
Unveiling Key Genes and Crucial Pathways in Goose Muscle Satellite Cell Biology Through Integrated Transcriptomic and Metabolomic Analyses

 ,
,
Abstract
1. Introduction
2. Results
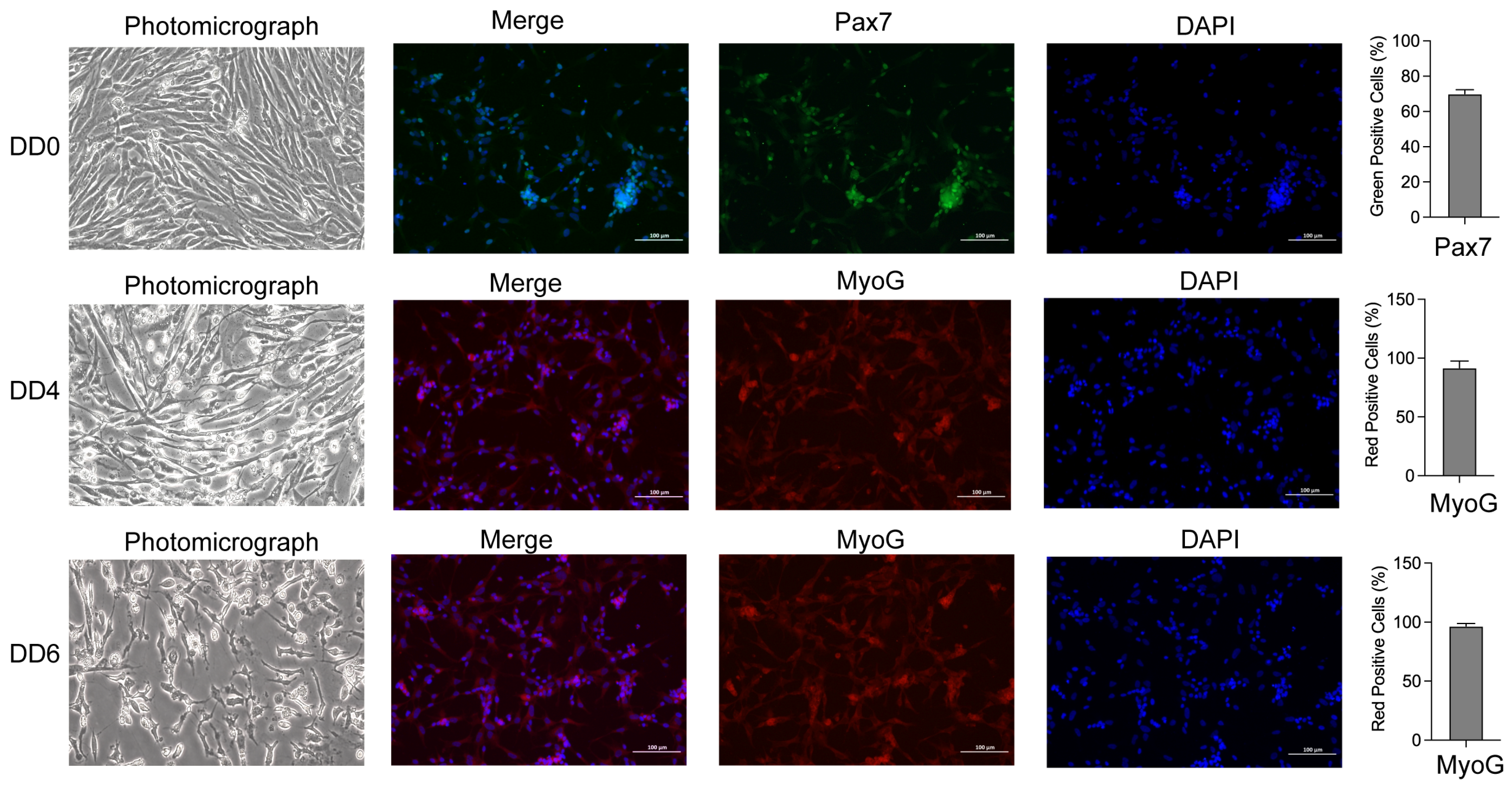
2.1. Morphological Observation, Identification, and Differentiation of Goose SMSCs
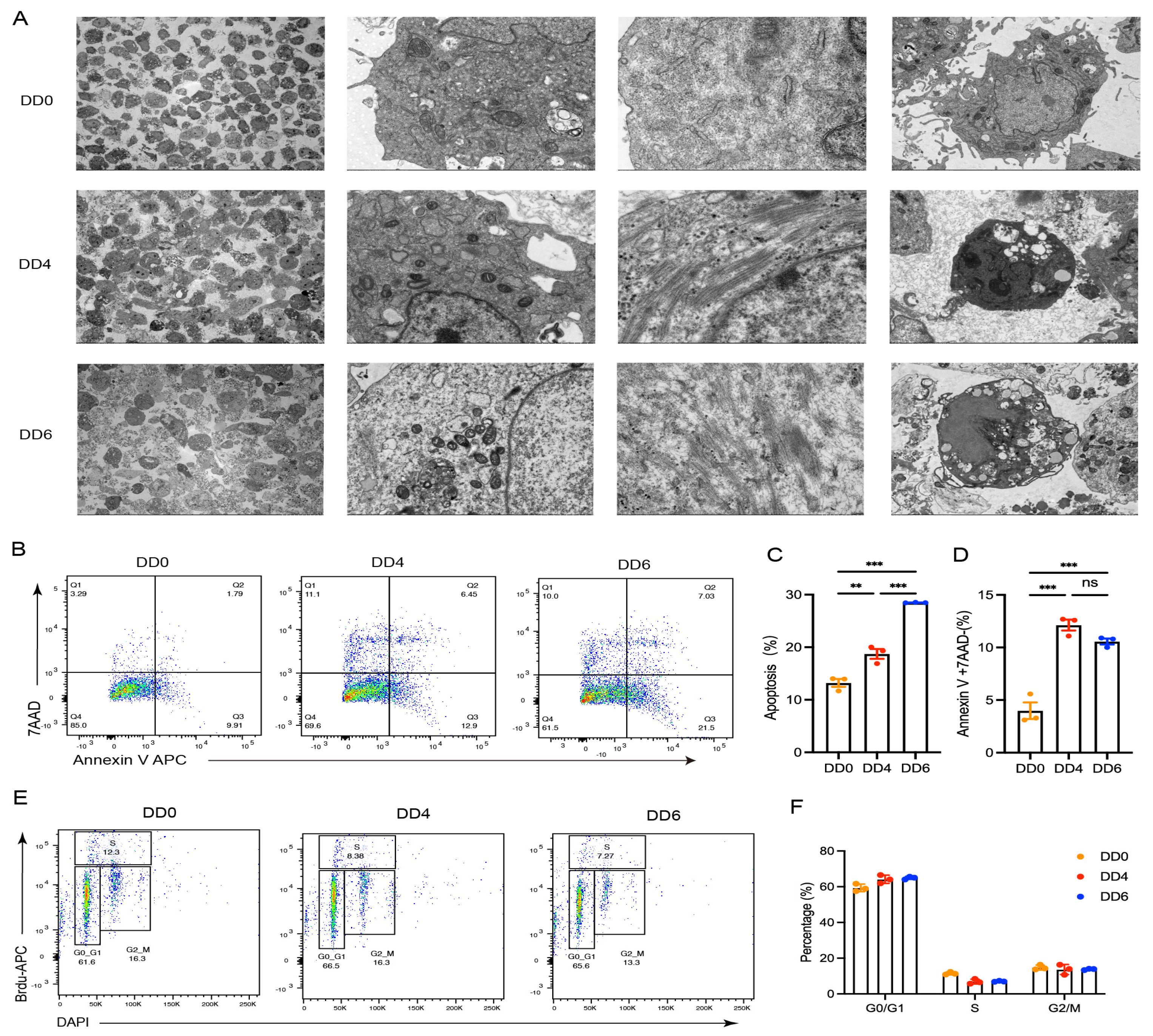
2.2. Cellular Structure Changes and Apoptosis Following the Induced Differentiation of Goose SMSCs
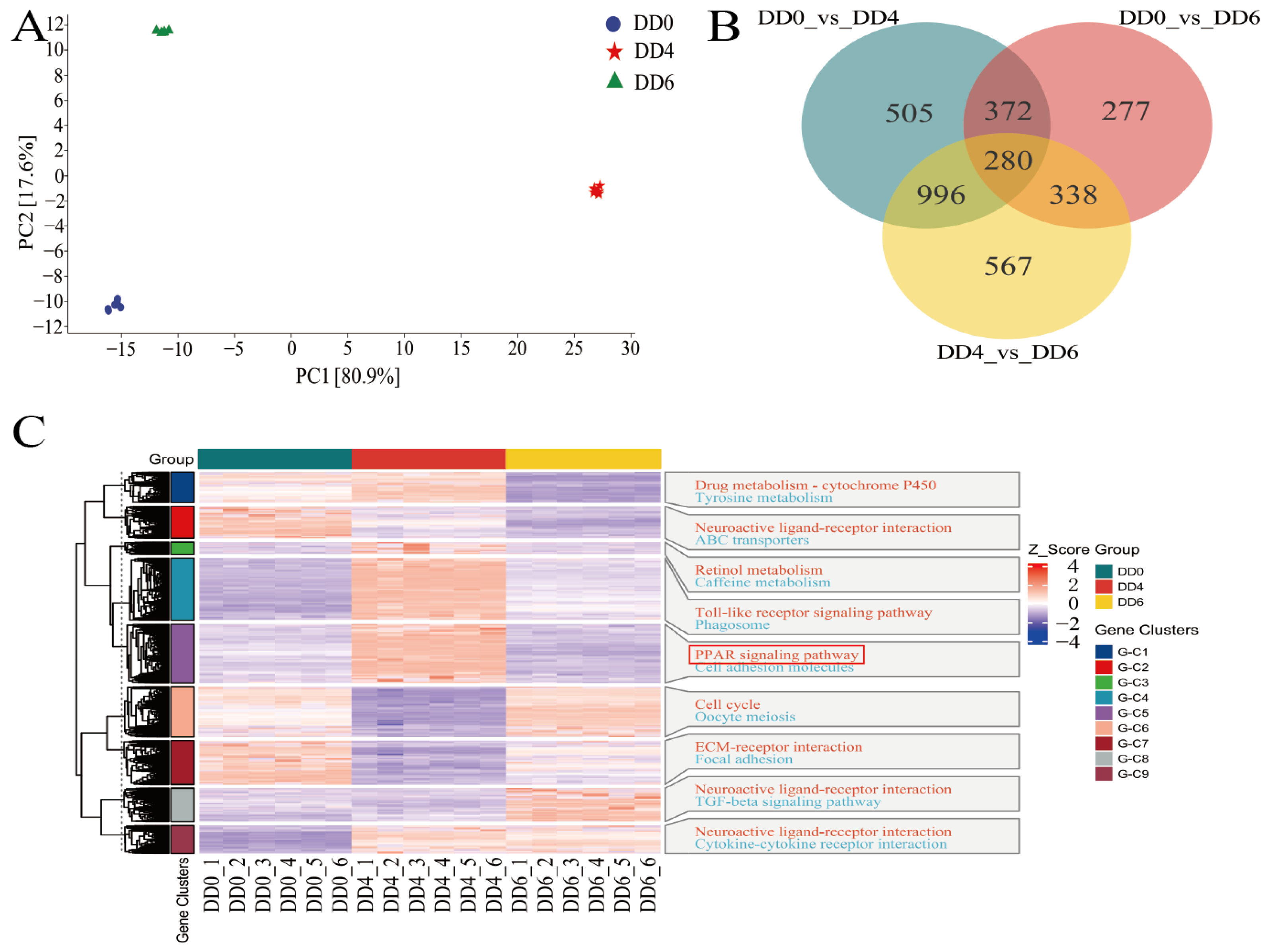
2.3. Analysis of DEGs in SMSCs in Different Periods
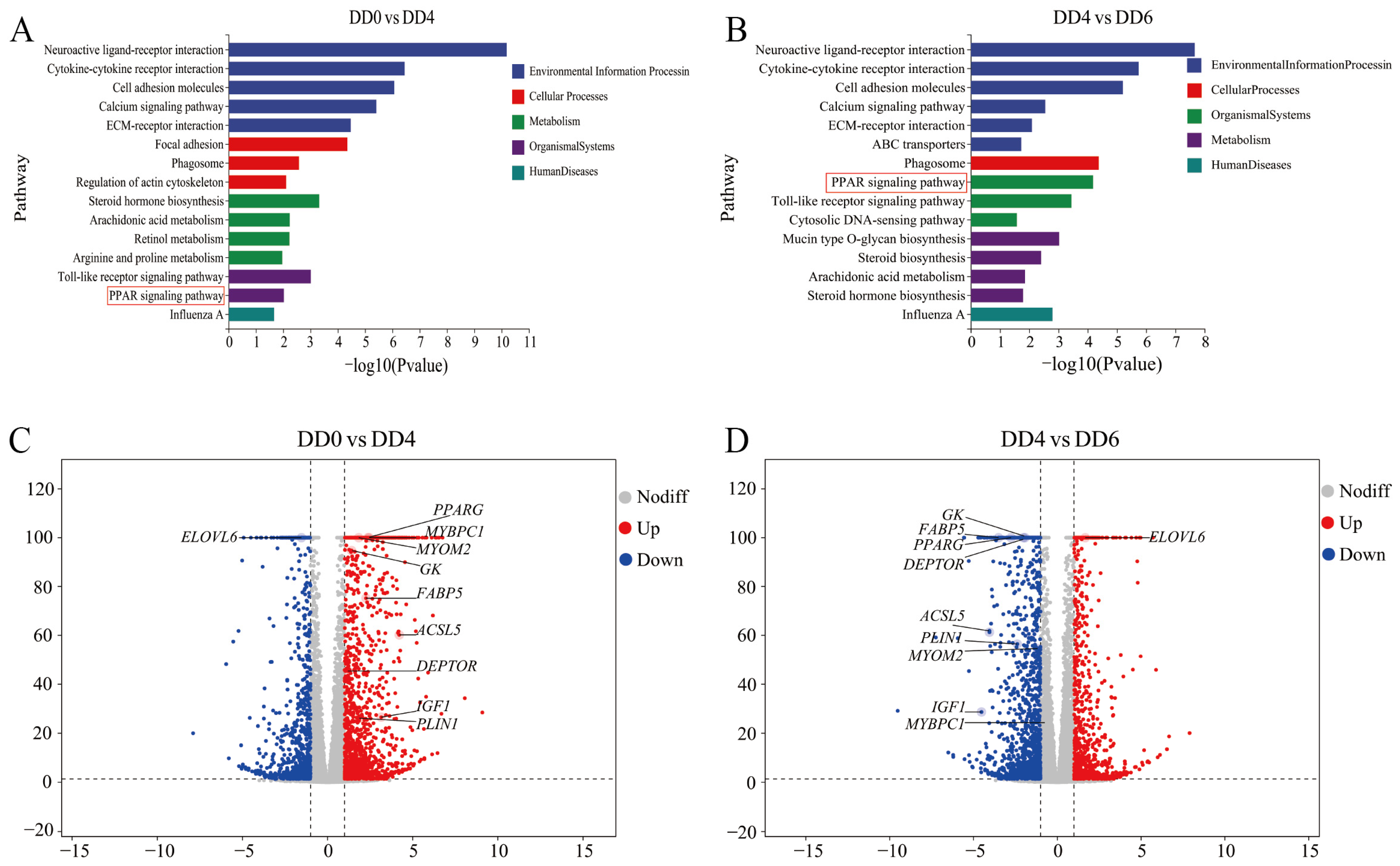
2.4. Identification of Key Regulatory Genes Associated with the PPAR Signaling Pathway
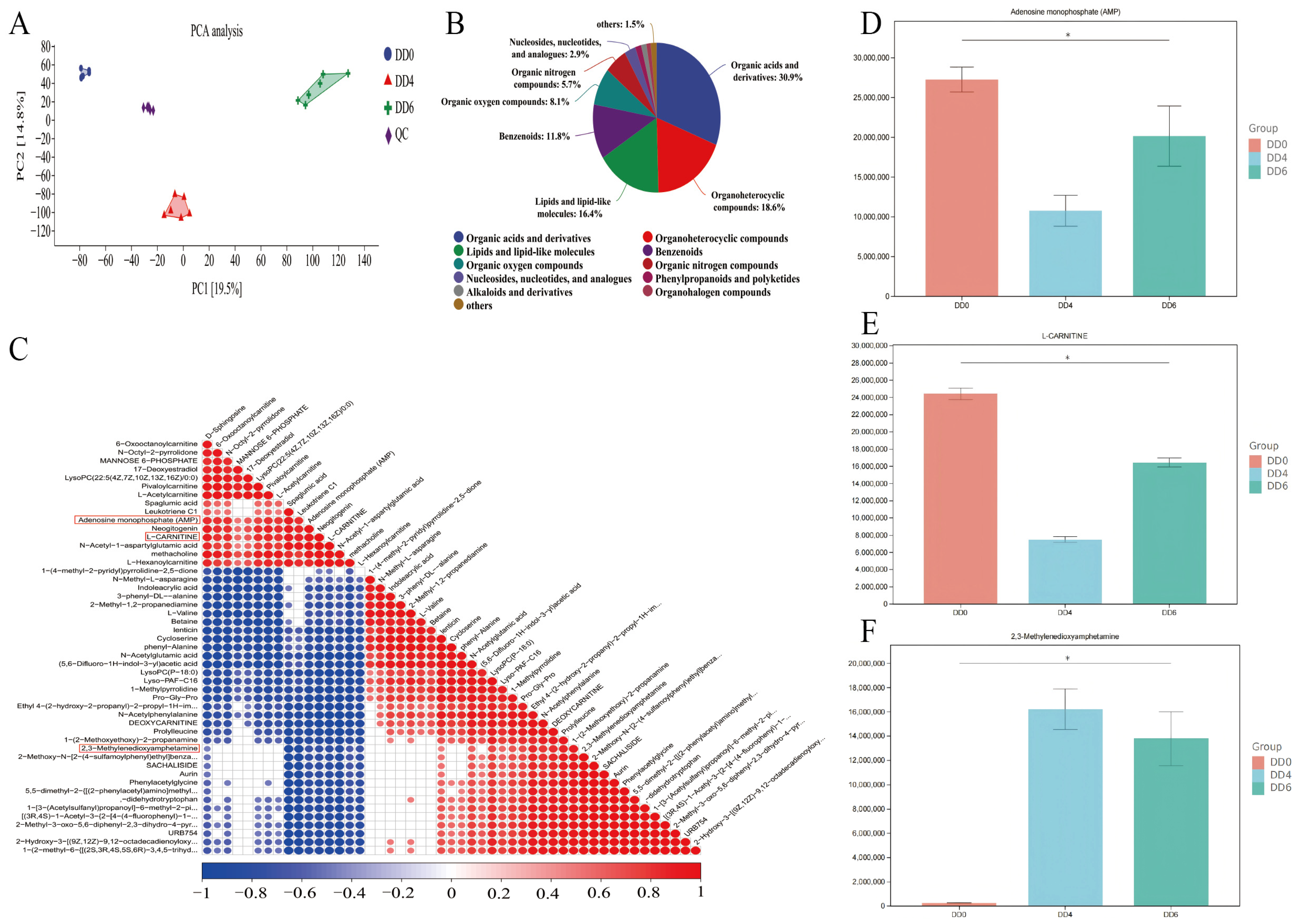
2.5. Metabolome Analysis and Identification of Key Metabolite in SMSCs in Different Periods
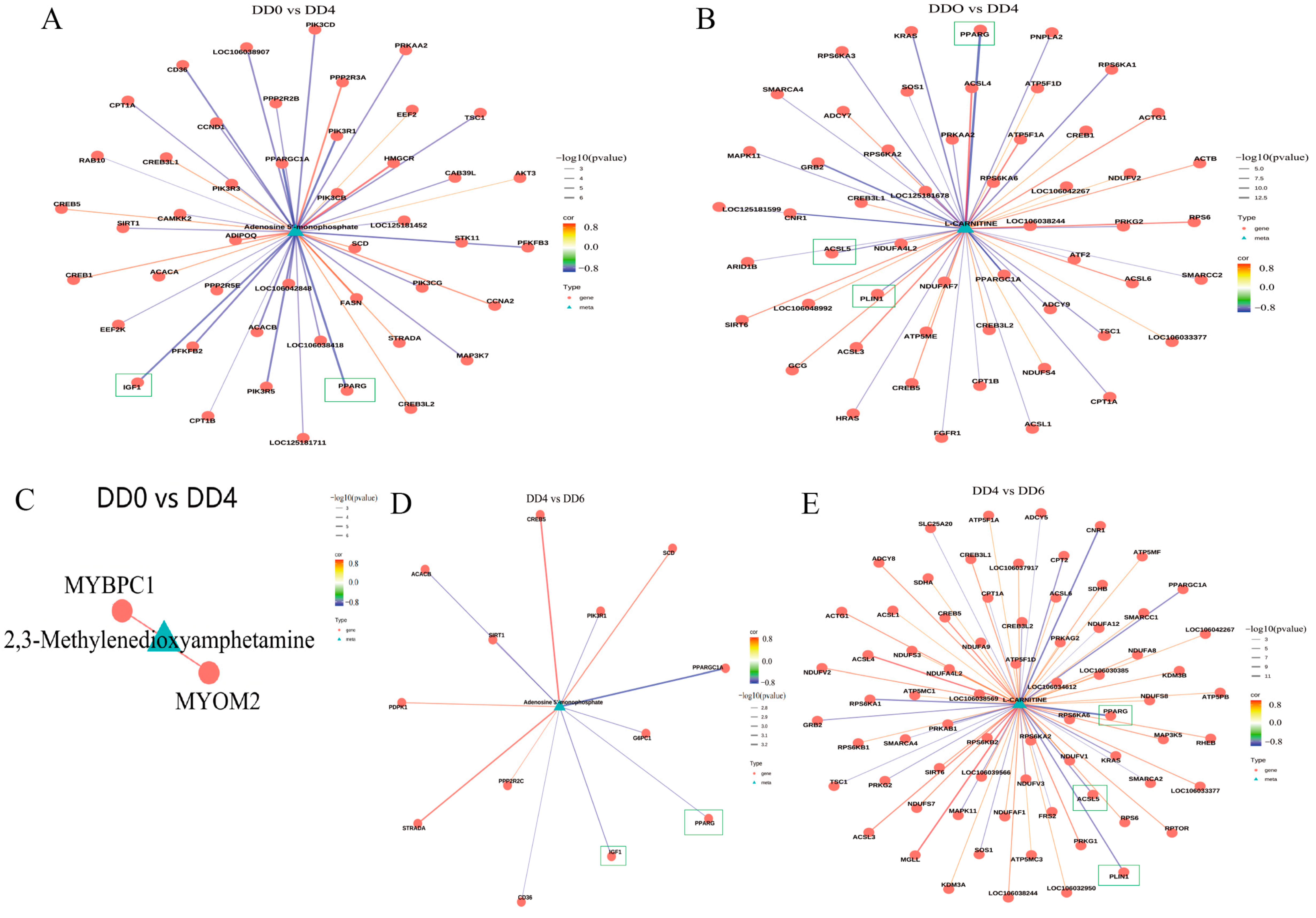
2.6. Analysis of the Correlations Between Transcriptome and Metabolome
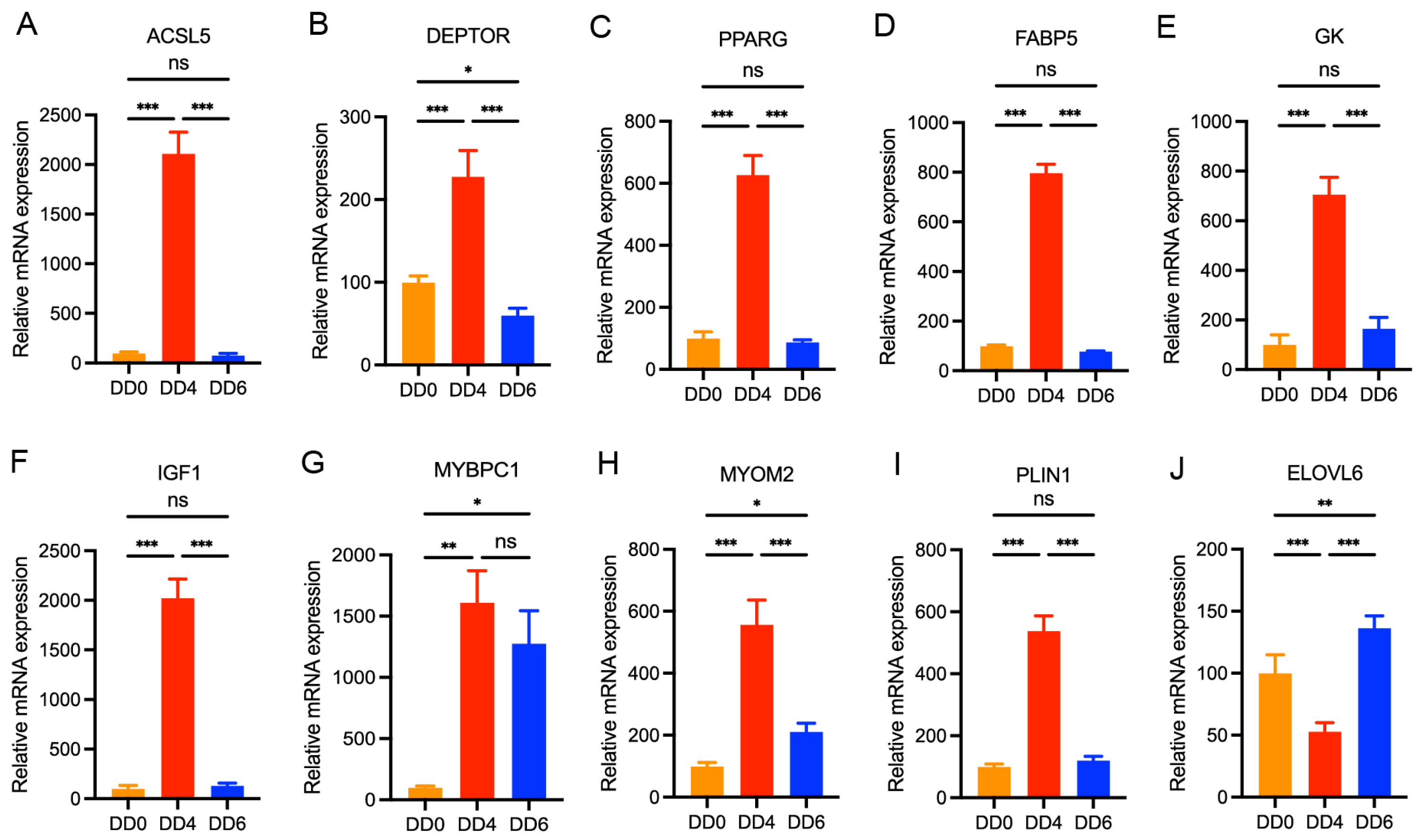
2.7. Validation of DEGs by qPCR
3. Discussion
4. Materials and Methods
4.1. Validation Isolation, Culture, and Differentiation of Goose SMSCs
4.2. Immunofluorescence Staining
4.3. Cell Cycle and Apoptosis Detection
4.4. TEM Staining for Cells
4.5. Total RNA Isolation and Transcriptome Sequencing
4.6. Transcriptome Analysis
4.7. Metabolite Extraction and Detection
4.8. Metabolome Analysis
4.9. Analysis of the Correlations Between the Transcriptome and Metabolome
4.10. qRT-PCR Validation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Güller, I.; Russell, A.P. MicroRNAs in skeletal muscle: Their role and regulation in development, disease and function. J. Physiol. 2010, 588, 4075–4087. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yu, Z.; Ai, N.; Liufu, S.; Liu, X.; Chen, B.; Li, X.; Jiang, J.; Zhang, Y.; Ma, H.; et al. Molecular Mechanism of MYL4 Regulation of Skeletal Muscle Development in Pigs. Genes 2023, 14, 1267. [Google Scholar] [CrossRef]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.D.; Lunt, A.I.; Parry, D.J.; Partridge, T.A. Culturing satellite cells from living single muscle fiber explants. Vitr. Cell Dev. Biol. Anim. 1995, 31, 773–779. [Google Scholar] [CrossRef]
- Blau, H.M.; Webster, C. Isolation and characterization of human muscle cells. Proc. Natl. Acad. Sci. USA 1981, 78, 5623–5627. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.V.; Martin, E.L.; Brannon, M.A.; Mathison, B.A.; McFarland, D.C. Optimization of bovine satellite cell-derived myotube formation in vitro. Tissue Cell 1987, 19, 159–166. [Google Scholar] [CrossRef]
- Wu, H.; Ren, Y.; Li, S.; Wang, W.; Yuan, J.; Guo, X.; Liu, D.; Cang, M. In vitro culture and induced differentiation of sheep skeletal muscle satellite cells. Cell Biol. Int. 2012, 36, 579–587. [Google Scholar] [CrossRef]
- Bennett, V.D.; Cowles, E.; Husic, H.D.; Suelter, C.H. Muscle cell cultures from chicken breast muscle have increased specific activities of creatine kinase when incubated at 41 degrees C compared with 37 degrees C. Exp. Cell Res. 1986, 164, 63–70. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, J.; Fan, B.; Hu, Y.; Yang, Y.; Wu, Y.; Li, F.; Ju, H. Different levels of autophagy induced by transient serum starvation regulate metabolism and differentiation of porcine skeletal muscle satellite cells. Sci. Rep. 2023, 13, 13153. [Google Scholar] [CrossRef]
- Liu, H.H.; Li, L.; Chen, X.; Cao, W.; Zhang, R.P.; Yu, H.Y.; Xu, F.; He, H.; Wang, J.W. Characterization of in vitro cultured myoblasts isolated from duck (Anas platyrhynchos) embryo. Cytotechnology 2011, 63, 399–406. [Google Scholar] [CrossRef]
- Wang, H.; He, K.; Zeng, X.; Zhou, X.; Yan, F.; Yang, S.; Zhao, A. Isolation and identification of goose skeletal muscle satellite cells and preliminary study on the function of C1q and tumor necrosis factor-related protein 3 gene. Anim. Biosci. 2021, 34, 1078–1087. [Google Scholar] [CrossRef]
- Gonzalez, M.L.; Busse, N.I.; Waits, C.M.; Johnson, S.E. Satellite cells and their regulation in livestock. J. Anim. Sci. 2020, 98, skaa081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Rudnicki, M.A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 2011, 13, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, R.P.; Fernyhough, M.E.; Liu, X.; McFarland, D.C.; Velleman, S.G.; Hausman, G.J.; Dodson, M.V. Extrinsic regulation of domestic animal-derived myogenic satellite cells II. Domest. Anim. Endocrinol. 2009, 36, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Loreti, M.; Savvo, A. Thejam session between Muscle stem cells and the extracellular matrix in the tissue microenvironment. NPJ Regen. Med. 2022, 7, 16. [Google Scholar] [CrossRef]
- Su, Y.; Yu, Y.Y.; Liu, C.C.; Zhang, Y.; Liu, C.; Ge, M.; Li, L.; Lan, M.; Wang, T.; Li, M.; et al. Fate decision of Satellite cell differentiation and self-renewal by miR-31-IL34axis. Cell Death Differ. 2020, 27, 949–965. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-mediated regulation of skeletal muscle Hypertrophy and atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Ma, N.; Chen, D.; Lee, J.H.; Kuri, P.; Hernandez, E.B.; Kocan, J.; Mahmood, H.; Tichy, E.D.; Rompolas, P.; Mourkioti, F. Piezo1 regulates the regenerative capacity of skeletal muscles via orchestration of stem cell morphological states. Sci. Adv. 2022, 8, eabn0485. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; García-Prat, L.; Muñoz-Cánoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 2022, 23, 204–226. [Google Scholar] [CrossRef]
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Chen, B.; You, W.; Wang, Y.; Shan, T. The regulatory role of Myomaker and Myomixer-Myomerger-Minion in muscle development and regeneration. Cell Mol. Life Sci. 2020, 77, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Rudnicki, M.A. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 2000, 218, 115–124. [Google Scholar] [CrossRef]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- Zammit, P.S.; Partridge, T.A.; Yablonka-Reuveni, Z. The skeletal muscle satellite cell: The stem cell that came in from the cold. J. Histochem. Cytochem. 2006, 54, 1177–1191. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef]
- Perruchot, M.H.; Ecolan, P.; Sorensen, I.L.; Oksbjerg, N.; Lefaucheur, L. In vitro characterization of proliferation and differentiation of pig satellite cells. Differentiation 2012, 84, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Gregg, C.R.; Hutson, B.L.; Flees, J.J.; Starkey, C.W.; Starkey, J.D. Effect of standard and physiological cell culture temperatures on in vitro proliferation and differentiation of primary broiler chicken pectoralis major muscle satellite cells. Front. Physiol. 2023, 14, 1288809. [Google Scholar] [CrossRef]
- Flees, J.J.; Starkey, C.W.; Starkey, J.D. Effect of Different Basal Culture Media and Sera Type Combinations on Primary Broiler Chicken Muscle Satellite Cell Heterogeneity during Proliferation and Differentiation. Animals 2022, 12, 1425. [Google Scholar] [CrossRef]
- Buckingham, M.; Rigby, P.W. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell 2014, 28, 225–238. [Google Scholar] [CrossRef]
- Ehrenborg, E.; Krook, A. Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol. Rev. 2009, 61, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Angione, A.R.; Jiang, C.; Pan, D.; Wang, Y.; Kuang, S. PPARδ regulates satellite cell proliferation and skeletal muscle regeneration. Skelet. Muscle 2011, 1, 33. [Google Scholar] [CrossRef]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Chung, K.Y.; Johnson, B.J.; Go, G.W.; Kim, K.H.; Choi, C.W.; Smith, S.B. Co-culture of bovine muscle satellite cells with preadipocytes increases PPARγ and C/EBPβ gene expression in differentiated myoblasts and increases GPR43 gene expression in adipocytes. J. Nutr. Biochem. 2012, 24, 539–543. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Dammone, G.; Karaz, S.; Lukjanenko, L.; Winkler, C.; Sizzano, F.; Jacot, G.; Migliavacca, E.; Palini, A.; Desvergne, B.; Gilardi, F.; et al. PPARγ Controls Ectopic Adipogenesis and Cross-Talks with Myogenesis During Skeletal Muscle Regeneration. Int. J. Mol. Sci. 2018, 19, 2044. [Google Scholar] [CrossRef]
- Gu, H.; Zhou, Y.; Yang, J.; Li, J.; Peng, Y.; Zhang, X.; Miao, Y.; Jiang, W.; Bu, G.; Hou, L.; et al. Targeted overexpression of PPARγ in skeletal muscle by random insertion and CRISPR/Cas9 transgenic pig cloning enhances oxidative fiber formation and intramuscular fat deposition. FASEB J. 2021, 35, e21308. [Google Scholar] [CrossRef]
- Yi, L.; Li, Q.; Zhu, J.; Cheng, W.; Xie, Y.; Huang, Y.; Zhao, H.; Hao, M.; Wei, H.; Zhao, S. Single-nucleus RNA sequencing and lipidomics reveal characteristics of transcriptional and lipid composition in porcine longissimus dorsi muscle. BMC Genom. 2024, 25, 622. [Google Scholar] [CrossRef]
- Chakravarthy, M.V.; Abraha, T.W.; Schwartz, R.J.; Fiorotto, M.L.; Booth, F.W. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3′-kinase/Akt signaling pathway. J. Biol. Chem. 2000, 275, 35942–35952. [Google Scholar] [CrossRef]
- Engert, J.C.; Berglund, E.B.; Rosenthal, N. Proliferation precedes differentiation in IGF-I-stimulated myogenesis. J. Cell Biol. 1996, 135, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A.; Rosenthal, N. Maturation of the myogenic program is induced by postmitotic expression of insulin-like growth factor I. Mol. Cell. Biol. 1999, 19, 3115–3124. [Google Scholar] [CrossRef]
- Theret, M.; Gsaier, L.; Schaffer, B.; Juban, G.; Ben Larbi, S.; Weiss-Gayet, M.; Bultot, L.; Collodet, C.; Foretz, M.; Desplanches, D.; et al. AMPKα1-LDH pathway regulates muscle stem cell self-renewal by controlling metabolic homeostasis. EMBO J. 2017, 36, 1946–1962. [Google Scholar] [CrossRef]
- Mounier, R.; Theret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef]
- Vaz, F.M.; Wanders, R.J. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; Sartorelli, V. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Lamber, E.P.; Guicheney, P.; Pinotsis, N. The role of the M-band myomesin proteins in muscle integrity and cardiac disease. J. Biomed. Sci. 2022, 29, 18. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Su, J.; Huang, T.; Lian, L.; Nie, Q.; Zhang, X.; Li, J.; Wang, Y. Genome-wide mapping of the binding sites of myocyte enhancer factor 2A in chicken primary myoblasts. Poult. Sci. 2024, 103, 104097. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, Y.; Zhao, Z. Investigation of single nucleotide polymorphisms in differentially expressed genes and proteins reveals the genetic basis of skeletal muscle growth differences between Tibetan and Large White pigs. Anim. Biosci. 2024, 37, 2021–2032. [Google Scholar] [CrossRef]
- Zuloaga, R.; Aravena-Canales, D.; Aedo, J.E.; Osorio-Fuentealba, C.; Molina, A.; Valdés, J.A. Effect of 11-Deoxycorticosterone in the Transcriptomic Response to Stress in Rainbow Trout Skeletal Muscle. Genes 2023, 14, 512. [Google Scholar] [CrossRef]
- Geist, J.; Kontrogianni-Konstantopoulos, A. MYBPC1, an Emerging Myopathic Gene: What We Know and What We Need to Learn. Front. Physiol. 2016, 7, 410. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Gene Ontol. Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–51550. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2012, 8, 17–32. [Google Scholar] [CrossRef]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.C.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H. Evaluation of O2PLS in Omics data integration. BMC Bioinform. 2016, 17 (Suppl. S2), S11. [Google Scholar] [CrossRef] [PubMed]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2011, 28, 373–380. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequences (5′–3′) | Annealing Temperature | Size of Target Fragments |
|---|---|---|---|
| CHD | F: TGCAGAAGCAATATTACAAGT | 60 °C | 466/326 bp |
| R: AATTCATTATCATCTGGTGG | |||
| FABP5 | F: ACAATCACCGTAAAAACAGAAA | 60 °C | 186 bp |
| R: AAGTTTCCGTGTTATTATGGTC | |||
| PPARG | F: CAGGAGCAGAACAAAGAGGTAG | 60 °C | 185 bp |
| R: GAAGCCAGGAGAGTATATATGA | |||
| DEPTOR | F: CACGAGGAGAAGGTCATTAAGG | 60 °C | 128 bp |
| R: TTAATTGCTGTCTCTCGGTCGG | |||
| ACSL5 | F: GGAAAGACCCCATGTGTGAAGA | 60 °C | 175 bp |
| R: ACACAATGCAAAGATCTTCAGG | |||
| MYOM2 | F: AAGGATCCGGTTTGCCAGTGAG | 60 °C | 160 bp |
| R: AGCTCGACTTATTCTTTCCTCA | |||
| MYBPC1 | F: CTGAAAAGGGCAAAGATGAAGA | 60 °C | 162 bp |
| R: CGACGAATAAGGTGGATCTCTG | |||
| ELOVL6 | F: ACTGTACGCTGCCTTTATATTT | 60 °C | 115 bp |
| R: AAGTATTCTGAAGACGGCAAGG | |||
| PLIN1 | F: GAGGGCTATGAGGCGACCAAGA | 60 °C | 165 bp |
| R: CTTCTGATCTGCTTCCTCGTCC | |||
| GK | F: AAGAAGGATGGGTGGAACAAGA | 60 °C | 195 bp |
| R: ACACAATTGCGTTATAAAGAGG | |||
| IGF1 | F: TTCTTCTACCTTGGCCTGTGTT | 60 °C | 237 bp |
| R: AGCACAGTACATCTCCAGCCTC | |||
| β-actin | F: TCCGTGACATCAAGGAGAAG | 60 °C | 224 bp |
| R: CATGATGGAGTTGAAGGTGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wang, C.; Li, M.; Yang, Y.; Wang, H.; Chen, S.; He, D. Unveiling Key Genes and Crucial Pathways in Goose Muscle Satellite Cell Biology Through Integrated Transcriptomic and Metabolomic Analyses. Int. J. Mol. Sci. 2025, 26, 3710. https://doi.org/10.3390/ijms26083710
Liu Y, Wang C, Li M, Yang Y, Wang H, Chen S, He D. Unveiling Key Genes and Crucial Pathways in Goose Muscle Satellite Cell Biology Through Integrated Transcriptomic and Metabolomic Analyses. International Journal of Molecular Sciences. 2025; 26(8):3710. https://doi.org/10.3390/ijms26083710
Chicago/Turabian StyleLiu, Yi, Cui Wang, Mingxia Li, Yunzhou Yang, Huiying Wang, Shufang Chen, and Daqian He. 2025. "Unveiling Key Genes and Crucial Pathways in Goose Muscle Satellite Cell Biology Through Integrated Transcriptomic and Metabolomic Analyses" International Journal of Molecular Sciences 26, no. 8: 3710. https://doi.org/10.3390/ijms26083710
APA StyleLiu, Y., Wang, C., Li, M., Yang, Y., Wang, H., Chen, S., & He, D. (2025). Unveiling Key Genes and Crucial Pathways in Goose Muscle Satellite Cell Biology Through Integrated Transcriptomic and Metabolomic Analyses. International Journal of Molecular Sciences, 26(8), 3710. https://doi.org/10.3390/ijms26083710