1. Introduction
Mitochondrial disorders (MDs) are a group of genetic conditions caused by mutations in mitochondrial or nuclear DNA resulting in impairment mitochondrial oxidative phosphorylation (OXPHOS) [
1,
2].
The mitochondrial respiratory chain is composed of multi-heteromeric complexes (I–V) that, functioning in concert, generate an electrochemical proton gradient across the internal mitochondrial membrane (complexes I–IV), the energy of which is eventually exploited to perform the condensation of ADP + P
i into ATP (complex V). Due to the essential function of OXPHOS for energy production in virtually all cells, MDs often exhibit a multi-systemic involvement, heterogeneous phenotypes, and a broad spectrum of clinical presentations, although the tissues with high energy demands, such as brain and cardiac or skeletal muscles are the most affected [
3].
An important subgroup amongst MDs comprises the mitochondrial complex III (CIII) deficiencies. CIII is a key component of the mitochondrial respiratory chain, since it catalyzes the transfer of electrons from coenzyme Q10 to cytochrome c, liberating energy exploited by the same complex III to pump protons from the mitochondrial matrix to the intermembrane space, thus sustaining the creation of the mitochondrial respiratory proton gradient. CIII consists of 11 subunits, most of which are encoded by nuclear genes, while only cytochrome b is encoded by mitochondrial DNA.
Pathogenic variants in genes encoding the core subunits of CIII and the assembly factors specific to complex III can cause mitochondrial CIII deficiency. Mutations in the
BCS1L gene, encoding for the BCS1L protein, are the most frequent [
4].
BCS1L is a homo-heptameric transmembrane AAA-ATPase belonging to the superfamily of AAA proteins. It acts as a chaperone, allowing the translocation of the folded Fe
2S
2 iron–sulfur Rieske (RISP) protein across the inner mitochondrial membrane, and its incorporation in the CIII precomplex [
5,
6,
7,
8].
The
BCS1L phenotypic spectrum ranges from Björnstad syndrome (BJS, OMIM #262000), characterized by noncongenital sensorineural hearing loss (SNHL) and
pili torti, to GRACILE syndrome (Growth Retardation, Aminoaciduria, Cholestasis, Iron Overload, and Lactic Acidosis, OMIM #603358) at the most severe end of the spectrum, through several different clinical manifestations in between, including proximal renal tubular acidosis (RTA), SNHL, and developmental delay [
9,
10].
Genetic mutations in
BCS1L may affect BCS1L protein levels and its mitochondrial import, the assembly/stability of CIII and supercomplexes, as well as the mitochondrial network [
5,
6]. However, the molecular mechanisms underlying the pathogenicity of specific variants remain incompletely understood.
This study investigates the pathogenicity of a BCS1L variant found in a child with a phenotype suggestive of an MD through functional assays in both yeast models and patient-derived human dermal fibroblasts (HDFs).
2. Results
2.1. Clinical Report
The patient, 27 months at the last follow-up, was the first-born offspring of healthy unrelated Italian parents of Caucasian ethnicity coming from a small city in northern Italy. He was born at term by vaginal delivery after spontaneous normal pregnancy. Family history was unremarkable. Apgar scores were 9 and 10 at 1 and 5 min. His birth weight was 2680 g (−2.04 SDS, small for gestational age, SGA), birth length 48 cm (−1.61 SDS), and occipitofrontal circumference 33 cm (−1.56 SDS). At 35 weeks of gestation, a prenatal ultrasound detected an anomaly in the renal regions. Following birth, a renal ultrasound confirmed bilateral renal ectopy, with both kidneys located in the right pelvic cavity. A subsequent evaluation of renal function revealed normal results, suggesting that this might represent a finding unrelated to his pathology.
He failed the newborn hearing screening, and as recommended by guidelines worldwide, a CMV diagnosis was ruled out using urine PCR analysis. At the age of 2 months, bilateral moderate-severe neurosensorial hearing loss was diagnosed as a result of otoemissions absence and pathological acoustic evoked potentials (V wave identifiable up to a stimulation threshold of 60 dB nHL bilaterally). Because of sensorineural hearing loss, trio whole exome sequencing (WES) was performed.
Following a reverse phenotyping approach, at the age of 18 months, the proband was found to be affected by proximal renal tubular acidosis, liver damage with high levels of plasma transaminases (4-fold normal values), and failure to thrive (weight 10.5 Kg, −0.37 SDS; length 71 cm, −4.17 SDS; BMI 20.83 Kg/m2, 3.02 SDS). Brain MRI and internal auditory structures were normal, whereas slight opacities of the lens were identified at eye examination. Echocardiogram was normal. He exhibited woolly and hypopigmented/brittle hair; however, pili torti were excluded at hair analysis by optical microscope.
At 23 months of age, the proband’s growth parameters were weight 10.7 Kg (−1.02 SDS), height 77.5 cm (−3.24 SDS), and BMI 17.81 Kg/m2 (0.79 SDS). A stimulation test with arginine was performed to assess growth hormone levels, which were found to be within the normal range (peak of 13.6 ng/mL). To correct the metabolic acidosis, oral supplementation with bicarbonate was started. However, despite a progressive incrementation of oral bicarbonates from 5 mEq/kg/die to 15 mEq/kg/die, blood tests showed persistent low levels of serum bicarbonates and severe acidosis. Management of the therapy was also complex due to poor patient compliance and a percutaneous endoscopic gastrostomy (PEG) was placed obtaining acid base balance by enteral administration of bicarbonates and other electrolytes.
He exhibited a delay in psychomotor development: he sat at 5 months, stood alone at around 18 months, and walked unassisted at 25 months. At the age of 18 months, his development was measured by Bayley III to be age-appropriate for the cognitive area (Development Quotient, DQ, 95) with weakness in the language skills (DQ 59), both receptive and expressive; however, the motor area was below the norm with deficit in gross motor, and static and dynamic balance skills, DQ 70. He was able to say his first words at 23 months and to combine two words at 27 months, exhibiting a sudden ‘lexical explosion’. At a speech and language assessment at 27 months, supported by administering standardized questionnaires (i.e., Little Ears, IT_MAIS and MacArthur-Bates CDI), language skills were measured to be slightly delayed. However, the patient had useful non-verbal communication to express needs and mood/emotions. Scores of the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS) 0–24 months are shown in
Supplementary Table S1. Items contributing the scores refer to developmental delay, in which gross motor skills were more impaired than language delay, fitting with the Bayley III results, and renal disease.
2.2. Genetic Evaluation
The WES analysis showed homozygous for the
BCS1L variant (NM_004328.5): c.38A>G, p.(Asn13Ser). Both parents were carriers of the same variant (
Figure 1A).
The p.(Asn13Ser) missense variant was of uncertain significance (VUS) according to the ACGS/ACMG criteria PM1 and PM2_moderate, PS3_supporting [
11] (
https://wintervar.wglab.org, accessed on 2 January 2024). The variant was absent from the gnomAD population database (
https://gnomad.broadinstitute.org, accessed on 2 January 2024).
It was previously described in a male affected by ventilation insufficiency, SNHL, tubulopathy, and hepatopathy; he died at 11 months (personal communication from Prof. Saskia B. Wortmann) [
10]. Indeed, the
BCS1L variant (NM_004328.5): c.38A>G, p.(Asn13Ser) was reported as pathogenic in Varsome (
https://varsome.com, accessed on 2 January 2024) and in HGMD
® Professional (
https://digitalinsights.qiagen.com, accessed on 2 January 2024) (
Figure 1B).
The Asn13 amino acid position is highly conserved from human to
Saccharomyces cerevisiae. It is located in the putative mitochondrial targeting signal, which is not cleaved in the case of
BCS1L [
12,
13].
2.3. Yeast Complementation Assay Confirmed BCS1L c.38A>G Variant Pathogenicity
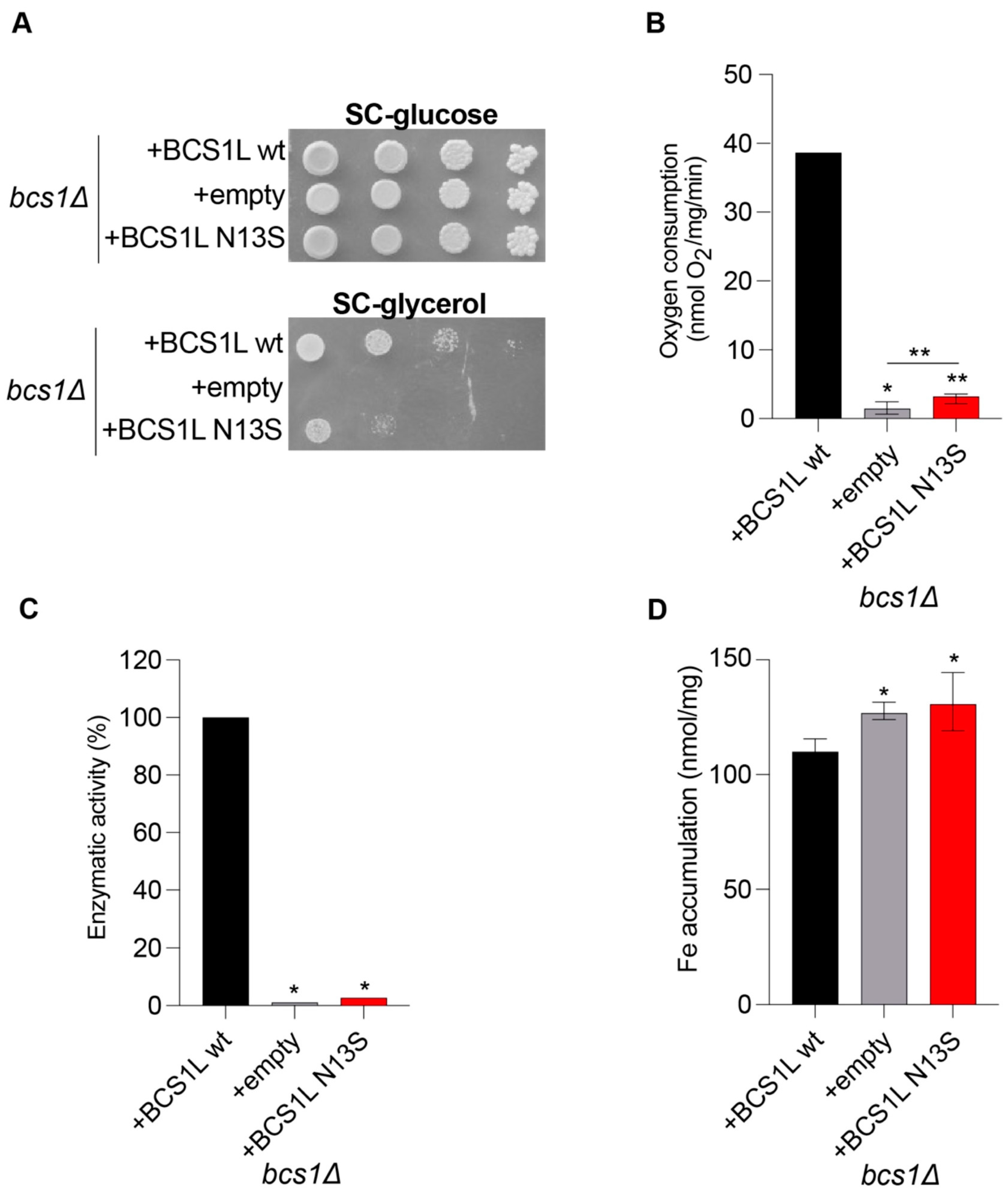
To study the functional effect of the p.Asn13Ser variant identified in the patient, we exploited the yeast S. cerevisiae ortholog gene BCS1.
As it was previously demonstrated that human
BCS1L cDNA is able to complement the defect of the yeast
bcs1Δ null mutant [
6,
14,
15], we used a homologous and a heterologous complementation approach, the latter consisting of the expression of the human
BCS1L gene (wild-type or harboring the Asn13Ser variant), cloned into the pYEX expression vector, in the yeast
bcs1Δ strain.
Data obtained by spot assay analyses and oxygen consumption measurements show a strong, albeit partial, oxidative growth defect and respiratory activity reduction for the strain carrying the mutant
bcs1lN13S allele (
Figure 2A,B). To better characterize the mechanisms underlying the mitochondrial impairment, we also showed that the
bcs1lN13S variant determines a strong CIII activity defect (
Figure 2C).
To evaluate if the mutation Asn13Ser is associated with iron accumulation, a quantitative determination of iron levels after growth with 2 mM ferrous sulfate was performed by a colorimetric assay that relies on the formation of colored iron complexes with BPS after nitric acid digestion of yeast cells [
16]. A significant increase in iron levels was observed in the strain carrying bcs1l
N13S (
Figure 2D).
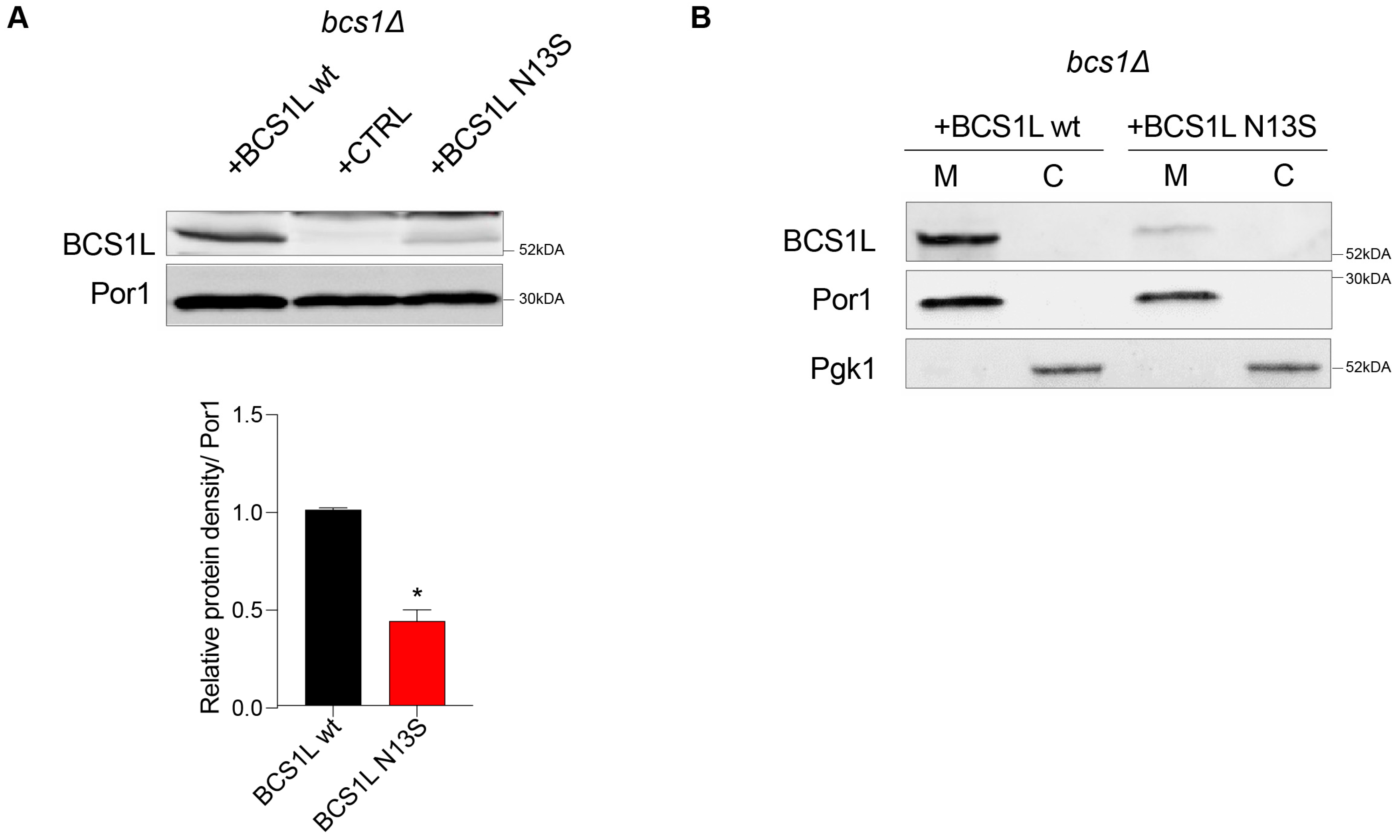
Furthermore, we assessed the effect of the variant on protein quantity, by measuring the steady-state level of the bcs1l wild-type and mutant protein through Western blot analysis. A significant reduction in the protein quantity was observed in the strain carrying
bcs1lN13S (43% ± 6 relative to the wild type), suggesting that the variant severely alters protein stability (
Figure 3A).
Due to the mutation position in the putative mitochondrial target sequence (MTS) [
7], we tested whether the aminoacidic change could inhibit the correct protein localization into mitochondria. Separation of cytosolic and mitochondrial proteins followed by Western blot analyses was employed to verify this hypothesis. The antibodies against porin/VDAC1 (Por1) and phosphoglycerate kinase (Pgk1) were used as markers for the mitochondrial and cytosolic fractions, respectively. Both the wild-type bcs1l and mutant bcs1l
N13S proteins correctly localized into the mitochondria as demonstrated by the signal in the mitochondrial fraction (
Figure 3B). Due to differences in the mitochondrial targeting sequence between yeast and humans and therefore possibly in the mechanism of protein import, we also analyzed protein localization of the yeast Bcs1
N49S mutant protein. As no commercial antibody against yeast Bcs1 exists, the HA epitope was added by mutagenic overlap PCR at the 3′ end. This data indicate that the mutation does not interfere with protein import but rather affects protein function and/or quantity.
In addition, a homologous complementation approach, using the yeast
BCS1L ortholog gene
BCS1, was used. As shown by protein alignment (
Supplementary Figure S1A), the human residue Asn13 is conserved and corresponds to the yeast residue Asn49. Thus, the corresponding codon was mutagenized, producing the mutant allele
bcs1N49S. The
BCS1 wild-type allele or the mutant allele, cloned into the centromeric vector pFL38, were transformed into the yeast
bcs1Δ null mutant, thus obtaining the strains
bcs1Δ/
bcs1 and
bcs1Δ/
bcs1N49S, respectively. The
bcs1Δ strain was also transformed with the empty vector as a control, obtaining
bcs1Δ/
pFL38. The latter strain could not grow on non-fermentable carbon sources (e.g., glycerol); however, re-expression of the wild-type
BCS1 rescued the growth defect (
Supplementary Figure S1B). The expression of the
bcs1N49S mutant allele resulted in a severe oxidative growth defect, but a partial ability to grow on oxidative carbon sources was retained (
Supplementary Figure S1B).
To better characterize the oxidative defect, mitochondrial respiratory activity was assessed by measuring the oxygen consumption rate; the strain expressing the wild-type
BCS1 allele consumed 42.55 ± 7.33 nmol O
2/mg/min. Instead, the strain expressing the mutant
bcs1N49S allele showed a strong reduction of oxygen consumption (4.87 ± 1.08 nmol O
2/mg/min), but a partial respiratory activity was retained. In fact, a significant difference with the null mutant strain (2.38 ± 0.67 nmol O
2/mg/min) was recorded (
Supplementary Figure S1C). Together, the results obtained show that the yeast Asn49Ser variant, equivalent to the human mutation Asn13Ser, is deleterious in yeast, supporting its pathogenicity in humans.
Altogether, data obtained in the yeast of the BCS1L Asn13Ser variant affecting oxidative growth and mitochondrial respiratory activity demonstrate its pathogenicity.
2.4. BCS1L N13S Affects Mitochondrial Respiration and CIII Activity in Human Dermal Fibroblasts (HDF)
The variant’s pathogenicity shown in the yeast model prompted us to extend the analysis in human cell models.
We isolated primary human dermal fibroblasts (HDFs) from the patient’s skin punch biopsy. In HDF cells, we first performed high-resolution respirometry analysis by Oroboros O2k oxygraphy showing a reduction of oxygen consumption in BCS1L Asn13Ser HDFs cells as compared to control wild-type cells (
Figure 4A). In detail, “routine respiration” was similar between the controls and the patient. Similarly, the non-phosphorylating resting state (“LEAK respiration”) stimulated by the addition of glutamate, and the OXPHOS capacity investigated through the oxidation of NADH at the level of CI stimulated by the addition of ADP and malate, did not display significant changes between control and patient-derived HDF cells, even though a slight reduction was observed. However, the electron transfer capacity (noncoupled electron transfer-state) activated by CCCP was lower in the patient’s cells as compared to a control, indicating an electron chain transport impairment. Similar results on oxygen consumption were obtained upon replacement of glucose with galactose in the culture media to force cells to rely upon OXPHOS for ATP production (
Supplementary Figure S2C); indeed, BCS1L Asn13Ser HDF cells grown in galactose show a strong reduction of proliferation as compared to the control, as well as to cells grown in glucose-rich medium (
Supplementary Figure S2D).
Taken together, the results clearly indicate impairment in electron transport. We then analyzed the activities of the mitochondrial respiratory chain enzymes, NADH dehydrogenase (complex I), DBH2:cytochrome oxidoreductase reductase (complex III), and cytochrome c oxidase (complex IV). Spectrophotometric kinetic measurement showed a strong reduction of the CIII enzymatic activity (
Figure 4B,
p = 0.04) (
Supplementary Figure S2A).
Altogether, these data demonstrate that the BCS1L Asn13Ser variant causes a reduction of oxygen consumption and a severe decrease of complex III activity in patient-derived fibroblasts confirming the deleterious impact of the variant as suggested by the yeast model.
2.5. BCS1L N13S Alters Mitochondrial Morphology and Network in HDFs
In addition, previous reports showed that some BCS1L protein variants might induce cytoplasmic protein mislocalization as well as abnormal mitochondrial morphologies secondary to the OXPHOS deficiency [
4,
15].
To verify the impact of the Asn13Ser variant, we performed immunofluorescence of HDFs derived either from the patient or control, finding that both BCS1L, either wild-type or Asn13Ser, specifically localized at the mitochondria, as indicated by Mitotracker Red mitochondrial marker co-staining (
Supplementary Figure S2B).
In addition, in patient fibroblasts, we assessed mitochondrial morphology by Mitotracker staining and BCS1L immunofluorescence, showing punctate mitochondria, shorter tubules, and reduced branches (
Figure 4C). Interestingly, we did not find any significant differences in mitochondrial count (
Figure 4D), while the average area and form factor (that reflects the complexity and branching aspect of mitochondria) were reduced in patient cells (
Figure 4E,F), suggesting smaller and more fragmented mitochondria. In addition, analyses of the mitochondrial network showed a reduction in the number, junction, and length of mitochondrial branches in BCS1L Asn13Ser HDF cells (
Figure 4G–I).
Altogether, these data suggest an impairment of the mitochondrial network in patient’s fibroblasts harboring the BCS1L Asn13Ser variant.
2.6. BCS1L N13S Affects CIII Assembly
Importantly, data obtained in yeast suggest that the BCS1L Asn13Ser variant affects protein levels. To validate this effect in human cells, we performed an immunoblot of BCS1L in lysates obtained from control- or patient-derived HDF cells, finding a significantly strong reduction of BCS1L levels (around 50%,
p = 0.004), without altering the levels of RISP protein, while cytochrome-b was slightly reduced (25%,
p = 0.04) (
Figure 4A), suggesting that the whole complex III holoenzyme is reduced. Of note, differences found in BCS1L protein levels does not rely on
BCS1L gene expression changes since mRNA levels in HDFs are unaltered (
Supplementary Figure S3B).
To understand the molecular mechanism underlying the BCS1L Asn13Ser-dependent biochemical alterations, we evaluated its impact on complex CIII assembly.
As a first step, we assessed the interaction between BCS1L and CIII by proximity ligation assays; unlike what was expected, the binding of BCS1 to the CIII complex was increased with the BCS1 Asn13Ser mutant form in HDFs as compared to the control (
Figure 5B).
To avoid genetic background effects, plasmids overexpressing FLAG-tagged BCS1L, either wild-type or a N13S mutant, were transfected into HDF cells from patients and controls and a proximity ligation assay was performed. The results showed that, regardless of the genetic background, BCS1L N13S interacts more with CIII (as revealed by anti- FLAG and cyt-b antibodies) compared to wild-type BCS1L, in both control and patient-derived fibroblasts (
Supplementary Figure S3B).
Thus, we next characterize the CIII assembly in patient cells by WB analysis on blue native gel electrophoresis. Western blot analysis with the anti-Cyt-B antibody showed a marked reduction of the CIII-CIV complex band in patient-derived cell lysates relative to the control; accordingly, the RISP protein is less present in CIII and CIII-CIV complexes. Taken together, these results indicate a reduction of the complete assembled CIII complex. Surprisingly, mutant BCS1L protein showed an accumulation in CIII complex in fibroblasts from the patient (
Figure 5C).
Altogether, these results suggest that BCS1L is unable to properly transfer the RISP protein on the nascent CIII complex remaining attached to it, resulting in a reduction of CIII holoenzyme and CIII-CIV supercomplex assembling, and the RISP protein (unaltered in total levels) is only partly bound to BCS1L remaining unincorporated.
3. Discussion
BCS1L is a homo-heptameric transmembrane AAA-ATPase that acts as a chaperone for CIII holoenzyme. Mutations in the
BCS1L gene might affect BCS1L protein levels and its mitochondrial import, assembly/stability of CIII and supercomplexes, and the mitochondrial network [
5,
6].
In this work, we identified a homozygous c.38A>G variant in the BCS1L gene (p.Asn13Ser) in a 27-month-old male showing proximal renal tubular acidosis, sensorineural hearing loss, hypopigmented hair, and developmental delay [
4]. By this clinical picture, the patient did not fulfill clinical criteria to perform classical GRACILE or Björnstad syndrome diagnosis. Although exhibiting clear symptoms of mitochondriopathy, its specific disease requires reclassification. Indeed, BCS1L-related diseases have a wide range of clinical symptoms that manifests as a continuum spectrum, instead of being distinct clinical entities [
10], and is referred to as mitochondrial complex III deficiency, nuclear type 1 (MC3DN1, OMIM #124000).
The p.Asn13Ser missense mutation localizes in the N-terminus of the BCS1L protein. The N-ter of BCS1L consists of three specialized regions involved in targeting and sorting of the protein in the mitochondria, namely the transmembrane domain (TMD), the mitochondrial targeting sequence (MTS), and the import auxiliary sequence (IAS) [
12,
13]. The residue 13 localizes in a portion of the protein that does not form any structural domain and lies in the intermembrane space. Thus, in analogy to other reported variants in the N-ter of
BCS1L, the mutation might affect BCS1L proper mitochondrial localization; however, in HDF cells, by BCS1L immunostaining and mitochondrial counterstaining, we showed that the BCS1L Asn13Ser does not influence it, excluding this as the pathogenic mechanism induced by the variant. Conversely, the mutant form of BCS1L seems to be less stable than the wild-type counterpart since the levels of BCS1L protein are reduced while its mRNA levels are unchanged. These results were supported by data in yeast models, that recapitulate a reduction on protein levels without affecting the mitochondrial localization; these results support an essential, evolutionary conserved role of the pAsn13 residue in the stability of the BCS1L protein.
Functional analysis, both in yeast models and in patient-derived human dermal fibroblasts, further unveiled the pathogenicity of this BCS1L variant showing the impairment of the mitochondrial respiratory function and the decrement of CIII activity, accompanied also by disruption of the mitochondrial network.
BCS1L is an assembly factor required for the insertion of the Rieske Fe
2S
2 protein into the CIII precomplex to complete CIII assembly. Thus, defects in BCS1L indeed might cause iron overload as the yeast model confirmed for the reported variant [
17].
Importantly, in patient-derived cells, we found that the levels of RISP were unaltered, despite the reduction of cytochrome-b levels and CIII activity, suggesting a deleterious impact on CIII complex assembly and stability.
To shed light on this molecular mechanism, we evaluated the interaction between the BCS1L and CIII complex by proximity ligation assay (PLA). PLA is based on immunodetection of two different proteins close to each other at a distance of 20 nanometers or less. This experiment shows a marked increase of interaction between CIII and mutant BCS1L, compared to the wild-type protein, suggesting that the binding between CIII and BCS1L, which is normally transient in controls, remains stalled in the Asn13Ser BCS1L variant. WB on native gel electrophoresis confirmed an impairment of CIII holoenzyme assembly and in the formation of the CIII-CIV supercomplex. The mutant BCS1L was unable to load RISP into the nascent complex III to complete its assembly, thus resulting in the accumulation of a BCS1L-containing nonfunctional preCIII. These findings give new insights into the pathogenic mechanisms of BCS1L mutant protein.
Interestingly, the same variant was previously described in a patient with worse clinical manifestation who died at 11 months old, suggesting the influence of the genetic background and the existence of compensatory mechanisms [
10].
Besides the role on CIII activity, the exact mechanism underlying the BCS1L-related phenotype remains unclear.
Even though the role of BCS1L as a chaperone on CIII is solid, its impact in OXPHOS in different tissue is less specific, as
BCS1L pathogenetic variants were identified in patients exhibiting normal respiratory chain function in the skeletal muscle, skin fibroblasts, and liver [
10]. This evidence suggests that other downstream cellular mechanisms might contribute to the pathomechanism and disease severity.
BCS1L pathogenic variants affecting the assembly or stability of respiratory complexes CIII and the supercomplex assembly between CI, CIII, and CIV might lead to ROS production, cell death, and structural alterations in the mitochondrial network. Upon mitochondrial dysfunction, cells are able to trigger compensatory responses, relying on retrograde signals from the mitochondria to the nucleus [
18]. Only recently, it has been reported that the BCS1L mutant causes DNA damage, cellular senescence, and systemic progeroid phenotype by triggering c-MYC upregulation, starting to shed light of the role of BCS1L in these processes [
19]. Identification of such pathways might ameliorate the clinical management of the secondary symptoms caused by
BCS1L pathogenic variants; that, however, goes further beyond the scope of this study.
Indeed, the mechanism by which this set of phenotypes is acquired is not known. More importantly, except for the Finnish variant (c.232A>G p.Ser78Gly) which always causes GRACILE syndrome with a well-defined natural history and outcome [
20,
21], for the other specific variants the pathogenicity remains incompletely understood without any clear correlation between the location of the mutations in the gene and the severity and clinical presentation of
BCS1L-related disease.
In this study, we report on the biochemical and metabolic consequences of a previously uncharacterized BCS1L variant, resulting in mitochondrial morphology and bioenergetic impairment. Unexpectedly, we reported the increased interaction between BCS1L and CIII, indicating the accumulation of BCS1L-containing nonfunctional preCIII unable to load RISP protein and complete CIII assembly. These data characterized the mechanism of pathogenicity and expand the phenotypic spectrum of BCS1L c.38A>G variant.
Understanding these pathogenic mechanisms contributes to the broader knowledge of mitochondrial disorders, possibly allowing for targeted interventions, finally improving the clinical outcomes of mitochondrial patients with CIII deficiency, which remains untreatable to date. Ultimately, these efforts could expand awareness of OXPHOS defects, deepen our understanding of their complex phenotypes, and pave the way for better treatment approaches in the future.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}