nicotinic acetylcholine receptor membrane localization in iPSCs-derived dopaminergic neurons from two Parkinson’s disease patients carrying the LRRK2 G2019S mutation. Neurobiol. Aging 2021, 99, 65–78. [Google Scholar] [CrossRef]
- Wickremaratchi, M.M.; Knipe, M.D.; Sastry, B.S.; Morgan, E.; Jones, A.; Salmon, R.; Weiser, R.; Moran, M.; Davies, D.; Ebenezer, L.; et al. The motor phenotype of Parkinson’s disease in relation to age at onset. Mov. Disord. 2011, 26, 457–463. [Google Scholar] [CrossRef]
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and alpha-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Sultan, A.; Platzer, J.; Dolatabadi, N.; Soldner, F.; McClatchy, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Ambasudhan, R.; Nakamura, T.; et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017, 21, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, K.; Funato, M.; Kato, Z.; Seki, J.; Kawase, C.; Tamai, Y.; Ono, Y.; Nagahara, Y.; Noda, Y.; Kameyama, T.; et al. Established Stem Cell Model of Spinal Muscular Atrophy Is Applicable in the Evaluation of the Efficacy of Thyrotropin-Releasing Hormone Analog. Stem Cells Transl. Med. 2016, 5, 152–163. [Google Scholar] [CrossRef]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef]
- Angelique di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-specific iPSC-Derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Mamais, A.; Kluss, J.H.; Bonet-Ponce, L.; Landeck, N.; Langston, R.G.; Smith, N.; Beilina, A.; Kaganovich, A.; Ghosh, M.C.; Pellegrini, L.; et al. Mutations in LRRK2 linked to Parkinson disease sequester Rab8a to damaged lysosomes and regulate transferrin-mediated iron uptake in microglia. PLoS Biol. 2021, 19, e3001480, Erratum in PLoS Biol. 2022, 20, e3001621. [Google Scholar] [CrossRef]
- Ramos-Gonzalez, P.; Mato, S.; Chara, J.C.; Verkhratsky, A.; Matute, C.; Cavaliere, F. Astrocytic atrophy as a pathological feature of Parkinson’s disease with LRRK2 mutation. NPJ Park. Dis. 2021, 7, 31. [Google Scholar] [CrossRef]
- Panagiotakopoulou, V.; Ivanyuk, D.; De Cicco, S.; Haq, W.; Arsic, A.; Yu, C.; Messelodi, D.; Oldrati, M.; Schondorf, D.C.; Perez, M.J.; et al. Interferon-gamma signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat. Commun. 2020, 11, 5163. [Google Scholar] [CrossRef]
- Haenseler, W.; Zambon, F.; Lee, H.; Vowles, J.; Rinaldi, F.; Duggal, G.; Houlden, H.; Gwinn, K.; Wray, S.; Luk, K.C.; et al. Excess alpha-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep. 2017, 7, 9003. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Botta-Orfila, T.; Morato, X.; Calatayud, C.; Ferrer-Lorente, R.; Marti, M.J.; Fernández, M.; Gaig, C.; Raya, A.; Consiglio, A.; et al. MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson disease patients. Neurobiol. Aging 2018, 69, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Fossepre, M.; Qing, X.; Jarazo, J.; Scancar, J.; Moreno, E.L.; Nickels, S.L.; Wasner, K.; Ouzren, N.; Walter, J.; et al. 3D Cultures of Parkinson’s Disease-Specific Dopaminergic Neurons for High Content Phenotyping and Drug Testing. Adv. Sci. 2019, 6, 1800927. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Park. Dis. 2019, 5, 5. [Google Scholar] [CrossRef]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef]
- Qing, X.; Walter, J.; Jarazo, J.; Arias-Fuenzalida, J.; Hillje, A.L.; Schwamborn, J.C. CRISPR/Cas9 and piggyBacmediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-Synuclein modulation in dopaminergic neurons. Stem Cell Res. 2017, 24, 44–50. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Hanss, Z.; Boussaad, I.; Jarazo, J.; Schwamborn, J.C.; Kruger, R. Quality control strategy for CRISPR-Cas9-based gene editing complicated by a pseudogene. Front. Genet. 2020, 10, 1297. [Google Scholar] [CrossRef]
- Ahfeldt, T.; Ordureau, A.; Bell, C.; Sarrafha, L.; Sun, C.; Piccinotti, S.; Grass, T.; Parfitt, G.M.; Paulo, J.A.; Yanagawa, F.; et al. Pathogenic pathways in early-onset autosomal recessive Parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Rep. 2020, 14, 75–90. [Google Scholar] [CrossRef]
- Chen, C.X.-Q.; You, Z.; Abdian, N.; Sirois, J.; Shlaifer, I.; Tabatabaei, M.; Boivin, M.N.; Gaborieau, L.; Karamchandani, J.; Beitel, L.K.; et al. Generation of homozygous PRKN, PINK1 and double PINK1/PRKN knockout cell lines from healthy induced pluripotent stem cells using CRISPR/Cas9 editing. Stem Cell Res. 2022, 62, 102806. [Google Scholar] [CrossRef]
- Harjuhaahto, S.; Rasila, T.S.; Molchanova, S.M.; Woldegebriel, R.; Kvist, J.; Konovalova, S.; Sainio, M.T.; Pennonen, J.; Torregrosa-Muñumer, R.; Ibrahim, H.; et al. ALS and Parkinson’s disease genes CHCHD10 and CHCHD2 modify synaptic transcriptomes in human iPSC-derived motor neurons. Neurobiol. Dis. 2020, 141, 104940. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.L.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef]
- Mattis, V.B.; Tom, C.; Akimov, S.; Saeedian, J.; Østergaard, M.E.; Southwell, A.L.; Doty, C.N.; Ornelas, L.; Sahabian, A.; Lenaeus, L.; et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet. 2015, 24, 3257–3271. [Google Scholar] [CrossRef]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef]
- Jeon, I.; Lee, N.; Li, J.Y.; Park, I.H.; Park, K.S.; Moon, J.; Shim, S.H.; Choi, C.; Chang, D.J.; Kwon, J.; et al. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 2012, 30, 2054–2062. [Google Scholar] [CrossRef]
- Zhang, N.; An, M.C.; Montoro, D.; Ellerby, L.M. Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. 2010, 2, RRN1193. [Google Scholar] [CrossRef]
- Oura, S.; Noda, T.; Morimura, N.; Hitoshi, S.; Nishimasu, H.; Nagai, Y.; Nureki, O.; Ikawa, M. Precise CAG repeat contraction in a Huntington’s disease mouse model is enabled by gene editing with SpCas9-NG. Commun. Biol. 2021, 4, 771. [Google Scholar] [CrossRef]
- Shin, J.W.; Kim, K.-H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Kawakami, H.; Kitajima, H.; Nishiyama, A.; Sasai, Y.; Inoue, H.; Muguruma, K. Vulnerability of Purkinje cells generated from spinocerebellar ataxia type 6 patient-derived iPSCs. Cell Rep. 2016, 17, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tuting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef]
- Ou, Z.; Luo, M.; Niu, X.; Chen, Y.; Xie, Y.; He, W.; Song, B.; Xian, Y.; Fan, D.; OuYang, S.; et al. Autophagy Promoted the degradation of mutant ATXN3 in neurally differentiated spinocerebellar Ataxia-3 Human induced pluripotent stem cells. Biomed. Res. Int. 2016, 2016, 6701793. [Google Scholar] [CrossRef]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Fuller, H.R.; Mandefro, B.; Shirran, S.L.; Gross, A.R.; Kaus, A.S.; Botting, C.H.; Morris, G.E.; Sareen, D. Spinal Muscular atrophy patient iPSC-Derived motor neurons have reduced expression of proteins important in neuronal development. Front. Cell. Neurosci. 2015, 9, 506. [Google Scholar] [CrossRef]
- Lin, X.; Li, J.J.; Qian, W.J.; Zhang, Q.J.; Wang, Z.F.; Lu, Y.Q.; Dong, E.L.; He, J.; Wang, N.; Ma, L.X.; et al. Modeling the differential phenotypes of spinal muscular atrophy with high-yield generation of motor neurons from human induced pluripotent stem cells. Oncotarget 2017, 8, 42030–42042. [Google Scholar] [CrossRef]
- Yoshida, M.; Kitaoka, S.; Egawa, N.; Yamane, M.; Ikeda, R.; Tsukita, K.; Amano, N.; Watanabe, A.; Morimoto, M.; Takahashi, J.; et al. Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs. Stem Cell Rep. 2015, 4, 561–568. [Google Scholar] [CrossRef]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.A.; Stucky, C.L.; Ebert, A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 2013, 61, 1418–1428. [Google Scholar] [CrossRef]
- Kondo, T.; Funayama, M.; Miyake, M.; Tsukita, K.; Era, T.; Osaka, H.; Ayaki, T.; Takahashi, R.; Inoue, H. Modeling Alexander disease with patient iPSCs reveals cellular and molecular pathology of astrocytes. Acta Neuropathol. Commun. 2016, 4, 69. [Google Scholar] [CrossRef]
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Sun, G.; Huang, Y.; Warden, C.D.; et al. GFAP Mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of alexander disease. Cell Stem Cell 2018, 23, 239–251.e6. [Google Scholar] [CrossRef]

{kind=link}
| Methods | Advantages | Disadvantages | References |
|---|---|---|---|
| Retroviral and lentiviral vectors | Highly efficient and robust | Risk of transgene reactivation | [12] |
| Adenoviral vectors | Lower risk of transgene reactivation | Low reprogramming efficiency; unsuitable for clinical use | [11,13] |
| Sendai virus | Higher efficiency; RNA virus can be completely removed | Still involves challenges in clinical application | [11,13] |
| PiggyBac | Lower risk of genomic instability and mutations | Low reprogramming efficiency; limited cell sources | [14,15,16] |
| Minicircle vectors | Lower risk of genomic instability and mutations | Low reprogramming efficiency; limited cell sources | [14,15,16] |
| Episomal plasmids | No genomic integration risk; cost-effective and easy to use | Requires daily transfection; moderate efficiency | [17,18] |
| RNA Delivery | Lower mutagenic risk; high efficiency | Limited to specific cell types (e.g., fibroblasts, peripheral blood cells) | [19,20,21] |
| Factors | Functions | References | |
|---|---|---|---|
| Yamanaka factors | OCT3/4, SOX2, KLF4, c-MYC | OCT3/4, SOX2, and KLF4 maintain pluripotency and inhibit differentiation c-MYC enhances reprogramming efficiency and promotes cell proliferation | [23] |
| Alternative factor combinations | OCT3/4, SOX2, NANOG, LIN28 | NANOG maintains self-renewal of stem cells LIN28 regulates RNA modification and expression | [24] |
| Enhancement or complementary factors | GLIS1, NR5A2, SALL4 | Substitute for c-MYC or OCT3/4 to improve reprogramming efficiency and stabilize cell states | [25,26,27] |
| Chemical cocktails | (1) CHALP cocktail: CHIR99021, PD0325901, LIF, A-83-01, bFGF, HA-100 | Enhances reprogramming efficiency | [2,28] |
| (2) Alternative chemical cocktail: cyclic pifithrin-α, A-83-01, CHIR99021, thiazovivin, NaB, PD0325901 | Significantly enhances reprogramming efficiency, particularly in hUCs | [2,29] | |
| Epigenetic modulators | DNA methyltransferase inhibitors, histone deacetylase inhibitors | Regulate DNA methylation, histone acetylation, and the expression of pluripotency-associated genes to enhance reprogramming efficiency | [30,31,32] |
| Cell Type | Sources | Advantages |
|---|---|---|
| Fibroblasts | Skin | Readily accessible and widely used |
| PBMCs | Blood | Non-invasive; useful for clinical applications |
| Keratinocytes | Skin or hair | Non-invasive and readily accessible |
| MSCs | Bone marrow, adipose tissue, teeth | Abundant and frequently used in regenerative research |
| Renal epithelial cells | Urine | Highly convenient and non-invasive |
| NSCs and NPCs | Brain tissue | Inherent pluripotency; useful for neurological applications |
| Liver cells | Liver tissue | Expands potential applications |
| Stomach cells | Stomach tissue | Expands potential applications |
| Cord blood cells | Cord blood | Readily available from umbilical cord; useful in neonatal research |
| Differentiated Cell/Organoid Type | Differentiation Methods | Limitations | References |
|---|---|---|---|
| NSCs/NPCs |
| Requires intermediate steps; limited differentiation efficiency | [3,56,57] |
| Neurons (e.g., dopaminergic, GABAergic, glutamatergic) |
| Complex protocols; variability in differentiation outcomes; phenotypic instability with direct conversion | [3,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] |
| Astrocytes |
| Significant plasticity; variability across protocols; in vitro astrocytes may not fully replicate in vivo properties | [72,75,76,77,78,79,80,81,82,83,84] |
| Microglia |
| High technical complexity; multiple steps; phenotypic and functional differences from in vivo counterparts | [85,86,87,88,89,90,91,92] |
| Oligodendrocytes |
| Time-consuming; phenotypic instability with direct transcription factor induction; challenges in achieving purity | [93,94,95,96,97,98,99,100,101,102,103] |
| Brain organoids (e.g., cortical, hippocampal, midbrain, cerebellar) | Non-guided (intrinsic morphogenetic potential or guided (dual SMAD inhibition, region-specific factors such as SHH, RA, WNT inhibitors) | Lack of vascularization and heterogeneity; difficulty in precisely controlling differentiation and region specificity | [104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125] |
| iPSC-Derived Model | AD’s Pathological Features | References |
|---|---|---|
| Neurons |
| [128,131,132,133,134,135,136,137,138,139,140] |
| NSCs/NPCs |
| [131,141,142] |
| Microglia |
| [85,143,144] |
| Astrocytes |
| [145,146,147,148] |
| Oligodendrocytes |
| [149] |
| 3D brain organoids |
| [150,151,152,153,154,155] |
| iPSC-Derived Model | PD’s Pathological Features | References |
|---|---|---|
| DA neurons |
| [2,5,105,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188] |
| Astrocytes |
| [189,190,191] |
| Microglia |
| [191,192,193] |
| Neural stem cells |
| [194,195] |
| 3D brain organoids |
| [104,196,197] |
| iPSC-Derived Model | HD’s Pathological Features | References |
|---|---|---|
| Neural progenitor/stem cells (NPCs/NSCs) |
| [206,207,208] |
| Forebrain neurons |
| [206] |
| Striatal-like GABAergic neurons (MSNs) |
| [209,210,211] |
| Astrocytes |
| [207,208] |
| Glial cells (general) |
| [208] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Wang, X.; Wang, J.; Ma, M.; Ren, Q. Current Development of iPSC-Based Modeling in Neurodegenerative Diseases. Int. J. Mol. Sci. 2025, 26, 3774. https://doi.org/10.3390/ijms26083774
Guo X, Wang X, Wang J, Ma M, Ren Q. Current Development of iPSC-Based Modeling in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2025; 26(8):3774. https://doi.org/10.3390/ijms26083774
Chicago/Turabian StyleGuo, Xiangge, Xumeng Wang, Jiaxuan Wang, Min Ma, and Qian Ren. 2025. "Current Development of iPSC-Based Modeling in Neurodegenerative Diseases" International Journal of Molecular Sciences 26, no. 8: 3774. https://doi.org/10.3390/ijms26083774
APA StyleGuo, X., Wang, X., Wang, J., Ma, M., & Ren, Q. (2025). Current Development of iPSC-Based Modeling in Neurodegenerative Diseases. International Journal of Molecular Sciences, 26(8), 3774. https://doi.org/10.3390/ijms26083774