
The Advanced Glycation End-Products (AGE)–Receptor for AGE System (RAGE): An Inflammatory Pathway Linking Obesity and Cardiovascular Diseases

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract

1. Introduction
2. Obesity and Cardiovascular Risk
2.1. Adipose Tissue Expansion in Obesity
2.2. How Can Obesity Affect the Heart and the Vascular System?
3. Advanced Glycation End-Products (AGE): From Synthesis to Mechanisms of Action
3.1. AGE Synthesis and Classification
3.2. AGE Accumulation
3.3. RAGE-Independent and RAGE-Mediated Effects
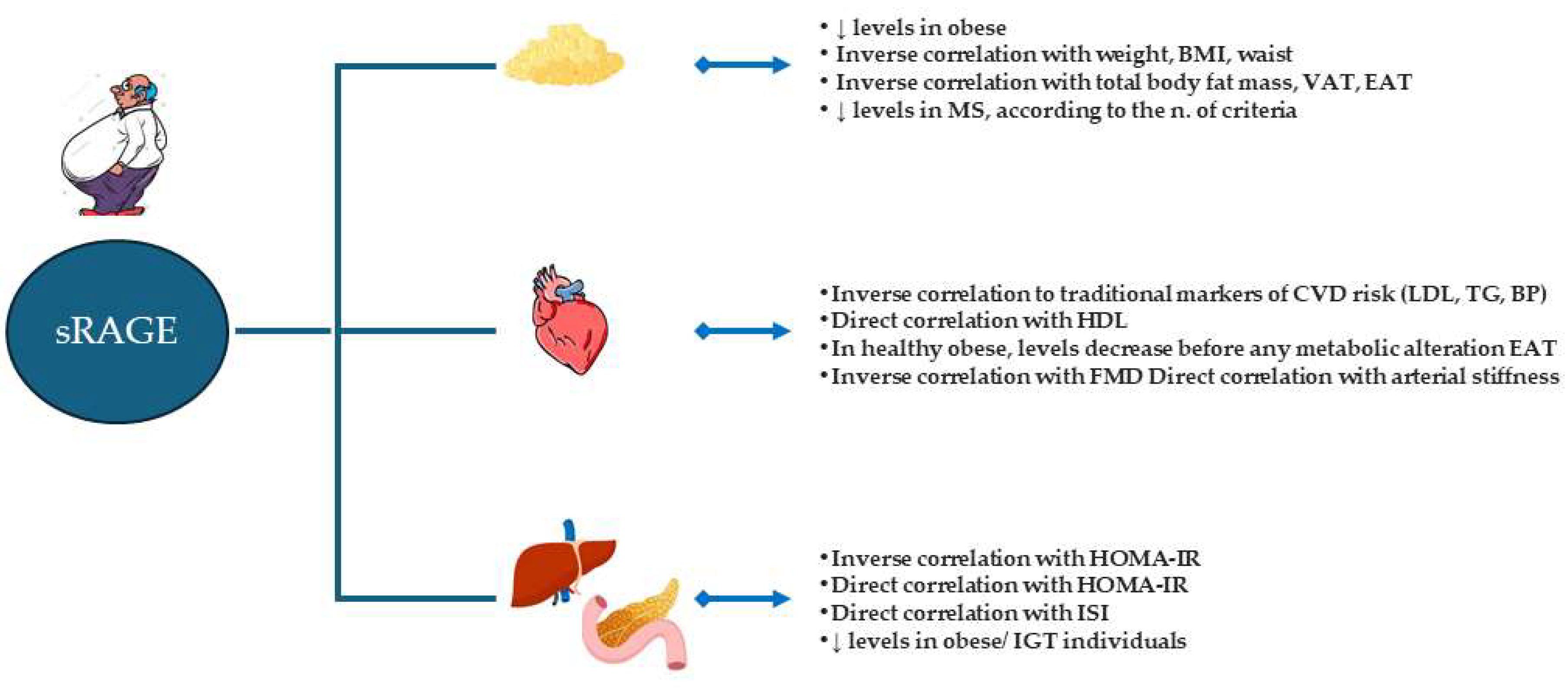
3.4. sRAGE as a Biomarker
4. Adipose Tissue: Source and Target of AGE
4.1. AGE Accumulation in Adipose Tissue
4.2. AGE Effects in the Adipose Tissue
4.3. How Can the AGE and RAGE System Contribute to CVD?
5. AGE and sRAGE: Role as Early Biomarkers in Obesity and CVD Risk
6. Potential Strategies for Reducing AGE–RAGE System Activation: From Preclinical to Clinical Studies

{kind=link}
{kind=link}
{kind=link}
| Mechanisms | Effects | Ref. |
|---|---|---|
| ↓ AGE INTAKE | - Moderate CR and physical activity by decreasing inflammation reduces EN-RAGE, but not sRAGE and sRAGE - A modest weight reduction is unlikely to improve decreased sRAGE - A 3-month diet did not affect sRAGE forms, but sRAGE predicts energy expenditure - CR diets and bariatric surgery reduced serum AGE - Weight loss induced by bariatric surgery increased sRAGE | [155] [156] [157] [158] [159] |
| ↓ AGE | - tRES and HESP, at concentrations achieved clinically, synergized to increase Glo1 expression - Streptozotocin-induced diabetic rats lycopene increased Glo-1 - PGG could be an effective agent to block Glu/MGO-triggered glycation in vitro - AAP-2S inhibited AGE synthesis in vitro - DSP supplementation reduced in human pentosidine levels - In mice, Liraglutide treatment reduced serum AGE levels | [165] [166] [167] [168] [169] [184] |
| ↑ sRAGE | - DSP supplementation increased sRAGE in humans | [169] |
| - Enalapril/lercanidipine increased sRAGE in humans | [174] | |
| - In diabetic rats, ACEi reduced the accumulation of AGE in DM partly by increasing the production and secretion of sRAGE into plasma | [175] | |
| - In animal and human, Vitamin D treatment increased sRAGE levels, particularly in vitamin D-deficient situations | [176] | |
| BLOCKING AGE | - sRAGE protected against liver fibrosis - In pigs, RAGE antagonism and sRAGE decreased lung inflammation - sRAGE inhibits I/R-induced apoptosis, both in the hearts of mice and cardiomyocytes - RAGE administration prevented renal tubular damage in models of ischemia/reperfusion-induced AKI - sRAGE effectively lessened microcirculation impairment and vascular injury after SAH - Empagliflozin reduced the AGE–RAGE-oxidative stress- induced inflammatory reactions in the adipose tissues of db/db mice | [177] [178] [179] [180] [181] [188] |
| INHIBITING RAGE EXPRESSION/ RAGE ACTIVATION | - PPARgamma agonists inhibit RAGE expression in vitro | [182] |
| - In mice, losartan attenuated hepatic I/R-induced RAGE expression | [183] | |
| - In mice, liraglutide reduced the expression of RAGE in the aorta | [184] | |
| - In vitro, statins decreased RAGE expression | [185] | |
| - In mice, liraglutide downregulated kidney RAGE | [186] | |
| - In vitro, liraglutide reduced the number of intact RAGE on the cell surface by promoting its shedding | [187] | |
| - The RAGE inhibitor azeliragon had promising results on slowing the loss of cognition in preclinical AD models and in a Phase2b study | [194] | |
| - The RAGE inhibitor azeliragon significantly inhibited tumor growth in a pancreatic cancer xenograft model | [195] | |
| - The RAGE inhibitor azeliragon suppressed metastasis in triple-negative breast cancer | [196] | |
| - Small-molecule RAGE antagonists blocked suPAR signaling suPAR-mediated inflammatory responses in vitro podocytes | [198] | |
| - The RAGE inhibitor azeliragon ameliorated streptozotocin-induced diabetic neuropathy | [199] | |
| BLOCKING RAGE SIGNALING | - In mice, antagonism of RAGE signaling by RAGE229 optimized healthy body mass and composition and metabolic fitness - Antagonism of RAGE signaling by small-molecule competitive inhibitors regulated signaling networks involved in inflammation and cell migration in vitro and in vivo - Targeting RAGE signaling with RAGE229 mitigated diabetic complications in rodents by attenuating inflammatory signaling | [189] [190] [192] |
7. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hemat Jouy, S.; Mohan, S.; Scichilone, G.; Mostafa, A.; Mahmoud, A.M. Adipokines in the Crosstalk between Adipose Tissues and Other Organs: Implications in Cardiometabolic Diseases. Biomedicines 2024, 12, 2129. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. An overview of obesity-related complications: The epidemiological evidence linking body weight and other markers of obesity to adverse health outcomes. Diabetes Obes. Metab. 2025. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M. Adipose Tissue Dynamics, Thermogenesis, and Interorgan Connections for Preventing Obesity and Metabolic Disorders. JMA J. 2024, 7, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Koyama, H.; Shoji, T.; Monden, M.; Fukumoto, S.; Tanaka, S.; Otsuka, Y.; Mima, Y.; Morioka, T.; Mori, K.; et al. Receptor for advanced glycation end-products (RAGE) regulation of adiposity and adiponectin is associated with atherogenesis in apoE-deficient mouse. Atherosclerosis 2011, 211, 431–436. [Google Scholar] [CrossRef]
- Monden, M.; Koyama, H.; Otsuka, Y.; Morioka, T.; Mori, K.; Shoji, T.; Mima, Y.; Motoyama, K.; Fukumoto, S.; Shioi, A.; et al. Receptor for advanced glycation end products regulates adipocyte hypertrophy and insulin sensitivity in mice: Involvement of Toll-like receptor 2. Diabetes 2013, 62, 478–489. [Google Scholar] [CrossRef]
- Takata, T.; Inoue, S.; Masauji, T.; Miyazawa, K.; Motoo, Y. Generation and Accumulation of Various Advanced Glycation End-Products in Cardiomyocytes May Induce Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 7319. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Erlanson-Albertsson, C. Adipose cell size: Importance in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R284–R295. [Google Scholar] [CrossRef]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W., 2nd; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef]
- Cho, C.H.; Koh, Y.J.; Han, J.; Sung, H.K.; Jong Lee, H.; Morisada, T.; Schwendener, R.A.; Brekken, R.A.; Kang, G.; Oike, Y.; et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ. Res. 2007, 100, e47–e57. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Petkova, A.P.; Granneman, J.G. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013, 18, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Lim, H.W.; Kim, Y.H.; Ho, W.Y.; Foong, Y.H.; Nelson, V.L.; Nguyen, H.C.B.; Chegireddy, K.; Kim, J.; Habertheuer, A.; et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 2018, 115, E5096–E5105. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698. [Google Scholar] [CrossRef]
- Haase, J.; Weyer, U.; Immig, K.; Kloting, N.; Bluher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Galvez, I.; Hinchado, M.D.; Martin-Cordero, L.; Moran-Plata, F.J.; Graham, G.; Francisco-Morcillo, J.; Ortega, E. The anti-inflammatory and bioregulatory effects of habitual exercise in high-fat diet-induced obesity involve crown-like structures and MCP-1 in white adipose tissue. Exerc. Immunol. Rev. 2023, 29, 111–120. [Google Scholar]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Morris, D.L.; Singer, K.; Lumeng, C.N. Adipose tissue macrophages: Phenotypic plasticity and diversity in lean and obese states. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 341–346. [Google Scholar] [CrossRef]
- Li, X.; Ren, Y.; Chang, K.; Wu, W.; Griffiths, H.R.; Lu, S.; Gao, D. Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Front. Immunol. 2023, 14, 1153915. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Cereijo, R.; Gavalda-Navarro, A.; Villarroya, J.; Giralt, M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J. Intern. Med. 2018, 284, 492–504. [Google Scholar] [CrossRef]
- Bluher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best. Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 163–177. [Google Scholar] [CrossRef]
- Henderson, G.C. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients 2021, 13, 2590. [Google Scholar] [CrossRef]
- Wieder, N.; Fried, J.C.; Kim, C.; Sidhom, E.H.; Brown, M.R.; Marshall, J.L.; Arevalo, C.; Dvela-Levitt, M.; Kost-Alimova, M.; Sieber, J.; et al. FALCON systematically interrogates free fatty acid biology and identifies a novel mediator of lipotoxicity. Cell Metab. 2023, 35, 887–905.e811. [Google Scholar] [CrossRef]
- Fang, Y.; Gonzales-Nieves, S.; Cifarelli, V.; Imoukhuede, P.I. Sex Differences in VEGF and PDGF Ligand and Receptor Protein Expression during Adipose Tissue Expansion and Regression. bioRxiv 2025. online ahead of print. [Google Scholar] [CrossRef]
- Kunes, J.; Hojna, S.; Mrazikova, L.; Montezano, A.; Touyz, R.M.; Maletinska, L. Obesity, Cardiovascular and Neurodegenerative Diseases: Potential Common Mechanisms. Physiol. Res. 2023, 72 (Suppl. S2), S73–S90. [Google Scholar] [CrossRef]
- Zeljkovic, A.; Vekic, J.; Stefanovic, A. Obesity and dyslipidemia in early life: Impact on cardiometabolic risk. Metabolism 2024, 156, 155919. [Google Scholar] [CrossRef]
- Dal, N.; Bilici, S. Dietary Modulations in Preventing Cardiometabolic Risk in Individuals with Type 2 Diabetes. Curr. Nutr. Rep. 2024, 13, 412–421. [Google Scholar] [CrossRef]
- Tomlinson, B.; Chan, P. Effects of glucose-lowering drugs on cardiovascular outcomes in patients with type 2 diabetes: An update. Expert. Opin. Drug Metab. Toxicol. 2024, 20, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Ouwens, D.M.; Diamant, M.; Fodor, M.; Habets, D.D.J.; Pelsers, M.; El Hasnaoui, M.; Dang, Z.C.; van den Brom, C.E.; Vlasblom, R.; Rietdijk, A.; et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007, 50, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Banke, N.H.; Wende, A.R.; Leone, T.C.; O’Donnell, J.M.; Abel, E.D.; Kelly, D.P.; Lewandowski, E.D. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 2010, 107, 233–241. [Google Scholar] [CrossRef]
- Bucci, M.; Borra, R.; Nagren, K.; Parkka, J.P.; Del Ry, S.; Maggio, R.; Tuunanen, H.; Viljanen, T.; Cabiati, M.; Rigazio, S.; et al. Trimetazidine reduces endogenous free fatty acid oxidation and improves myocardial efficiency in obese humans. Cardiovasc. Ther. 2012, 30, 333–341. [Google Scholar] [CrossRef]
- Young, M.E.; Guthrie, P.H.; Razeghi, P.; Leighton, B.; Abbasi, S.; Patil, S.; Youker, K.A.; Taegtmeyer, H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 2002, 51, 2587–2595. [Google Scholar] [CrossRef]
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.; Marsala, J.; Meyer, T.; et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004, 109, 2191–2196. [Google Scholar] [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef]
- Dozio, E.; Vianello, E.; Bandera, F.; Longhi, E.; Brizzola, S.; Nebuloni, M.; Corsi Romanelli, M.M. Soluble Receptor for Advanced Glycation End Products: A Protective Molecule against Intramyocardial Lipid Accumulation in Obese Zucker Rats? Mediat. Inflamm. 2019, 2019, 2712376. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Dadsena, S.; Holthuis, J.C.M. A switchable ceramide transfer protein for dissecting the mechanism of ceramide-induced mitochondrial apoptosis. FEBS Lett. 2020, 594, 3739–3750. [Google Scholar] [CrossRef] [PubMed]
- Choi, R.H.; Tatum, S.M.; Symons, J.D.; Summers, S.A.; Holland, W.L. Ceramides and other sphingolipids as drivers of cardiovascular disease. Nat. Rev. Cardiol. 2021, 18, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109 (Suppl. S1), II2-10. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Fantuzzi, G.; Mazzone, T. Adipose tissue and atherosclerosis: Exploring the connection. Arter. Thromb. Vasc. Biol. 2007, 27, 996–1003. [Google Scholar] [CrossRef]
- Emerging Risk Factors, C.; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar] [CrossRef]
- Chen, C.Y.; Zhang, J.Q.; Li, L.; Guo, M.M.; He, Y.F.; Dong, Y.M.; Meng, H.; Yi, F. Advanced Glycation End Products in the Skin: Molecular Mechanisms, Methods of Measurement, and Inhibitory Pathways. Front. Med. 2022, 9, 837222. [Google Scholar] [CrossRef]
- Cho, S.J.; Roman, G.; Yeboah, F.; Konishi, Y. The road to advanced glycation end products: A mechanistic perspective. Curr. Med. Chem. 2007, 14, 1653–1671. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Dozio, E.; Caldiroli, L.; Molinari, P.; Castellano, G.; Delfrate, N.W.; Romanelli, M.M.C.; Vettoretti, S. Accelerated AGEing: The Impact of Advanced Glycation End Products on the Prognosis of Chronic Kidney Disease. Antioxidants 2023, 12, 584. [Google Scholar] [CrossRef] [PubMed]
- Twarda-Clapa, A.; Olczak, A.; Bialkowska, A.M.; Koziolkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 54. [Google Scholar] [CrossRef]
- Kikuchi, C.; Sakasai-Sakai, A.; Okimura, R.; Tanaka, H.; Takata, T.; Takeuchi, M.; Matsunaga, T. Accumulation of Toxic Advanced Glycation End-Products Induces Cytotoxicity and Inflammation in Hepatocyte-Like Cells Differentiated from Human Induced Pluripotent Stem Cells. Biol. Pharm. Bull. 2021, 44, 1399–1402. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular Toxic Advanced Glycation End-Products May Induce Cell Death and Suppress Cardiac Fibroblasts. Metabolites 2022, 12, 615. [Google Scholar] [CrossRef]
- Wang, Y.X.; Xu, H.; Liu, X.; Liu, L.; Wu, Y.N.; Gong, Z.Y. Studies on mechanism of free Nepsilon-(carboxymethyl)lysine-induced toxic injury in mice. J. Biochem. Mol. Toxicol. 2019, 33, e22322. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Sun, Z. Dietary N(epsilon)-(carboxymethyl) lysine affects cardiac glucose metabolism and myocardial remodeling in mice. World J. Diabetes 2022, 13, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, L.; Yin, K.; Zang, G.; Qian, Y.; Mao, X.; Li, L.; Jing, Q.; Wang, Z. SIRT3-and FAK-mediated acetylation-phosphorylation crosstalk of NFATc1 regulates N(epsilon)-carboxymethyl-lysine-induced vascular calcification in diabetes mellitus. Atherosclerosis 2023, 377, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Burke, G.A.; Chen, F.; McMullen, C.B.; Vlassara, H. Advanced glycation end-product receptor interactions on microvascular cells occur within caveolin-rich membrane domains. FASEB J. 2000, 14, 2390–2392. [Google Scholar] [CrossRef]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef]
- Busch, M.; Franke, S.; Ruster, C.; Wolf, G. Advanced glycation end-products and the kidney. Eur. J. Clin. Investig. 2010, 40, 742–755. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- Vlassara, H.; Fuh, H.; Donnelly, T.; Cybulsky, M. Advanced glycation endproducts promote adhesion molecule (VCAM-1, ICAM-1) expression and atheroma formation in normal rabbits. Mol. Med. 1995, 1, 447–456. [Google Scholar] [CrossRef]
- Vlassara, H.; Li, Y.M.; Imani, F.; Wojciechowicz, D.; Yang, Z.; Liu, F.T.; Cerami, A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): A new member of the AGE-receptor complex. Mol. Med. 1995, 1, 634–646. [Google Scholar] [CrossRef]
- Iacobini, C.; Amadio, L.; Oddi, G.; Ricci, C.; Barsotti, P.; Missori, S.; Sorcini, M.; Di Mario, U.; Pricci, F.; Pugliese, G. Role of galectin-3 in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. S3), S264–S270. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Charonis, A.S.; Reger, L.A.; Dege, J.E.; Kouzi-Koliakos, K.; Furcht, L.T.; Wohlhueter, R.M.; Tsilibary, E.C. Laminin alterations after in vitro nonenzymatic glycosylation. Diabetes 1990, 39, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.E.; Gerbitz, K.D.; Olgemoller, B.; Schleicher, E.D. In-vitro carboxymethylation of low density lipoprotein alters its metabolism via the high-affinity receptor. Horm. Metab. Res. 1993, 25, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Giardino, I.; Edelstein, D.; Brownlee, M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J. Clin. Investig. 1994, 94, 110–117. [Google Scholar] [CrossRef]
- Basta, G.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 2004, 63, 582–592. [Google Scholar] [CrossRef]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef]
- Strieder-Barboza, C.; Baker, N.A.; Flesher, C.G.; Karmakar, M.; Neeley, C.K.; Polsinelli, D.; Dimick, J.B.; Finks, J.F.; Ghaferi, A.A.; Varban, O.A.; et al. Advanced glycation end-products regulate extracellular matrix-adipocyte metabolic crosstalk in diabetes. Sci. Rep. 2019, 9, 19748. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Deepu, V.; Rai, V.; Agrawal, D.K. Quantitative Assessment of Intracellular Effectors and Cellular Response in RAGE Activation. Arch. Intern. Med. Res. 2024, 7, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Parkin, E.; Harris, B. A disintegrin and metalloproteinase (ADAM)-mediated ectodomain shedding of ADAM10. J. Neurochem. 2009, 108, 1464–1479. [Google Scholar] [CrossRef]
- Lee, A.C.; Lam, J.K.; Shiu, S.W.; Wong, Y.; Betteridge, D.J.; Tan, K.C. Serum Level of Soluble Receptor for Advanced Glycation End Products Is Associated with A Disintegrin and Metalloproteinase 10 in Type 1 Diabetes. PLoS ONE 2015, 10, e0137330. [Google Scholar] [CrossRef]
- Revuelta-Lopez, E.; Castellano, J.; Roura, S.; Galvez-Monton, C.; Nasarre, L.; Benitez, S.; Bayes-Genis, A.; Badimon, L.; Llorente-Cortes, V. Hypoxia induces metalloproteinase-9 activation and human vascular smooth muscle cell migration through low-density lipoprotein receptor-related protein 1-mediated Pyk2 phosphorylation. Arter. Thromb. Vasc. Biol. 2013, 33, 2877–2887. [Google Scholar] [CrossRef]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef]
- Selvin, E.; Halushka, M.K.; Rawlings, A.M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. sRAGE and risk of diabetes, cardiovascular disease, and death. Diabetes 2013, 62, 2116–2121. [Google Scholar] [CrossRef]
- Momma, H.; Niu, K.; Kobayashi, Y.; Huang, C.; Chujo, M.; Otomo, A.; Tadaura, H.; Miyata, T.; Nagatomi, R. Higher serum soluble receptor for advanced glycation end product levels and lower prevalence of metabolic syndrome among Japanese adult men: A cross-sectional study. Diabetol. Metab. Syndr. 2014, 6, 33. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, H.B.; Luo, M.; Zheng, H. Serum soluble RAGE level inversely correlates with left ventricular hypertrophy in essential hypertension patients. Genet. Mol. Res. 2016, 15, 15. [Google Scholar] [CrossRef]
- Dozio, E.; Briganti, S.; Delnevo, A.; Vianello, E.; Ermetici, F.; Secchi, F.; Sardanelli, F.; Morricone, L.; Malavazos, A.E.; Corsi Romanelli, M.M. Relationship between soluble receptor for advanced glycation end products (sRAGE), body composition and fat distribution in healthy women. Eur. J. Nutr. 2017, 56, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Leurs, P.; Lindholm, B. The AGE-RAGE pathway and its relation to cardiovascular disease in patients with chronic kidney disease. Arch. Med. Res. 2013, 44, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Li, W.P.; Shen, X.H.; Guo, X.Y.; Hua, B.; Li, H.W. Dynamic fluctuations of advanced glycation end products and its C-terminal truncated receptor level in patients with acute ST-segment elevation myocardial infarction and undergoing diabetes or not: A retrospective study. Medicine 2018, 97, e11278. [Google Scholar] [CrossRef]
- Sabbatinelli, J.; Castiglione, S.; Macri, F.; Giuliani, A.; Ramini, D.; Vinci, M.C.; Tortato, E.; Bonfigli, A.R.; Olivieri, F.; Raucci, A. Circulating levels of AGEs and soluble RAGE isoforms are associated with all-cause mortality and development of cardiovascular complications in type 2 diabetes: A retrospective cohort study. Cardiovasc. Diabetol. 2022, 21, 95. [Google Scholar] [CrossRef]
- Jensen, L.J.; Flyvbjerg, A.; Bjerre, M. Soluble Receptor for Advanced Glycation End Product: A Biomarker for Acute Coronary Syndrome. Biomed. Res. Int. 2015, 2015, 815942. [Google Scholar] [CrossRef]
- Xu, B.; Chibber, R.; Ruggiero, D.; Kohner, E.; Ritter, J.; Ferro, A. Impairment of vascular endothelial nitric oxide synthase activity by advanced glycation end products. FASEB J. 2003, 17, 1289–1291. [Google Scholar] [CrossRef]
- Li, J.; Hou, F.; Guo, Z.; Shan, Y.; Zhang, X.; Liu, Z. Advanced glycation end products upregulate C-reactive protein synthesis by human hepatocytes through stimulation of monocyte IL-6 and IL-1 beta production. Scand. J. Immunol. 2007, 66, 555–562. [Google Scholar] [CrossRef]
- Gawlowski, T.; Stratmann, B.; Ruetter, R.; Buenting, C.E.; Menart, B.; Weiss, J.; Vlassara, H.; Koschinsky, T.; Tschoepe, D. Advanced glycation end products strongly activate platelets. Eur. J. Nutr. 2009, 48, 475–481. [Google Scholar] [CrossRef]
- Lee, B.W.; Chae, H.Y.; Kwon, S.J.; Park, S.Y.; Ihm, J.; Ihm, S.H. RAGE ligands induce apoptotic cell death of pancreatic beta-cells via oxidative stress. Int. J. Mol. Med. 2010, 26, 813–818. [Google Scholar]
- Plotkin, L.I.; Essex, A.L.; Davis, H.M. RAGE Signaling in Skeletal Biology. Curr. Osteoporos. Rep. 2019, 17, 16–25. [Google Scholar] [CrossRef]
- Dozio, E.; Vettoretti, S.; Lungarella, G.; Messa, P.; Corsi Romanelli, M.M. Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress. Biomedicines 2021, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Du, Z.; Shu, X.; Zhu, L.; Wu, J.; Gao, Q.; Wang, L.; Chen, N.; Li, Y.; Luo, M.; et al. Role of RAGE in obesity-induced adipose tissue inflammation and insulin resistance. Cell Death Discov. 2021, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; He, J.; Wang, J.; Liu, J.; Chen, Z.; Deng, B.; Wei, L.; Wu, H.; Liang, B.; Li, H.; et al. Knockout RAGE alleviates cardiac fibrosis through repressing endothelial-to-mesenchymal transition (EndMT) mediated by autophagy. Cell Death Dis. 2021, 12, 470. [Google Scholar] [CrossRef]
- Liu, B.; Sun, T.; Li, H.; Qiu, S.; Li, Y.; Zhang, D. Proximal tubular RAGE mediated the renal fibrosis in UUO model mice via upregulation of autophagy. Cell Death Dis. 2022, 13, 399. [Google Scholar] [CrossRef]
- Snelson, M.; Lucut, E.; Coughlan, M.T. The Role of AGE-RAGE Signalling as a Modulator of Gut Permeability in Diabetes. Int. J. Mol. Sci. 2022, 23, 1766. [Google Scholar] [CrossRef]
- Durak, S.; Yilmazer, Y.; Celik, F.; Yesiloglu, E.; Karakose, D.; Dincol, S.; Ucak, S.; Yaman, M.; Zeybek, U. Investigation of Advanced Glycation End Products in Liver, Adipose, and Renal Tissue of Mice on a High-Fat Diet. Cell Biochem. Biophys. 2024, 82, 1101–1108. [Google Scholar] [CrossRef]
- Tessier, F.J.; Niquet-Leridon, C.; Jacolot, P.; Jouquand, C.; Genin, M.; Schmidt, A.M.; Grossin, N.; Boulanger, E. Quantitative assessment of organ distribution of dietary protein-bound (13) C-labeled N(varepsilon) -carboxymethyllysine after a chronic oral exposure in mice. Mol. Nutr. Food Res. 2016, 60, 2446–2456. [Google Scholar] [CrossRef]
- Gaens, K.H.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Blaak, E.E.; et al. Nepsilon-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arter. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef]
- Son, K.H.; Son, M.; Ahn, H.; Oh, S.; Yum, Y.; Choi, C.H.; Park, K.Y.; Byun, K. Age-related accumulation of advanced glycation end-products-albumin, S100beta, and the expressions of advanced glycation end product receptor differ in visceral and subcutaneous fat. Biochem. Biophys. Res. Commun. 2016, 477, 271–276. [Google Scholar] [CrossRef]
- Song, F.; Hurtado del Pozo, C.; Rosario, R.; Zou, Y.S.; Ananthakrishnan, R.; Xu, X.; Patel, P.R.; Benoit, V.M.; Yan, S.F.; Li, H.; et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes 2014, 63, 1948–1965. [Google Scholar] [CrossRef]
- Du, Z.; Wu, J.; Feng, Z.; Ma, X.; Zhang, T.; Shu, X.; Xu, J.; Wang, L.; Luo, M.; Wu, J. RAGE displays sex-specific differences in obesity-induced adipose tissue insulin resistance. Biol. Sex. Differ. 2022, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Bujo, H.; Yamagishi, S.; Takeuchi, M.; Imaizumi, T.; Saito, Y. Advanced glycation end products attenuate cellular insulin sensitivity by increasing the generation of intracellular reactive oxygen species in adipocytes. Diabetes Res. Clin. Pr. 2007, 76, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Huang, H.W.; Huang, S.M.; Lin, J.A.; Yeh, C.T.; Yen, G.C. AGE-induced interference of glucose uptake and transport as a possible cause of insulin resistance in adipocytes. J. Agric. Food Chem. 2011, 59, 7978–7984. [Google Scholar] [CrossRef]
- Izgilov, R.; Naftaly, A.; Benayahu, D. Advanced Glycation End Products Effects on Adipocyte Niche Stiffness and Cell Signaling. Int. J. Mol. Sci. 2023, 24, 2261. [Google Scholar] [CrossRef]
- Fasshauer, M.; Klein, J.; Lossner, U.; Paschke, R. Interleukin (IL)-6 mRNA expression is stimulated by insulin, isoproterenol, tumour necrosis factor alpha, growth hormone, and IL-6 in 3T3-L1 adipocytes. Horm. Metab. Res. 2003, 35, 147–152. [Google Scholar] [CrossRef]
- Dozio, E.; Vianello, E.; Briganti, S.; Lamont, J.; Tacchini, L.; Schmitz, G.; Corsi Romanelli, M.M. Expression of the Receptor for Advanced Glycation End Products in Epicardial Fat: Link with Tissue Thickness and Local Insulin Resistance in Coronary Artery Disease. J. Diabetes Res. 2016, 2016, 2327341. [Google Scholar] [CrossRef]
- Hurtado Del Pozo, C.; Ruiz, H.H.; Arivazhagan, L.; Aranda, J.F.; Shim, C.; Daya, P.; Derk, J.; MacLean, M.; He, M.; Frye, L.; et al. A Receptor of the Immunoglobulin Superfamily Regulates Adaptive Thermogenesis. Cell Rep. 2019, 28, 773–791.e777. [Google Scholar] [CrossRef]
- Ding, Y.S.; Malik, N.; Mendoza, S.; Tuchman, D.; Del Pozo, C.H.; Diez, R.L.; Schmidt, A.M. PET imaging study of brown adipose tissue (BAT) activity in mice devoid of receptor for advanced glycation end products (RAGE). J. Biosci. 2019, 44, 93. [Google Scholar] [CrossRef]
- Collins, S. beta-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front. Endocrinol. 2011, 2, 102. [Google Scholar] [CrossRef]
- Tayyib, N.A.; Ramaiah, P.; Alshahrani, S.H.; Margiana, R.; Almalki, S.G.; Kareem, A.K.; Zabibah, R.S.; Shbeer, A.M.; Ali, S.H.J.; Mustafa, Y.F. Soluble receptor for advanced glycation end products (sRAGE) is associated with obesity rates: A systematic review and meta-analysis of cross-sectional study. BMC Endocr. Disord. 2023, 23, 275. [Google Scholar] [CrossRef]
- Davis, K.E.; Prasad, C.; Vijayagopal, P.; Juma, S.; Imrhan, V. Serum soluble receptor for advanced glycation end products correlates inversely with measures of adiposity in young adults. Nutr. Res. 2014, 34, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Dong, C.; Gardener, H.; Elkind, M.S.; Wright, C.B.; Goldberg, R.; Sacco, R.L.; Rundek, T. Serum levels of soluble receptor for advanced glycation end-products and metabolic syndrome: The Northern Manhattan Study. Metabolism 2014, 63, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- He, C.T.; Lee, C.H.; Hsieh, C.H.; Hsiao, F.C.; Kuo, P.; Chu, N.F.; Hung, Y.J. Soluble form of receptor for advanced glycation end products is associated with obesity and metabolic syndrome in adolescents. Int. J. Endocrinol. 2014, 2014, 657607. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.; Kamal, S.; Kholousi, S.; El-Bassyouni, H.T.; Yousef, W.; Reyad, H.; Mohamed, R.; Basha, W.A. Serum soluble receptor of advanced glycation end products and risk of metabolic syndrome in Egyptian obese women. EXCLI J. 2017, 16, 973–980. [Google Scholar] [CrossRef]
- Vega-Cardenas, M.; Vargas-Morales, J.M.; Portales-Perez, D.P.; Gomez-Ojeda, A.; Luevano-Contreras, C.; Aradillas-Garcia, C. Soluble receptor for advanced glycation end-products (sRAGE) in childhood obesity: Association with gene expression of RAGE and cardiometabolic markers. Nutr. Hosp. 2023, 40, 960–966. [Google Scholar] [CrossRef]
- Rodriguez-Mortera, R.; Luevano-Contreras, C.; Solorio-Meza, S.; Gomez-Ojeda, A.; Caccavello, R.; Bains, Y.; Gugliucci, A.; Garay-Sevilla, M.E. Soluble Receptor for Advanced Glycation End Products and Its Correlation with Vascular Damage in Adolescents with Obesity. Horm. Res. Paediatr. 2019, 92, 28–35. [Google Scholar] [CrossRef]
- Flores-Ramirez, A.G.; Ibarra-Reynoso, L.D.R.; Garay-Sevilla, M.E. Soluble receptor for advanced glycation end products and lipid profile ratio as cardiovascular risk markers in children with obesity. Gac. Med. Mex. 2023, 159, 10–16. [Google Scholar] [CrossRef]
- D’Adamo, E.; Giannini, C.; Chiavaroli, V.; de Giorgis, T.; Verrotti, A.; Chiarelli, F.; Mohn, A. What is the significance of soluble and endogenous secretory receptor for advanced glycation end products in liver steatosis in obese prepubertal children? Antioxid. Redox Signal 2011, 14, 1167–1172. [Google Scholar] [CrossRef]
- Miranda, E.R.; Somal, V.S.; Mey, J.T.; Blackburn, B.K.; Wang, E.; Farabi, S.; Karstoft, K.; Fealy, C.E.; Kashyap, S.; Kirwan, J.P.; et al. Circulating soluble RAGE isoforms are attenuated in obese, impaired-glucose-tolerant individuals and are associated with the development of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E631–E640. [Google Scholar] [CrossRef]
- Villegas-Rodriguez, M.E.; Uribarri, J.; Solorio-Meza, S.E.; Fajardo-Araujo, M.E.; Cai, W.; Torres-Graciano, S.; Rangel-Salazar, R.; Wrobel, K.; Garay-Sevilla, M.E. The AGE-RAGE Axis and Its Relationship to Markers of Cardiovascular Disease in Newly Diagnosed Diabetic Patients. PLoS ONE 2016, 11, e0159175. [Google Scholar] [CrossRef]
- Koborova, I.; Gurecka, R.; Csongova, M.; Volkovova, K.; Szoko, E.; Tabi, T.; Sebekova, K. Association between metabolically healthy central obesity in women and levels of soluble receptor for advanced glycation end products, soluble vascular adhesion protein-1, and the activity of semicarbazide-sensitive amine oxidase. Croat. Med. J. 2017, 58, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gurecka, R.; Koborova, I.; Csongova, M.; Sebek, J.; Sebekova, K. Correlation among soluble receptors for advanced glycation end-products, soluble vascular adhesion protein-1/semicarbazide-sensitive amine oxidase (sVAP-1) and cardiometabolic risk markers in apparently healthy adolescents: A cross-sectional study. Glycoconj. J. 2016, 33, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Mariscal, F.M.; Lopez-Moreno, A.; Torres-Pena, J.D.; Gomez-Luna, P.; Arenas-de Larriva, A.P.; Romero-Cabrera, J.L.; Luque, R.M.; Uribarri, J.; Perez-Martinez, P.; Delgado-Lista, J.; et al. Modulation of circulating levels of advanced glycation end products and its impact on intima-media thickness of both common carotid arteries: CORDIOPREV randomised controlled trial. Cardiovasc. Diabetol. 2024, 23, 361. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, T.; Qi, Y.; Luo, S.; Xia, Y.; Lang, B.; Zhang, B.; Zheng, S. AGE-RAGE Axis and Cardiovascular Diseases: Pathophysiologic Mechanisms and Prospects for Clinical Applications. Cardiovasc. Drugs Ther. 2024. [Google Scholar] [CrossRef]
- Dozio, E.; Massaccesi, L.; Corsi Romanelli, M.M. Glycation and Glycosylation in Cardiovascular Remodeling: Focus on Advanced Glycation End Products and O-Linked Glycosylations as Glucose-Related Pathogenetic Factors and Disease Markers. J. Clin. Med. 2021, 10, 4792. [Google Scholar] [CrossRef]
- Scavello, F.; Zeni, F.; Milano, G.; Macri, F.; Castiglione, S.; Zuccolo, E.; Scopece, A.; Pezone, G.; Tedesco, C.C.; Nigro, P.; et al. Soluble Receptor for Advanced Glycation End-products regulates age-associated Cardiac Fibrosis. Int. J. Biol. Sci. 2021, 17, 2399–2416. [Google Scholar] [CrossRef]
- Liang, B.; Zhou, Z.; Yang, Z.; Liu, J.; Zhang, L.; He, J.; Li, H.; Huang, Y.; Yang, Q.; Xian, S.; et al. AGEs-RAGE axis mediates myocardial fibrosis via activation of cardiac fibroblasts induced by autophagy in heart failure. Exp. Physiol. 2022, 107, 879–891. [Google Scholar] [CrossRef]
- Zheng, D.L.; Wu, Q.R.; Zeng, P.; Li, S.M.; Cai, Y.J.; Chen, S.Z.; Luo, X.S.; Kuang, S.J.; Rao, F.; Lai, Y.Y.; et al. Advanced glycation end products induce senescence of atrial myocytes and increase susceptibility of atrial fibrillation in diabetic mice. Aging Cell 2022, 21, e13734. [Google Scholar] [CrossRef]
- He, J.; Wei, L.; Tan, S.; Liang, B.; Liu, J.; Lu, L.; Wang, T.; Wang, J.; Huang, Y.; Chen, Z.; et al. Macrophage RAGE deficiency prevents myocardial fibrosis by repressing autophagy-mediated macrophage alternative activation. FASEB J. 2023, 37, e23259. [Google Scholar] [CrossRef]
- Tan, H.; Hu, J.; Zuo, W.; Huang, Y.; Cui, J.; Gong, F.; Bai, W. Activation of the High Mobility Group Box 1/Receptor for Advanced Glycation Endproducts /NOD-like Receptor Family Pyrin Domain-Containing 3 Axis Under Chronic Intermittent Hypoxia Induction Promotes the Progression of Atherosclerosis in ApoE−/− Mice. J. Am. Heart Assoc. 2023, 12, e024397. [Google Scholar] [CrossRef]
- Zhao, J.; Randive, R.; Stewart, J.A. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.H.; Son, W.R.; Lee, Y.S.; Lee, K.W. Glycolaldehyde-derived advanced glycation end products (glycol-AGEs)-induced vascular smooth muscle cell dysfunction is regulated by the AGES-receptor (RAGE) axis in endothelium. Cell Commun. Adhes. 2015, 22, 67–78. [Google Scholar] [CrossRef]
- Xiong, F.; Leonov, S.; Howard, A.C.; Xiong, S.; Zhang, B.; Mei, L.; McNeil, P.; Simon, S.; Xiong, W.C. Receptor for advanced glycation end products (RAGE) prevents endothelial cell membrane resealing and regulates F-actin remodeling in a beta-catenin-dependent manner. J. Biol. Chem. 2011, 286, 35061–35070. [Google Scholar] [CrossRef]
- de la Cruz-Ares, S.; Cardelo, M.P.; Gutierrez-Mariscal, F.M.; Torres-Pena, J.D.; Garcia-Rios, A.; Katsiki, N.; Malagon, M.M.; Lopez-Miranda, J.; Perez-Martinez, P.; Yubero-Serrano, E.M. Endothelial Dysfunction and Advanced Glycation End Products in Patients with Newly Diagnosed Versus Established Diabetes: From the CORDIOPREV Study. Nutrients 2020, 12, 238. [Google Scholar] [CrossRef]
- Dozio, E.; Tassistro, E.; Orlando, A.; Giussani, M.; Beba, G.; Patti, I.; Lieti, G.; Antolini, L.; Vianello, E.; Corsi Romanelli, M.M.; et al. The soluble receptor for advanced glycation end products is independently associated with systolic blood pressure values and hypertension in children. Nutr. Metab. Cardiovasc. Dis. 2025, 103862. [Google Scholar] [CrossRef]
- Singh, S.; Siva, B.V.; Ravichandiran, V. Advanced Glycation End Products: Key player of the pathogenesis of atherosclerosis. Glycoconj. J. 2022, 39, 547–563. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef]
- Turki Jalil, A.; Alameri, A.A.; Iqbal Doewes, R.; El-Sehrawy, A.A.; Ahmad, I.; Ramaiah, P.; Kadhim, M.M.; Kzar, H.H.; Sivaraman, R.; Romero-Parra, R.M.; et al. Circulating and dietary advanced glycation end products and obesity in an adult population: A paradox of their detrimental effects in obesity. Front. Endocrinol. 2022, 13, 966590. [Google Scholar] [CrossRef]
- Masania, J.; Malczewska-Malec, M.; Razny, U.; Goralska, J.; Zdzienicka, A.; Kiec-Wilk, B.; Gruca, A.; Stancel-Mozwillo, J.; Dembinska-Kiec, A.; Rabbani, N.; et al. Dicarbonyl stress in clinical obesity. Glycoconj. J. 2016, 33, 581–589. [Google Scholar] [CrossRef]
- Accacha, S.; Rosenfeld, W.; Jacobson, A.; Michel, L.; Schnurr, F.J.; Shelov, S.; Ten, S.; Boucher-Berry, C.; Carey, D.E.; Speiser, P.W.; et al. Plasma advanced glycation end products (AGEs), receptors for AGEs and their correlation with inflammatory markers in middle school-age children. Horm. Res. Paediatr. 2013, 80, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Vianello, E.; Briganti, S.; Fink, B.; Malavazos, A.E.; Scognamiglio, E.T.; Dogliotti, G.; Sigruener, A.; Schmitz, G.; Corsi Romanelli, M.M. Increased reactive oxygen species production in epicardial adipose tissues from coronary artery disease patients is associated with brown-to-white adipocyte trans-differentiation. Int. J. Cardiol. 2014, 174, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Vianello, E.; Dozio, E.; Arnaboldi, F.; Marazzi, M.G.; Martinelli, C.; Lamont, J.; Tacchini, L.; Sigruner, A.; Schmitz, G.; Corsi Romanelli, M.M. Epicardial adipocyte hypertrophy: Association with M1-polarization and toll-like receptor pathways in coronary artery disease patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Vianello, E.; Dozio, E.; Bandera, F.; Froldi, M.; Micaglio, E.; Lamont, J.; Tacchini, L.; Schmitz, G.; Corsi Romanelli, M.M. Correlative Study on Impaired Prostaglandin E2 Regulation in Epicardial Adipose Tissue and its Role in Maladaptive Cardiac Remodeling via EPAC2 and ST2 Signaling in Overweight Cardiovascular Disease Subjects. Int. J. Mol. Sci. 2020, 21, 520. [Google Scholar] [CrossRef]
- Kanikowska, D.; Kanikowska, A.; Strojny, Z.; Kawka, E.; Zawada, A.; Rutkowski, R.; Litwinowicz, M.; Sato, M.; Grzymislawski, M.; Breborowicz, A.; et al. Assessment of EN-RAGE, sRAGE, and its isoforms: cRAGE, esRAGE in obese patients treated by moderate caloric restriction combined with physical activity conducted in hospital condition. Cytokine 2024, 180, 156665. [Google Scholar] [CrossRef]
- Kanikowska, D.; Kanikowska, A.; Swora-Cwynar, E.; Grzymislawski, M.; Sato, M.; Breborowicz, A.; Witowski, J.; Korybalska, K. Soluble receptor for advanced glycation end products (sRAGE) correlates with obesity-related parameters, and it is not easy to be modified by moderate caloric restriction in obese humans. J. Physiol. Pharmacol. 2022, 73, 10–26402. [Google Scholar] [CrossRef]
- Popp, C.J.; Zhou, B.; Manigrasso, M.B.; Li, H.; Curran, M.; Hu, L.; St-Jules, D.E.; Aleman, J.O.; Vanegas, S.M.; Jay, M.; et al. Soluble Receptor for Advanced Glycation End Products (sRAGE) Isoforms Predict Changes in Resting Energy Expenditure in Adults with Obesity during Weight Loss. Curr. Dev. Nutr. 2022, 6, nzac046. [Google Scholar] [CrossRef]
- Tavares, J.F.; Ribeiro, P.V.M.; Coelho, O.G.L.; Silva, L.E.D.; Alfenas, R.C.G. Can advanced glycation end-products and their receptors be affected by weight loss? A systematic review. Obes. Rev. 2020, 21, e13000. [Google Scholar] [CrossRef]
- Brix, J.M.; Hollerl, F.; Kopp, H.P.; Schernthaner, G.H.; Schernthaner, G. The soluble form of the receptor of advanced glycation endproducts increases after bariatric surgery in morbid obesity. Int. J. Obes. 2012, 36, 1412–1417. [Google Scholar] [CrossRef]
- Parikh, M.; Chung, M.; Sheth, S.; McMacken, M.; Zahra, T.; Saunders, J.K.; Ude-Welcome, A.; Dunn, V.; Ogedegbe, G.; Schmidt, A.M.; et al. Randomized pilot trial of bariatric surgery versus intensive medical weight management on diabetes remission in type 2 diabetic patients who do NOT meet NIH criteria for surgery and the role of soluble RAGE as a novel biomarker of success. Ann. Surg. 2014, 260, 617–622, discussion 622–614. [Google Scholar] [CrossRef]
- Lorenzi, R.; Pattou, F.; Beuscart, J.B.; Grossin, N.; Lambert, M.; Fontaine, P.; Caiazzo, R.; Pigeyre, M.; Patrice, A.; Daroux, M.; et al. Anti-sRAGE autoimmunity in obesity: Downturn after bariatric surgery is independent of previous diabetic status. Diabetes Metab. 2014, 40, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Hagen, I.; Schulte, D.M.; Muller, N.; Martinsen, J.; Turk, K.; Hedderich, J.; Schreiber, S.; Laudes, M. Soluble receptor for advanced glycation end products as a potential biomarker to predict weight loss and improvement of insulin sensitivity by a very low calorie diet of obese human subjects. Cytokine 2015, 73, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.; Saunders, J.K.; Ude-Welcome, A.; Marie Schmidt, A.; Dunn, V.; Leon Pachter, H.; Parikh, M. Three-year follow-up comparing metabolic surgery versus medical weight management in patients with type 2 diabetes and BMI 30-35. The role of sRAGE biomarker as predictor of satisfactory outcomes. Surg. Obes. Relat. Dis. 2016, 12, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.R.; Fuller, K.N.Z.; Perkins, R.K.; Kroeger, C.M.; Trepanowski, J.F.; Varady, K.A.; Haus, J.M. Endogenous secretory RAGE increases with improvements in body composition and is associated with markers of adipocyte health. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 1155–1165. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef]
- Figueiredo, I.D.; Lima, T.F.O.; Carlstrom, P.F.; Assis, R.P.; Brunetti, I.L.; Baviera, A.M. Lycopene in Combination with Insulin Triggers Antioxidant Defenses and Increases the Expression of Components That Detoxify Advanced Glycation Products in Kidneys of Diabetic Rats. Nutrients 2024, 16, 1580. [Google Scholar] [CrossRef]
- Peng, J.; Liang, G.; Wen, W.; Qiu, Z.; Huang, W.; Wang, Q.; Xiao, G. Penta-O-galloyl-beta-d-glucose inhibits the formation of advanced glycation end-products (AGEs): A mechanistic investigation. Int. J. Biol. Macromol. 2023, 237, 124161. [Google Scholar] [CrossRef]
- Gong, P.; Pei, S.; Long, H.; Yang, W.; Yao, W.; Li, N.; Wang, J.; Zhao, Y.; Chen, F.; Xie, J.; et al. Potential inhibitory effect of Auricularia auricula polysaccharide on advanced glycation end-products (AGEs). Int. J. Biol. Macromol. 2024, 262 Pt 1, 129856. [Google Scholar] [CrossRef]
- Mohamadizadeh, M.; Dehghan, P.; Azizi-Soleiman, F.; Maleki, P. Effectiveness of date seed on glycemia and advanced glycation end-products in type 2 diabetes: A randomized placebo-controlled trial. Nutr. Diabetes 2024, 14, 37. [Google Scholar] [CrossRef]
- Tam, H.L.; Shiu, S.W.; Wong, Y.; Chow, W.S.; Betteridge, D.J.; Tan, K.C. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis 2010, 209, 173–177. [Google Scholar] [CrossRef]
- Lu, L.; Peng, W.H.; Wang, W.; Wang, L.J.; Chen, Q.J.; Shen, W.F. Effects of atorvastatin on progression of diabetic nephropathy and local RAGE and soluble RAGE expressions in rats. J. Zhejiang Univ. Sci. B 2011, 12, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Quade-Lyssy, P.; Kanarek, A.M.; Baiersdorfer, M.; Postina, R.; Kojro, E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J. Lipid Res. 2013, 54, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M.; Knani, I.; Bouzidi, H.; Berriche, O.; Hammami, M.; Kerkeni, M. Plasma Levels of Pentosidine, Carboxymethyl-Lysine, Soluble Receptor for Advanced Glycation End Products, and Metabolic Syndrome: The Metformin Effect. Dis. Markers 2016, 2016, 6248264. [Google Scholar] [CrossRef]
- Derosa, G.; Bonaventura, A.; Romano, D.; Bianchi, L.; Fogari, E.; D’Angelo, A.; Maffioli, P. Effects of enalapril/lercanidipine combination on some emerging biomarkers in cardiovascular risk stratification in hypertensive patients. J. Clin. Pharm. Ther. 2014, 39, 277–285. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorpe, S.R.; Thallas-Bonke, V.; Pete, J.; Thomas, M.C.; Deemer, E.R.; Bassal, S.; El-Osta, A.; Long, D.M.; Panagiotopoulos, S.; et al. Modulation of soluble receptor for advanced glycation end products by angiotensin-converting enzyme-1 inhibition in diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2363–2372. [Google Scholar] [CrossRef]
- Kheirouri, S.; Alizadeh, M. Vitamin D and advanced glycation end products and their receptors. Pharmacol. Res. 2020, 158, 104879. [Google Scholar] [CrossRef]
- Xia, P.; He, H.; Kristine, M.S.; Guan, W.; Gao, J.; Wang, Z.; Hu, J.; Han, L.; Li, J.; Han, W.; et al. Therapeutic effects of recombinant human S100A6 and soluble receptor for advanced glycation end products(sRAGE) on CCl4-induced liver fibrosis in mice. Eur. J. Pharmacol. 2018, 833, 86–93. [Google Scholar] [CrossRef]
- Audard, J.; Godet, T.; Blondonnet, R.; Joffredo, J.B.; Paquette, B.; Belville, C.; Lavergne, M.; Gross, C.; Pasteur, J.; Bouvier, D.; et al. Inhibition of the Receptor for Advanced Glycation End-Products in Acute Respiratory Distress Syndrome: A Randomised Laboratory Trial in Piglets. Sci. Rep. 2019, 9, 9227. [Google Scholar] [CrossRef]
- Dang, M.; Zeng, X.; Chen, B.; Wang, H.; Li, H.; Du, F.; Guo, C. Interferon-gamma mediates the protective effects of soluble receptor for advanced glycation end-product in myocardial ischemia/reperfusion. Lab. Investig. 2019, 99, 358–370. [Google Scholar] [CrossRef]
- Miyagawa, T.; Iwata, Y.; Oshima, M.; Ogura, H.; Sato, K.; Nakagawa, S.; Yamamura, Y.; Kamikawa, Y.; Miyake, T.; Kitajima, S.; et al. Soluble receptor for advanced glycation end products protects from ischemia- and reperfusion-induced acute kidney injury. Biol. Open 2022, 11, bio058852. [Google Scholar] [CrossRef]
- Yang, L.Y.; Tang, S.C.; Lee, J.E.; Chen, Y.R.; Chen, Y.T.; Chen, K.W.; Hsieh, S.T.; Wang, K.C. Recombinant soluble form of receptor for advanced glycation end products ameliorates microcirculation impairment and neuroinflammation after subarachnoid hemorrhage. Neurotherapeutics 2024, 21, e00312. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhou, Z.; Zhang, M.; Fan, L.; Forudi, F.; Zhou, X.; Qu, W.; Lincoff, A.M.; Schmidt, A.M.; Topol, E.J.; et al. Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J. Pharmacol. Exp. Ther. 2006, 317, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.J.; Yoon, S.J.; Lee, S.M. Losartan protects liver against ischaemia/reperfusion injury through PPAR-gamma activation and receptor for advanced glycation end-products down-regulation. Br. J. Pharmacol. 2013, 169, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Tang, Z.; Wang, L.; Feng, B. Glucagon-like peptide-1 analogue liraglutide ameliorates atherogenesis via inhibiting advanced glycation end product-induced receptor for advanced glycosylation end product expression in apolipoprotein-E deficient mice. Mol. Med. Rep. 2017, 16, 3421–3426. [Google Scholar] [CrossRef]
- Tsujinaka, H.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Shobatake, R.; Makino, M.; Masuda, N.; Hirai, H.; Takasawa, S.; Ogata, N. Statins decrease vascular epithelial growth factor expression via down-regulation of receptor for advanced glycation end-products. Heliyon 2017, 3, e00401. [Google Scholar] [CrossRef]
- Sourris, K.C.; Ding, Y.; Maxwell, S.S.; Al-Sharea, A.; Kantharidis, P.; Mohan, M.; Rosado, C.J.; Penfold, S.A.; Haase, C.; Xu, Y.; et al. Glucagon-like peptide-1 receptor signaling modifies the extent of diabetic kidney disease through dampening the receptor for advanced glycation end products-induced inflammation. Kidney Int. 2024, 105, 132–149. [Google Scholar] [CrossRef]
- Baek, C.H.; Kim, H.; Moon, S.Y.; Yang, W.S. Liraglutide, a glucagon-like peptide-1 receptor agonist, induces ADAM10-dependent ectodomain shedding of RAGE via AMPK activation in human aortic endothelial cells. Life Sci. 2022, 292, 120331. [Google Scholar] [CrossRef]
- Matsui, T.; Sotokawauchi, A.; Nishino, Y.; Koga, Y.; Yamagishi, S.I. Empagliflozin ameliorates renal and metabolic derangements in obese type 2 diabetic mice by blocking advanced glycation end product-receptor axis. Mol. Med. 2025, 31, 88. [Google Scholar] [CrossRef]
- Wilson, R.A.; Arivazhagan, L.; Ruiz, H.H.; Zhou, B.; Qian, K.; Manigrasso, M.B.; Bernadin, R.; Mangar, K.; Shekhtman, A.; Li, H.; et al. Pharmacological antagonism of receptor for advanced glycation end products signaling promotes thermogenesis, healthful body mass and composition, and metabolism in mice. Obesity 2023, 31, 1825–1843. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.B.; Rabbani, P.; Egana-Gorrono, L.; Quadri, N.; Frye, L.; Zhou, B.; Reverdatto, S.; Ramirez, L.S.; Dansereau, S.; Pan, J.; et al. Small-molecule antagonism of the interaction of the RAGE cytoplasmic domain with DIAPH1 reduces diabetic complications in mice. Sci. Transl. Med. 2021, 13, eabf7084. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef]
- Kong, W.; Zhu, L.; Li, T.; Chen, J.; Fan, B.; Ji, W.; Zhang, C.; Cai, X.; Hu, C.; Sun, X.; et al. Azeliragon inhibits PAK1 and enhances the therapeutic efficacy of AKT inhibitors in pancreatic cancer. Eur. J. Pharmacol. 2023, 948, 175703. [Google Scholar] [CrossRef]
- Magna, M.; Hwang, G.H.; McIntosh, A.; Drews-Elger, K.; Takabatake, M.; Ikeda, A.; Mera, B.J.; Kwak, T.; Miller, P.; Lippman, M.E.; et al. RAGE inhibitor TTP488 (Azeliragon) suppresses metastasis in triple-negative breast cancer. NPJ Breast Cancer 2023, 9, 59. [Google Scholar] [CrossRef]
- Wautier, M.P.; Guillausseau, P.J.; Wautier, J.L. Activation of the receptor for advanced glycation end products and consequences on health. Diabetes Metab. Syndr. 2017, 11, 305–309. [Google Scholar] [CrossRef]
- Kim, E.Y.; Dryer, S.E. RAGE and alphaVbeta3-integrin are essential for suPAR signaling in podocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166186. [Google Scholar] [CrossRef]
- Ma, S.; Nakamura, Y.; Hisaoka-Nakashima, K.; Morioka, N. Blockade of receptor for advanced glycation end-products with azeliragon ameliorates streptozotocin-induced diabetic neuropathy. Neurochem. Int. 2023, 163, 105470. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vianello, E.; Beltrami, A.P.; Aleksova, A.; Janjusevic, M.; Fluca, A.L.; Corsi Romanelli, M.M.; La Sala, L.; Dozio, E. The Advanced Glycation End-Products (AGE)–Receptor for AGE System (RAGE): An Inflammatory Pathway Linking Obesity and Cardiovascular Diseases. Int. J. Mol. Sci. 2025, 26, 3707. https://doi.org/10.3390/ijms26083707
Vianello E, Beltrami AP, Aleksova A, Janjusevic M, Fluca AL, Corsi Romanelli MM, La Sala L, Dozio E. The Advanced Glycation End-Products (AGE)–Receptor for AGE System (RAGE): An Inflammatory Pathway Linking Obesity and Cardiovascular Diseases. International Journal of Molecular Sciences. 2025; 26(8):3707. https://doi.org/10.3390/ijms26083707
Chicago/Turabian StyleVianello, Elena, Antonio P. Beltrami, Aneta Aleksova, Milijana Janjusevic, Alessandra L. Fluca, Massimiliano M. Corsi Romanelli, Lucia La Sala, and Elena Dozio. 2025. "The Advanced Glycation End-Products (AGE)–Receptor for AGE System (RAGE): An Inflammatory Pathway Linking Obesity and Cardiovascular Diseases" International Journal of Molecular Sciences 26, no. 8: 3707. https://doi.org/10.3390/ijms26083707
APA StyleVianello, E., Beltrami, A. P., Aleksova, A., Janjusevic, M., Fluca, A. L., Corsi Romanelli, M. M., La Sala, L., & Dozio, E. (2025). The Advanced Glycation End-Products (AGE)–Receptor for AGE System (RAGE): An Inflammatory Pathway Linking Obesity and Cardiovascular Diseases. International Journal of Molecular Sciences, 26(8), 3707. https://doi.org/10.3390/ijms26083707