
Integrating Machine Learning and Bulk and Single-Cell RNA Sequencing to Decipher Diverse Cell Death Patterns for Predicting the Prognosis of Neoadjuvant Chemotherapy in Breast Cancer

 and
and
Abstract
1. Introduction
2. Results
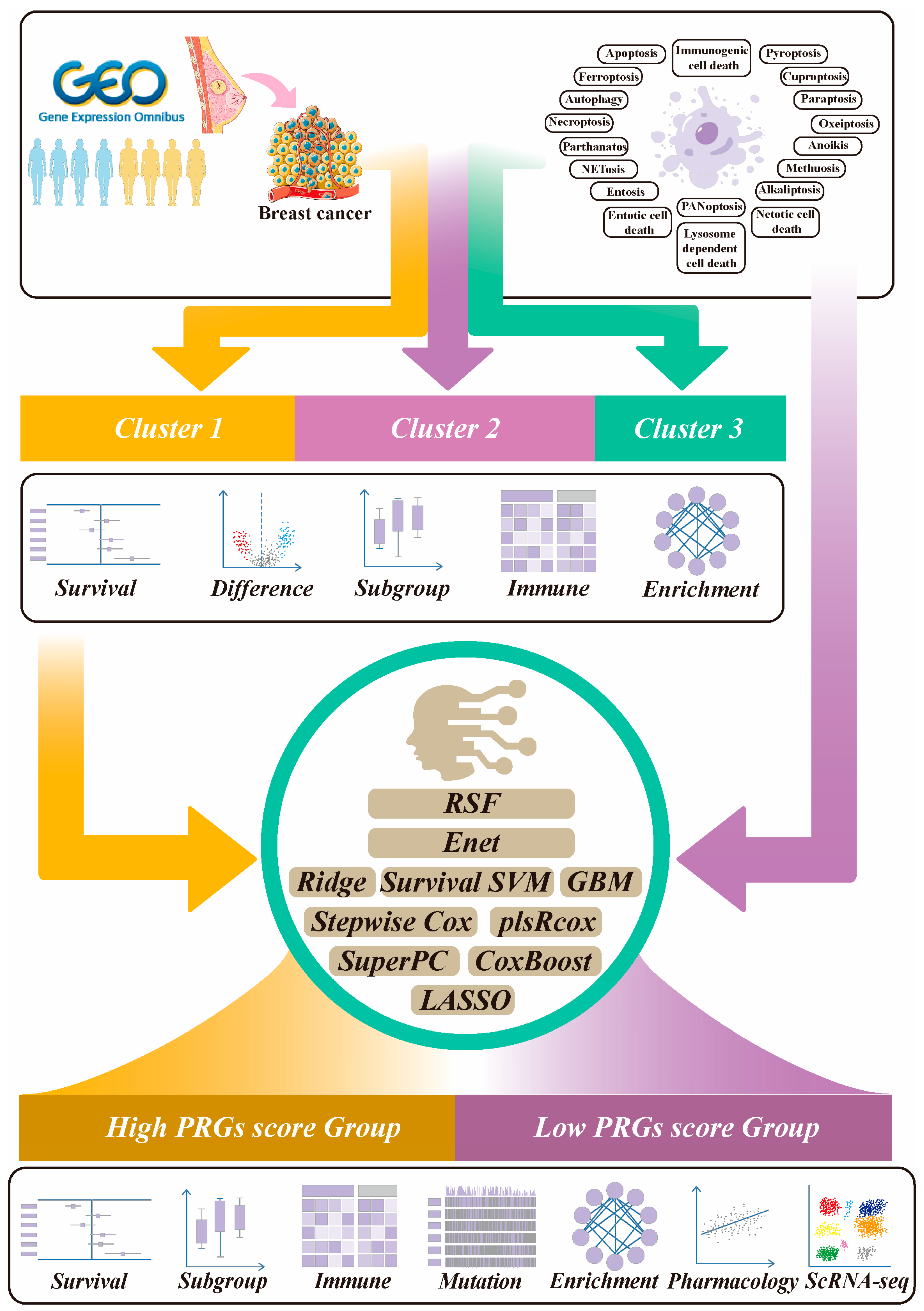
2.1. The Workflow of This Study
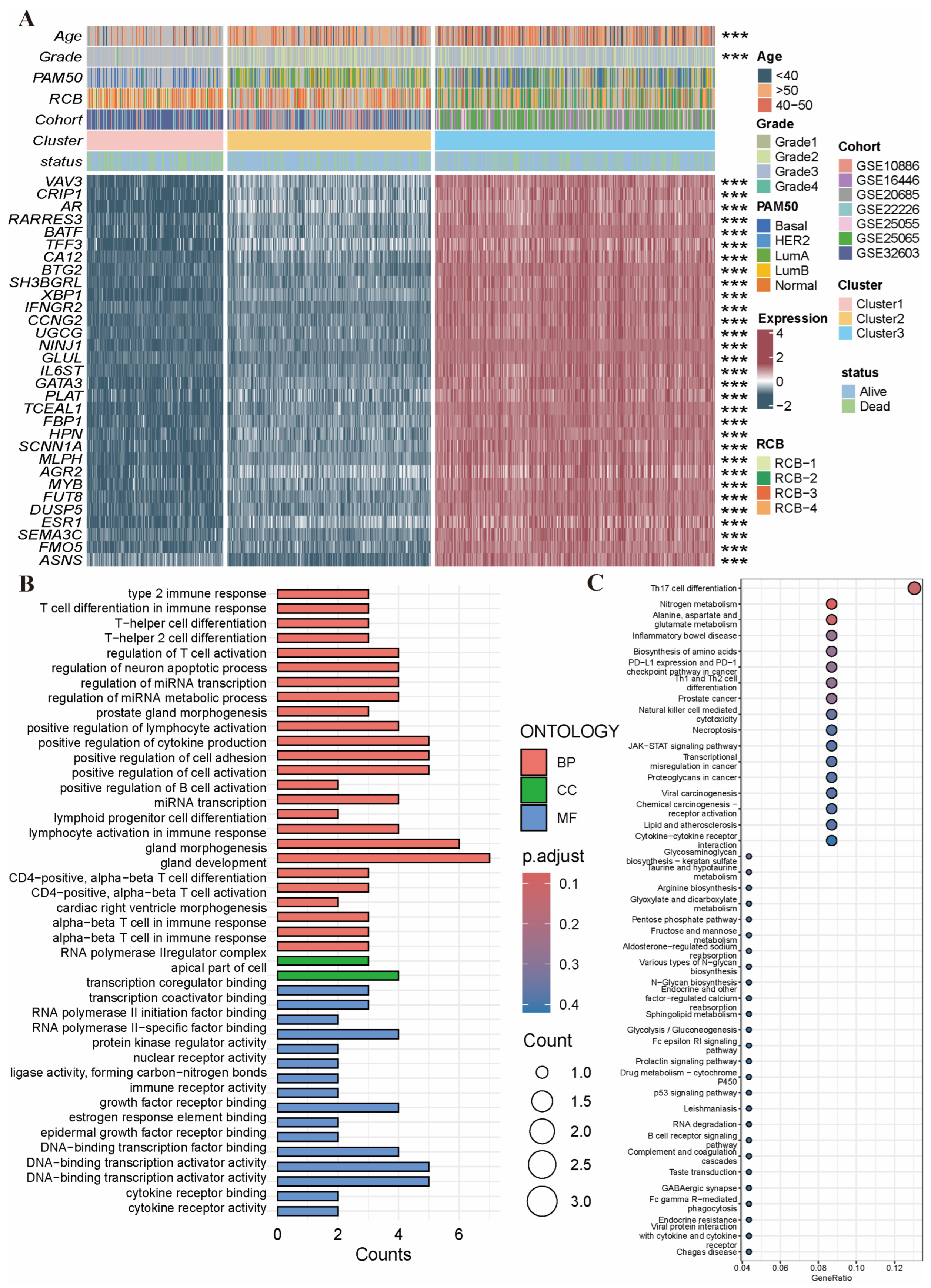
2.2. Identification of NAC-Related PRG Clusters
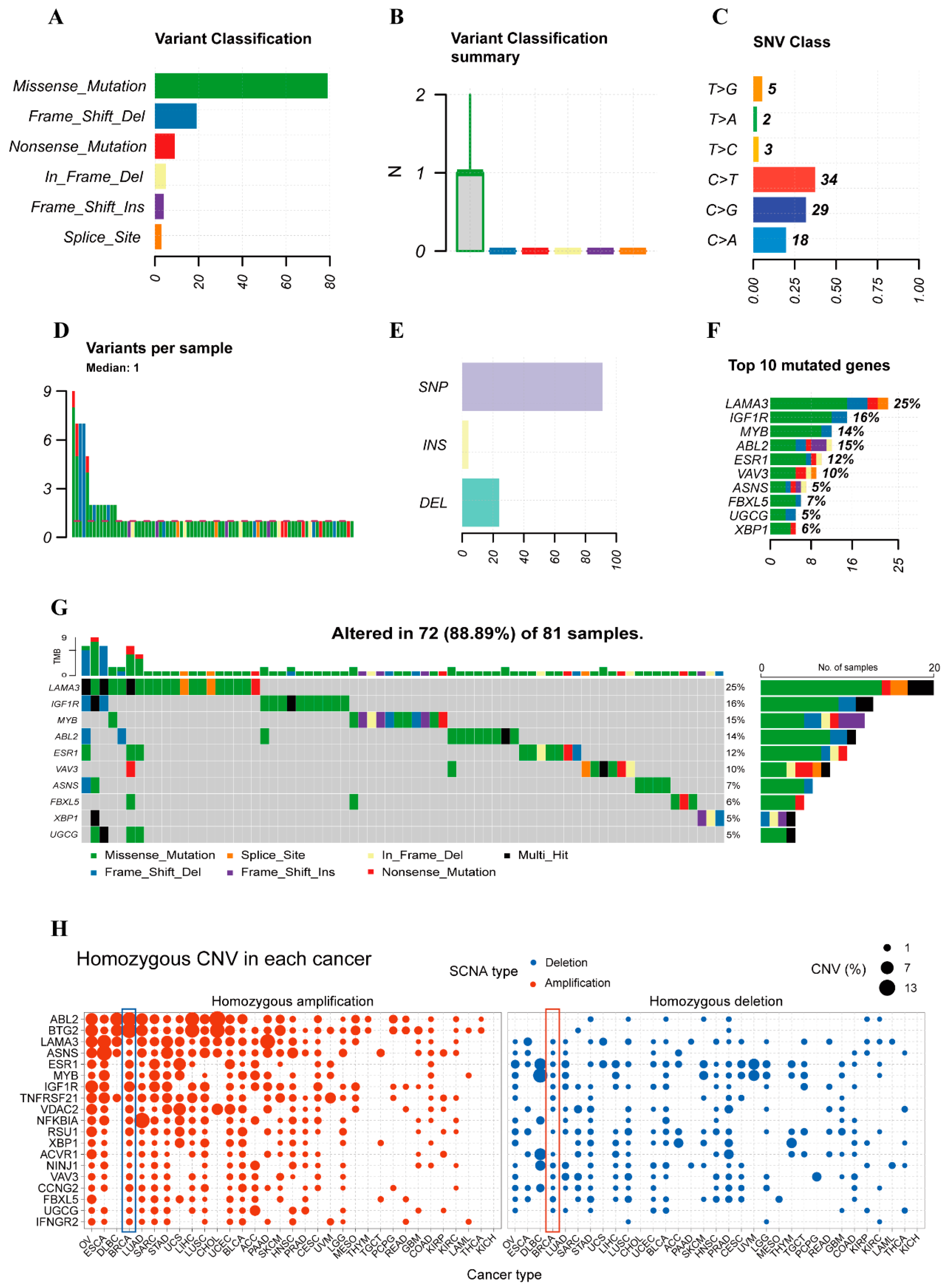
2.3. Variant Landscape of PCD-Related DEGs in BRCA Patients
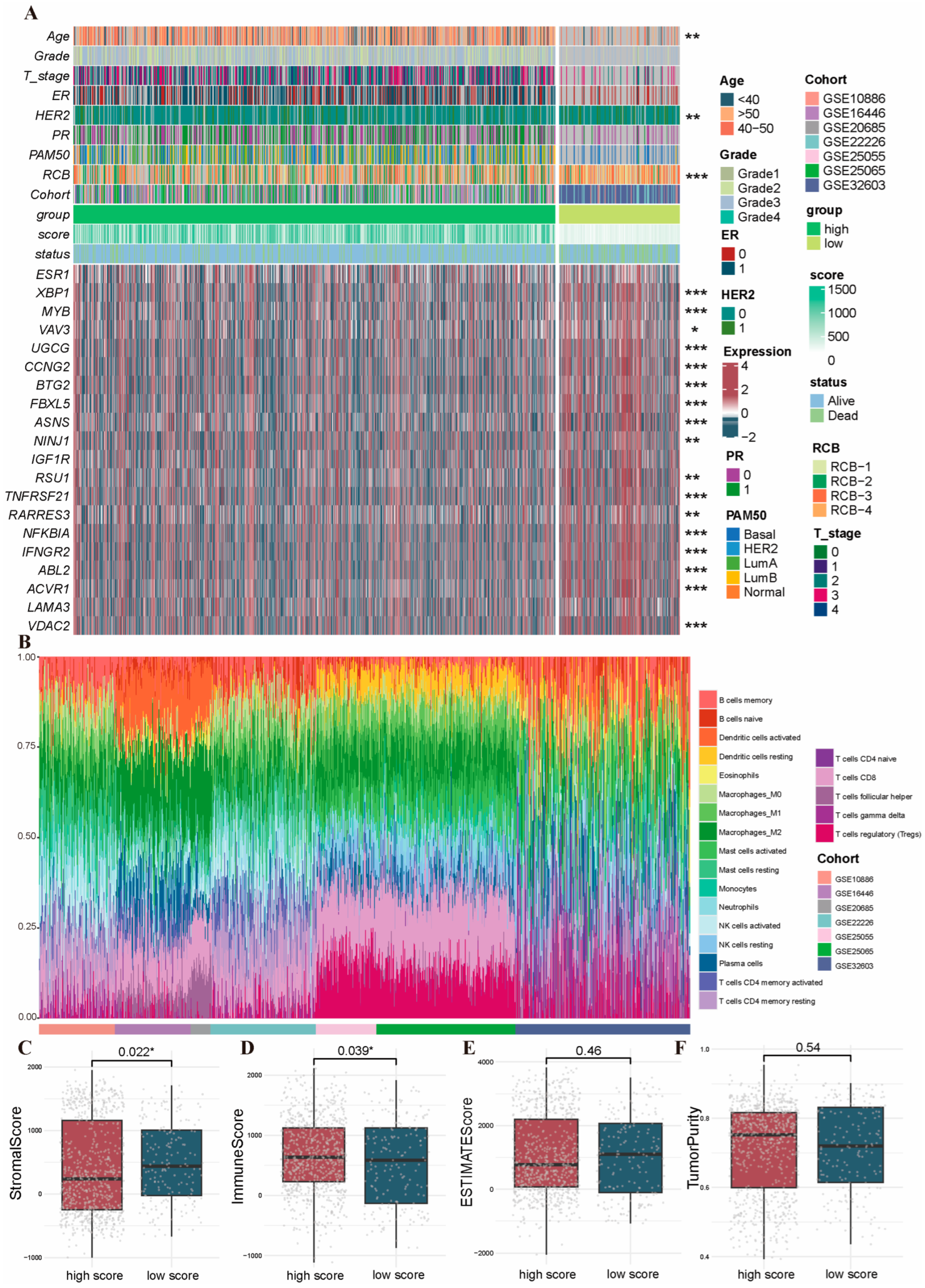
2.4. Immune Infiltration Characteristics in the Distinct PRG Clusters
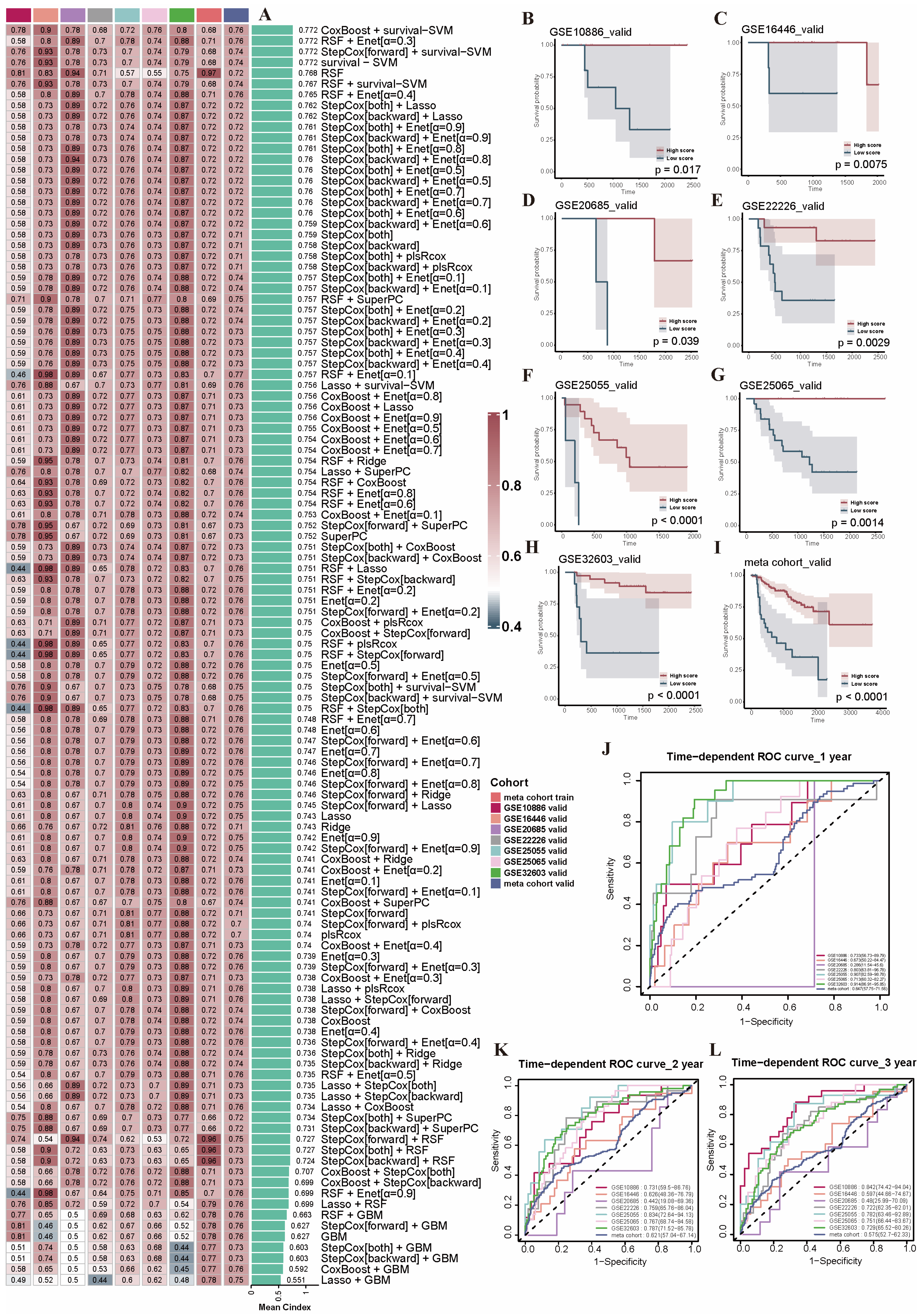
2.5. Construction and Evaluation of the PRGs Prognostic Risk Model via the Machine Learning-Based Integrative Procedure for BRCA Patients
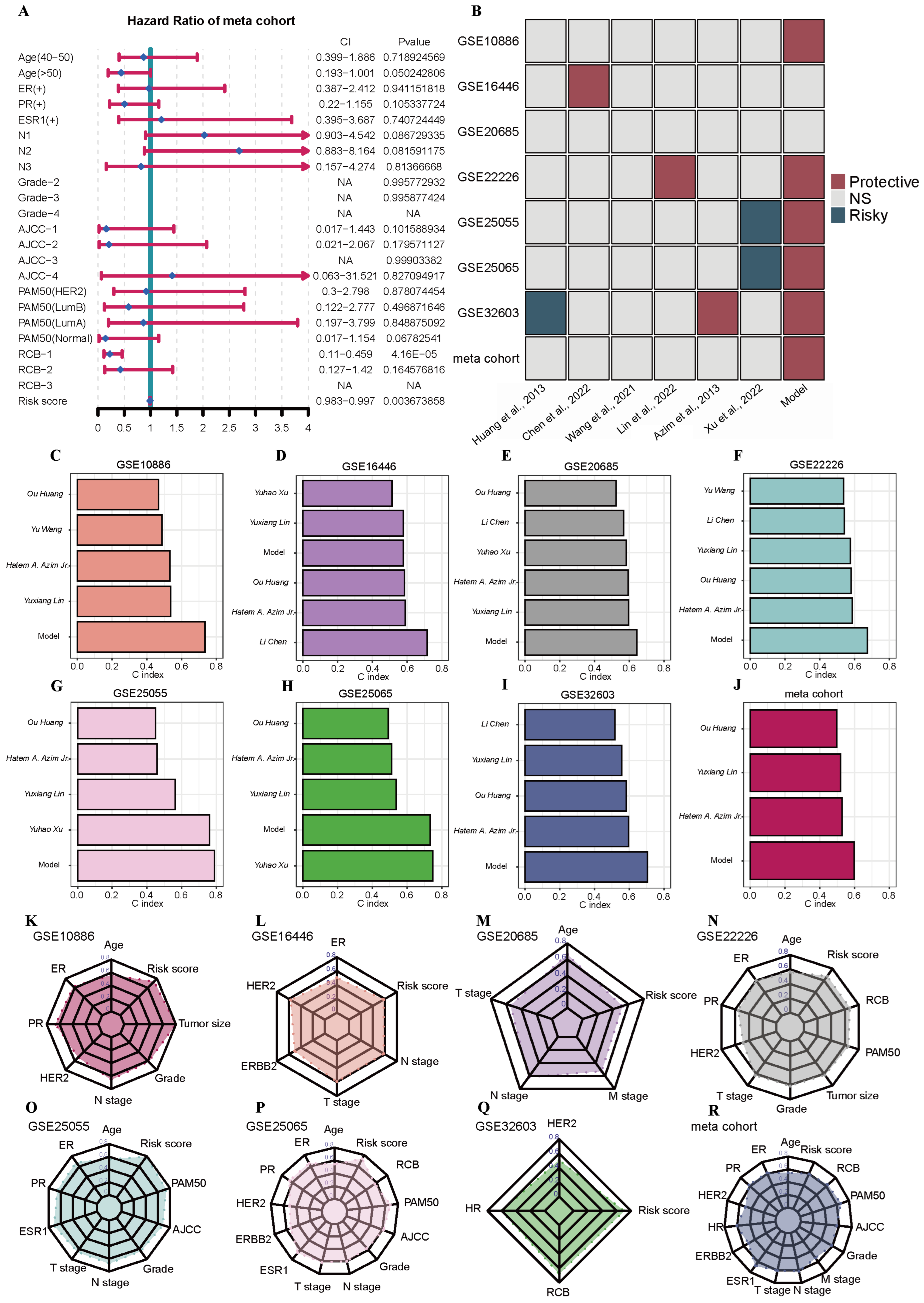
2.6. Clinicopathologic Features and TIME Analysis Based on the PRGs Prognostic Risk Model
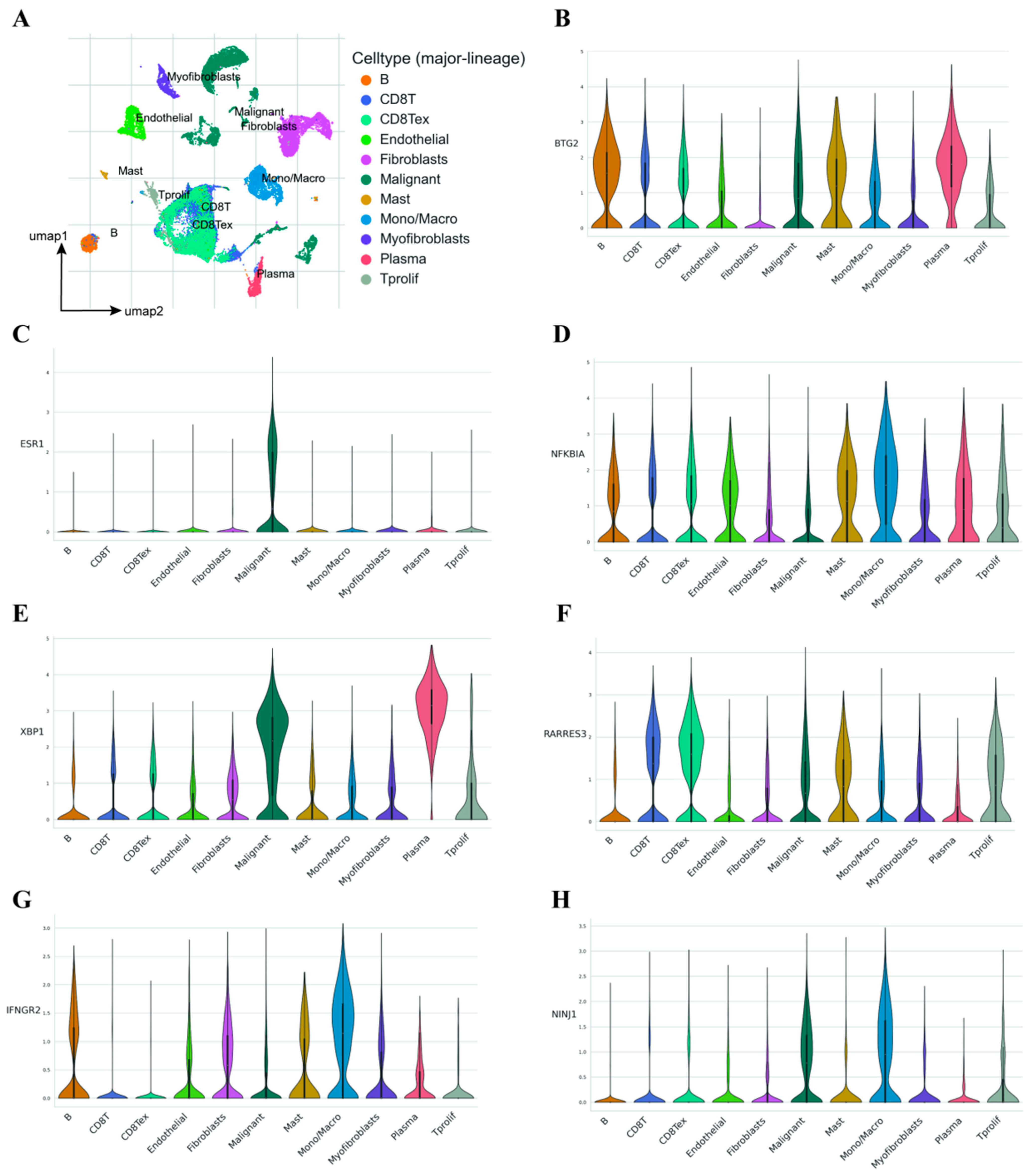
2.7. Single-Cell Transcriptome Analysis Demonstrates the Influence of Model Genes on the TME
2.8. Mutation Abundance Analysis
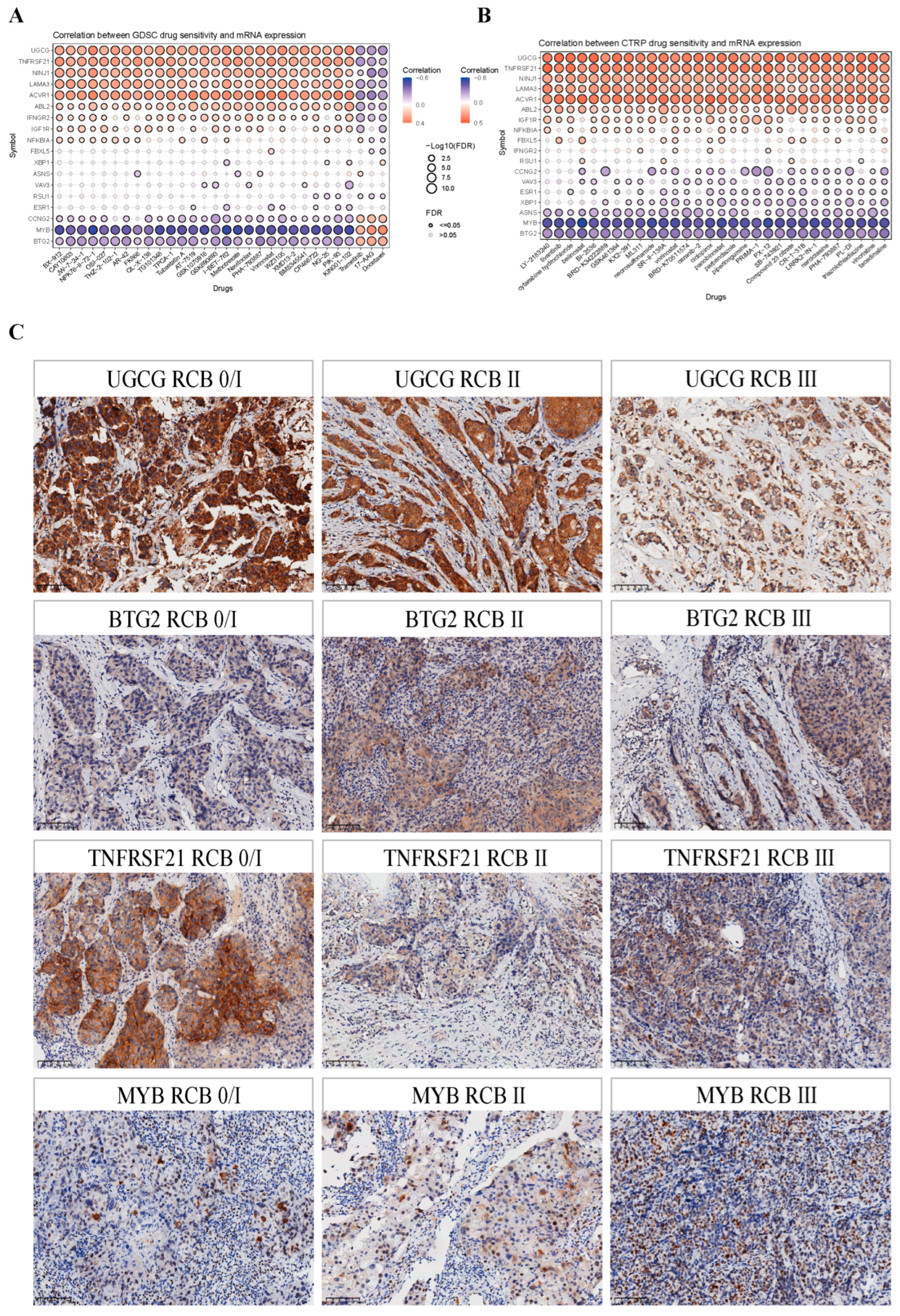
2.9. Validation of Immunohistochemical Staining of Model Genes from HPA Database
2.10. The Prognostic Model Is Associated with the Response to NAC in BRCA
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Identification of NAC-Related PRGs
4.3. Consensus Unsupervised Clustering Analysis
4.4. Functional Enrichment Analysis
4.5. Tumor Immune Microenvironment (TIME) Analysis
4.6. Construction of the PRGs Prognostic Risk Model by Machine Learning
4.7. Single-Cell Sequencing Analysis
4.8. Pharmacological Analysis
4.9. Mutation Analysis
4.10. Validation of Immunohistochemical Staining from the HPA Database
4.11. Clinical Sample Collection and Immunohistochemical Analysis of BRCA
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawiak, A. Molecular Research and Treatment of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 9617. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Peng, C.; Tang, H.; Liu, X.; Peng, F. New Advances in the Research of Resistance to Neoadjuvant Chemotherapy in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 9644. [Google Scholar] [CrossRef] [PubMed]
- Yau, C.; Osdoit, M.; van der Noordaa, M.; Shad, S.; Wei, J.; de Croze, D.; Hamy, A.S.; Laé, M.; Reyal, F.; Sonke, G.S.; et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: A multicentre pooled analysis of 5161 patients. Lancet Oncol. 2022, 23, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Vallon-Christersson, J.; Hakkinen, J.; Hegardt, C.; Saal, L.H.; Larsson, C.; Ehinger, A.; Lindman, H.; Olofsson, H.; Sjöblom, T.; Wärnberg, F.; et al. Cross comparison and prognostic assessment of breast cancer multigene signatures in a large population-based contemporary clinical series. Sci. Rep. 2019, 9, 12184. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar]
- Qin, H.; Abulaiti, A.; Maimaiti, A.; Abulaiti, Z.; Fan, G.; Aili, Y.; Ji, W.; Wang, Z.; Wang, Y. Integrated machine learning survival framework develops a prognostic model based on inter-crosstalk definition of mitochondrial function and cell death patterns in a large multicenter cohort for lower-grade glioma. J. Transl. Med. 2023, 21, 588. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Yan, H.; Luo, B.; Wu, X.; Guan, F.; Yu, X.; Zhao, L.; Ke, X.; Wu, J.; Yuan, J. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int. J. Biol. Sci. 2021, 17, 2606–2621. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Li, D.; Zhou, X.; Qin, Q.; Zhang, Q. Caspase-3-mediated GSDME induced Pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J. Cell. Mol. Med. 2021, 25, 8159–8168. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Shao, Z.; Zhong, X.; Ding, X.; Wu, L.; Chen, J.; He, P.; Cheng, Y.; Zhu, K.; et al. Pyrotinib and chrysin synergistically potentiate autophagy in HER2-positive breast cancer. Signal Transduct. Target. Ther. 2023, 8, 463. [Google Scholar] [CrossRef]
- Seo, M.J.; Kim, I.Y.; Lee, D.M.; Park, Y.J.; Cho, M.Y.; Jin, H.J.; Choi, K.S. Dual inhibition of thioredoxin reductase and proteasome is required for auranofin-induced paraptosis in breast cancer cells. Cell Death Dis. 2023, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Di Leo, A.; de Azambuja, E.; Larsimont, D.; Haibe-Kains, B.; Selleslags, J.; Delaloge, S.; Duhem, C.; Kains, J.P.; Carly, B.; et al. Multifactorial approach to predicting resistance to anthracyclines. J. Clin. Oncol. 2011, 29, 1578–1586. [Google Scholar] [CrossRef]
- Kao, K.J.; Chang, K.M.; Hsu, H.C.; Huang, A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef]
- Esserman, L.J.; Berry, D.A.; Cheang, M.C.; Yau, C.; Perou, C.M.; Carey, L.; DeMichele, A.; Gray, J.W.; Conway-Dorsey, K.; Lenburg, M.E.; et al. Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: Results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res. Treat. 2012, 132, 1049–1062. [Google Scholar] [CrossRef]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef]
- Magbanua, M.J.; Wolf, D.M.; Yau, C.; Davis, S.E.; Crothers, J.; Au, A.; Haqq, C.M.; Livasy, C.; Rugo, H.S.; I-SPY 1 TRIAL Investigators; et al. Serial expression analysis of breast tumors during neoadjuvant chemotherapy reveals changes in cell cycle and immune pathways associated with recurrence and response. Breast Cancer Res. 2015, 17, 73. [Google Scholar] [CrossRef]
- Huang, O.; Jiang, M.; Zhang, X.; Xie, Z.; Chen, X.; Wu, J.; Liu, H.; Shen, K. Grb14 as an independent good prognosis factor for breast cancer patients treated with neoadjuvant chemotherapy. Jpn J. Clin. Oncol. 2013, 43, 1064–1072. [Google Scholar] [CrossRef]
- Chen, L.; Huang, S.; Liu, Q.; Kong, X.; Su, Z.; Zhu, M.; Fang, Y.; Zhang, L.; Li, X.; Wang, J. PD-L1 Protein Expression Is Associated With Good Clinical Outcomes and Nomogram for Prediction of Disease Free Survival and Overall Survival in Breast Cancer Patients Received Neoadjuvant Chemotherapy. Front. Immunol. 2022, 13, 849468. [Google Scholar]
- Wang, Y.; Zhao, H.; Zhao, P.; Wang, X. Targeting PKM2 promotes chemosensitivity of breast cancer cells in vitro and in vivo. Cancer Biomark. 2021, 32, 221–230. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, E.; Li, Y.; Chen, X.; Chen, M.; Huang, J.; Guo, W.; Chen, L.; Wu, L.; Zhang, X.; et al. Thrombospondin 2 is a Functional Predictive and Prognostic Biomarker for Triple-Negative Breast Cancer Patients With Neoadjuvant Chemotherapy. Pathol. Oncol. Res 2022, 28, 1610559. [Google Scholar] [CrossRef]
- Azim, H.A., Jr.; Singhal, S.; Ignatiadis, M.; Desmedt, C.; Fumagalli, D.; Veys, I.; Larsimont, D.; Piccart, M.; Michiels, S.; Sotiriou, C. Association between SPARC mRNA expression, prognosis and response to neoadjuvant chemotherapy in early breast cancer: A pooled in-silico analysis. PLoS ONE 2013, 8, e62451. [Google Scholar] [CrossRef]
- Xu, Y.; Du, Y.; Zheng, Q.; Zhou, T.; Ye, B.; Wu, Y.; Xu, Q.; Meng, X. Identification of Ferroptosis-Related Prognostic Signature and Subtypes Related to the Immune Microenvironment for Breast Cancer Patients Receiving Neoadjuvant Chemotherapy. Front. Immunol. 2022, 13, 895110. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, M.; Li, Y.; Chen, D.; Wu, Y.; Hu, Y. Programmed Cell Death Tunes Tumor Immunity. Front. Immunol. 2022, 13, 847345. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; McGinn, O.; Wu, Y.; Bahreini, A.; Priedigkeit, N.M.; Ding, K.; Onkar, S.; Lampenfeld, C.; Sartorius, C.A.; Miller, L.; et al. ESR1 mutant breast cancers show elevated basal cytokeratins and immune activation. Nat. Commun. 2022, 13, 2011. [Google Scholar] [CrossRef]
- Arabkari, V.; Barua, D.; Hossain, M.M.; Webber, M.; Smith, T.; Gupta, A.; Gupta, S. miRNA-378 Is Downregulated by XBP1 and Inhibits Growth and Migration of Luminal Breast Cancer Cells. Int. J. Mol. Sci. 2023, 25, 186. [Google Scholar] [CrossRef]
- Ciciro, Y.; Sala, A. MYB oncoproteins: Emerging players and potential therapeutic targets in human cancer. Oncogenesis 2021, 10, 19. [Google Scholar] [CrossRef]
- Al-Hawary, S.; Alsalamy, A.; Gupta, R.; Alsaab, H.O.; Hjazi, A.; Edilboyev, U.; Ramadan, M.F.; Hussien, B.M.; Ahmed, M.; Hosseini-Fard, S.R. VAV3 in human cancers: Mechanism and clinical implication. Pathol. Res. Pract. 2023, 248, 154681. [Google Scholar] [CrossRef]
- Schomel, N.; Hancock, S.E.; Gruber, L.; Olzomer, E.M.; Byrne, F.L.; Shah, D.; Hoehn, K.L.; Turner, N.; Grösch, S.; Geisslinger, G.; et al. UGCG influences glutamine metabolism of breast cancer cells. Sci. Rep. 2019, 9, 15665. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, C.; Li, Z.; Li, Y.; Dai, D.; Han, K.; Lv, L.; Lu, Y.; Hou, L.; Wang, J. CCNG2 Overexpression Mediated by AKT Inhibits Tumor Cell Proliferation in Human Astrocytoma Cells. Front. Neurol. 2018, 9, 255. [Google Scholar] [CrossRef]
- Kim, S.H.; Jung, I.R.; Hwang, S.S. Emerging role of anti-proliferative protein BTG1 and BTG2. BMB Rep. 2022, 55, 380–388. [Google Scholar] [CrossRef]
- Terzi, E.M.; Sviderskiy, V.O.; Alvarez, S.W.; Whiten, G.C.; Possemato, R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 2021, 7, eabg4302. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, J.; Franco, F.; Parik, S.; Altea-Manzano, P.; Pane, A.A.; Broekaert, D.; van Elsen, J.; Di Conza, G.; Vermeire, I.; Schalley, T.; et al. CD8+ T cell metabolic rewiring defined by scRNA-seq identifies a critical role of ASNS expression dynamics in T cell differentiation. Cell Rep. 2022, 41, 111639. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Sha, N.; Hou, G.; Leng, Z.; Zhao, Q.; Zhang, L.; He, L.; Xu, M.; Jiang, Y.; Chen, T. IGF1R-phosphorylated PYCR1 facilitates ELK4 transcriptional activity and sustains tumor growth under hypoxia. Nat. Commun. 2023, 14, 6117. [Google Scholar] [CrossRef]
- Kim, Y.C.; Cutler, M.L. MicroRNA-Dependent Targeting of RSU1 and the IPP Adhesion Complex Regulates the PTEN/PI3K/AKT Signaling Pathway in Breast Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 5458. [Google Scholar] [CrossRef]
- Christou, C.; Christodoulou, M.I.; Zaravinos, A.; Gkretsi, V. Ras suppressor 1 long form (RSU1L) silencing promotes apoptosis in invasive breast cancer cells. Cell. Signal. 2023, 101, 110522. [Google Scholar] [CrossRef]
- Zhang, T.; Yu, J.; Wang, G.; Zhang, R. Amyloid precursor protein binds with TNFRSF21 to induce neural inflammation in Alzheimer’s Disease. Eur. J. Pharm. Sci. 2021, 157, 105598. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Yao, L.; He, R.; Man, C.; Fan, Y. Construction and validation of a RARRES3-based prognostic signature related to the specific immune microenvironment of pancreatic cancer. Front. Oncol. 2024, 14, 1246308. [Google Scholar] [CrossRef]
- Yang, W.; Li, J.; Zhang, M.; Yu, H.; Zhuang, Y.; Zhao, L.; Ren, L.; Gong, J.; Bi, H.; Zeng, L.; et al. Elevated expression of the rhythm gene NFIL3 promotes the progression of TNBC by activating NF-kappaB signaling through suppression of NFKBIA transcription. J. Exp. Clin. Cancer Res. 2022, 41, 67. [Google Scholar] [CrossRef]
- Singh, S.; Chakrabarti, R. Challenges of Using IFNgamma in Clinical Settings. Cancer Res. 2023, 83, 2093–2095. [Google Scholar] [CrossRef]
- Khatri, A.; Wang, J.; Pendergast, A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016, 129, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tangseefa, P.; Jin, H.; Zhang, H.; Ibáñez, C.F. Human ACVR1C missense variants that correlate with altered body fat distribution produce metabolic alterations of graded severity in knock-in mutant mice. Mol. Metab. 2024, 81, 101890. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, C.F. Regulation of metabolic homeostasis by the TGF-beta superfamily receptor ALK7. FEBS J. 2022, 289, 5776–5797. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.Y.; Huang, Y.Z.; Zhang, W.; Li, L. LAMA3 DNA methylation and transcriptome changes associated with chemotherapy resistance in ovarian cancer. J. Ovarian Res. 2021, 14, 67. [Google Scholar] [CrossRef]
- Yuan, Z.; Dewson, G.; Czabotar, P.E.; Birkinshaw, R.W. VDAC2 and the BCL-2 family of proteins. Biochem. Soc. Trans. 2021, 49, 2787–2795. [Google Scholar] [CrossRef]
- Jutte, H.; Reike, M.; Wirtz, R.M.; Kriegmair, M.; Erben, P.; Tully, K.; Weyerer, V.; Eckstein, M.; Hartmann, A.; Eidt, S.; et al. KRT20, KRT5, ESR1 and ERBB2 Expression Can Predict Pathologic Outcome in Patients Undergoing Neoadjuvant Chemotherapy and Radical Cystectomy for Muscle-Invasive Bladder Cancer. J. Pers. Med. 2021, 11, 473. [Google Scholar] [CrossRef]
- de Groot, S.; Charehbili, A.; van Laarhoven, H.W.; Mooyaart, A.L.; Dekker-Ensink, N.G.; van de Ven, S.; Janssen, L.G.; Swen, J.J.; Smit, V.T.; Heijns, J.B.; et al. Insulin-like growth factor 1 receptor expression and IGF1R 3129G > T polymorphism are associated with response to neoadjuvant chemotherapy in breast cancer patients: Results from the NEOZOTAC trial (BOOG 2010-01). Breast Cancer Res. 2016, 18, 3. [Google Scholar] [CrossRef]
- Spring, L.M.; Bar, Y.; Isakoff, S.J. The Evolving Role of Neoadjuvant Therapy for Operable Breast Cancer. J. Natl. Compr. Cancer Netw. 2022, 20, 723–734. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, W.; Liu, C.; Gao, Y.; Chen, D.; Wu, M.; Li, C.; Wang, X.; Song, X.; Yu, J.; et al. The residual cancer burden index as a valid prognostic indicator in breast cancer after neoadjuvant chemotherapy. BMC Cancer 2024, 24, 13. [Google Scholar] [CrossRef]
- Tao, X.; Han, J.; Li, Y.; Tian, Y.; Li, Z.j.; Li, J.; Guo, X.; Zhao, J. The Difference of RCB 0 and RCB I in Prognosis of Breast Cancer After Neoadjuvant Therapy: A Meta-Analysis. Clin. Breast Cancer 2024. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, C.; Chen, X.; Guan, G.; Zou, C.; Shen, S.; Wu, J.; Wang, Y.; Lin, Z.; Chen, L.; et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro -Oncol. 2022, 24, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, R.; Hu, D.; Zhang, C.; Cao, P.; Huang, J. Machine learning reveals diverse cell death patterns in lung adenocarcinoma prognosis and therapy. NPJ Precis. Oncol. 2024, 8, 49. [Google Scholar] [CrossRef]
- Lu, J.; Li, F.; Ye, M. PANoptosis and Autophagy-Related Molecular Signature and Immune Landscape in Ulcerative Colitis: Integrated Analysis and Experimental Validation. J. Inflamm. Res. 2024, 17, 3225–3245. [Google Scholar] [CrossRef]
- Mumme, H.; Thomas, B.E.; Bhasin, S.S.; Krishnan, U.; Dwivedi, B.; Perumalla, P.; Sarkar, D.; Ulukaya, G.B.; Sabnis, H.S.; Park, S.I.; et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat. Commun. 2023, 14, 6209. [Google Scholar] [CrossRef]
- Xiang, L.; Rao, J.; Yuan, J.; Xie, T.; Yan, H. Single-Cell RNA-Sequencing: Opening New Horizons for Breast Cancer Research. Int. J. Mol. Sci. 2024, 25, 9482. [Google Scholar] [CrossRef]
- Yan, H.; Ju, X.; Huang, A.; Yuan, J. Advancements in technology for characterizing the tumor immune microenvironment. Int. J. Biol. Sci. 2024, 20, 2151–2167. [Google Scholar] [CrossRef]
- Schomel, N.; Gruber, L.; Alexopoulos, S.J.; Trautmann, S.; Olzomer, E.M.; Byrne, F.L.; Hoehn, K.L.; Gurke, R.; Thomas, D.; Ferreirós, N.; et al. UGCG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Sci. Rep. 2020, 10, 8182. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhou, B.; Sun, R.Y.; Ai, Y.L.; Cheng, K.; Li, F.N.; Wang, B.R.; Liu, F.J.; Jiang, Z.H.; Wang, W.J.; et al. The metabolite alpha-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell. Res. 2021, 31, 980–997. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.I.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Sharma, A.; Seow, J.; Dutertre, C.A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020, 183, 377–394. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Cases (n = 56) | RCB-0 + RCB-I (n = 31) | RCB-II (n =12) | RCB-III (n = 13) | p |
|---|---|---|---|---|---|
| Age (years) | 0.330 a | ||||
| ≤50 | 31 | 18 | 8 | 5 | |
| >50 | 25 | 13 | 4 | 8 | |
| ER | 0.525 b | ||||
| Positive | 6 | 2 | 2 | 2 | |
| Negative | 50 | 29 | 10 | 11 | |
| PR | 0.642 b | ||||
| Positive | 7 | 3 | 2 | 2 | |
| Negative | 49 | 28 | 10 | 11 | |
| HER2 | 0.375 a | ||||
| Positive | 32 | 18 | 5 | 9 | |
| Negative | 24 | 13 | 7 | 4 | |
| UGCG | 0.019 b * | ||||
| Low | 10 | 2 | 3 | 5 | |
| High | 46 | 29 | 9 | 8 | |
| BTG2 | 0.012 a * | ||||
| Low | 33 | 23 | 3 | 7 | |
| High | 23 | 8 | 9 | 6 | |
| TNFRSF21 | <0.001 b * | ||||
| Negative | 20 | 7 | 10 | 3 | |
| Positive | 36 | 24 | 2 | 10 | |
| MYB | 0.029 a * | ||||
| Negative | 33 | 23 | 4 | 6 | |
| Positive | 23 | 8 | 8 | 7 | |
| Ki-67(%) | 0.244 b | ||||
| ≤20 | 3 | 1 | 0 | 2 | |
| >20 | 53 | 30 | 12 | 11 | |
| Molecular subtype | 0.524 b | ||||
| HR+ and HER2– | 2 | 2 | 0 | 0 | |
| HER2+ | 32 | 18 | 5 | 9 | |
| Triple-negative | 22 | 11 | 7 | 4 | |
| Lymph nodes metastasis | <0.001 a * | ||||
| No metastasis | 28 | 20 | 8 | 0 | |
| Metastasis | 28 | 11 | 4 | 13 |
| Characteristic | Age | ER | PR | HER2 | UGCG | BTG2 | TNFRSF21 | MYB | Ki-67 | Lymph Nodes Metastasis |
|---|---|---|---|---|---|---|---|---|---|---|
| r | −0.061 | −0.153 | −0.095 | 0.021 | 0.332 | −0.346 | 0.305 | −0.346 | 0.105 | −0.323 |
| p | 0.657 | 0.259 | 0.486 | 0.879 | 0.013 * | 0.009 * | 0.022 * | 0.009 * | 0.439 | 0.015 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, L.; Yang, J.; Rao, J.; Ma, A.; Liu, C.; Zhang, Y.; Huang, A.; Xie, T.; Xue, H.; Chen, Z.; et al. Integrating Machine Learning and Bulk and Single-Cell RNA Sequencing to Decipher Diverse Cell Death Patterns for Predicting the Prognosis of Neoadjuvant Chemotherapy in Breast Cancer. Int. J. Mol. Sci. 2025, 26, 3682. https://doi.org/10.3390/ijms26083682
Xiang L, Yang J, Rao J, Ma A, Liu C, Zhang Y, Huang A, Xie T, Xue H, Chen Z, et al. Integrating Machine Learning and Bulk and Single-Cell RNA Sequencing to Decipher Diverse Cell Death Patterns for Predicting the Prognosis of Neoadjuvant Chemotherapy in Breast Cancer. International Journal of Molecular Sciences. 2025; 26(8):3682. https://doi.org/10.3390/ijms26083682
Chicago/Turabian StyleXiang, Lingyan, Jiajun Yang, Jie Rao, Aolong Ma, Chen Liu, Yuqi Zhang, Aoling Huang, Ting Xie, Haochen Xue, Zhengzhuo Chen, and et al. 2025. "Integrating Machine Learning and Bulk and Single-Cell RNA Sequencing to Decipher Diverse Cell Death Patterns for Predicting the Prognosis of Neoadjuvant Chemotherapy in Breast Cancer" International Journal of Molecular Sciences 26, no. 8: 3682. https://doi.org/10.3390/ijms26083682
APA StyleXiang, L., Yang, J., Rao, J., Ma, A., Liu, C., Zhang, Y., Huang, A., Xie, T., Xue, H., Chen, Z., Yuan, J., & Yan, H. (2025). Integrating Machine Learning and Bulk and Single-Cell RNA Sequencing to Decipher Diverse Cell Death Patterns for Predicting the Prognosis of Neoadjuvant Chemotherapy in Breast Cancer. International Journal of Molecular Sciences, 26(8), 3682. https://doi.org/10.3390/ijms26083682