
The Revolution of Targeted Therapies in Thyroid Cancer Treatment: Present and Future Promising Anti-Cancer Drugs


Abstract
1. Introduction
2. Genomic Events in Thyroid Carcinogenesis Used as Targets
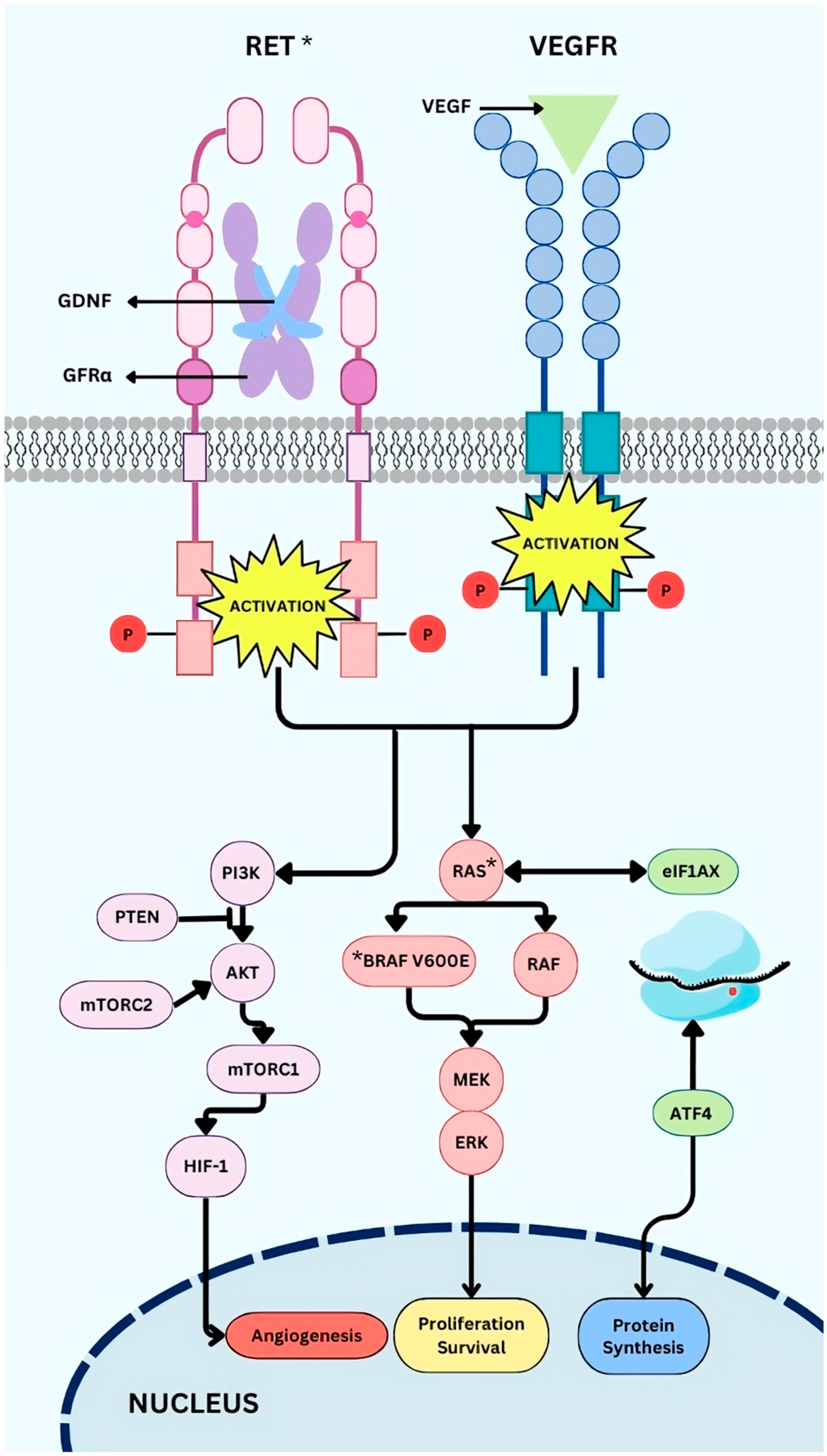
2.1. Driver Mutations
2.2. Other Alterations
3. Anti-Cancer Agents in TC
3.1. Multikinase Inhibitors (MKIs)
3.2. Combined Targeted Therapies
3.3. RET Selective Inhibitors
3.4. RAS Inhibitors
3.5. Other Potential Selective Inhibitors for TC
3.6. Immunotherapies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALK | Anaplastic lymphoma kinase |
| ATC | Anaplastic thyroid carcinoma |
| ATP | Adenosine triphosphate |
| CDK | Cyclin-dependent kinases |
| cMYC | Cardiac myosin-binding protein C |
| DHGTC | Differentiated high-grade thyroid carcinoma |
| DTC | Differentiated thyroid carcinoma |
| EGFR | Epidermal growth factor receptor |
| EIF1AX | Eukaryotic translation initiation factor 1A X-linked |
| EMA | European Medicines Agency |
| ERK | Extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| FGFR | Fibroblast growth factor receptor |
| FTase | Farnesyltransferase |
| HGF | Hepatocyte growth factor |
| HR | Hazard ratio |
| MEN2A/2B | Multiple endocrine neoplasia type 2A/2B |
| MEK | Mitogen-activated extracellular signal-regulated kinase |
| MKI | Multikinase inhibitor |
| MAPK | Mitogen-Activated Protein Kinase |
| MTC | Medullary thyroid carcinoma |
| NF1 | Neurofibromin |
| NIS | Sodium Iodide Symporter |
| NRG1 | Neuregulin 1 |
| NTRK | Neurotrophic tropomyosin-receptor kinase |
| PDGFR -β | Platelet-derived growth factor receptor-beta |
| PD-L1 | Programmed cell death ligand 1 |
| PD-1 | Programmed cell death protein 1 |
| PDTC | Poorly differentiated thyroid carcinoma |
| PFS | Progression-free survival |
| PIC | Preinitiation complex |
| PI3K | Phosphoinositide 3-kinase |
| PTC | Papillary thyroid carcinoma |
| RAIR | Radioactive iodine refractory |
| RET | REarranged during Transfection |
| RETi | RET inhibitor |
| RTK | Receptor tyrosine kinase |
| sMTC | Sporadic medullary thyroid carcinoma |
| TC | Thyroid cancer |
| TCGA | The Cancer Genome Atlas |
| TERT | Telomerase reverse transcriptase |
| VEGFR | Vascular endothelial growth factor receptor |
| WT | Wild type |
References
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid Cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Boucai, L.; Zafereo, M.; Cabanillas, M.E. Thyroid Cancer: A Review. JAMA 2024, 331, 425–435. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Series. WHO Classification of Tumours Editorial Board. Endocrine and Neuroendocrine Tumours, 5th ed.; WHO: Lyon, France, 2022; Volume 10.
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Mete, O.; Baloch, Z.W. The 2022 WHO Classification of Thyroid Tumors: Novel Concepts in Nomenclature and Grading. Endocr.-Relat. Cancer 2023, 30, e220293. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and Transcriptomic Hallmarks of Poorly Differentiated and Anaplastic Thyroid Cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Zeng, P.Y.F.; Prokopec, S.D.; Lai, S.Y.; Pinto, N.; Chan-Seng-Yue, M.A.; Clifton-Bligh, R.; Williams, M.D.; Howlett, C.J.; Plantinga, P.; Cecchini, M.J.; et al. The Genomic and Evolutionary Landscapes of Anaplastic Thyroid Carcinoma. Cell Rep. 2024, 43, 113826. [Google Scholar] [CrossRef]
- Wells, S.A.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association Guidelines for the Management of Medullary Thyroid Carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Stamatakos, M.; Paraskeva, P.; Stefanaki, C.; Katsaronis, P.; Lazaris, A.; Safioleas, K.; Kontzoglou, K. Medullary Thyroid Carcinoma: The Third Most Common Thyroid Cancer Reviewed. Oncol. Lett. 2011, 2, 49–53. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Ryder, M.; Jimenez, C. Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef]
- Landa, I.; Cabanillas, M.E. Genomic Alterations in Thyroid Cancer: Biological and Clinical Insights. Nat. Rev. Endocrinol. 2024, 20, 93–110. [Google Scholar] [CrossRef]
- Nannini, M.; Repaci, A.; Nigro, M.C.; Colapinto, A.; Vicennati, V.; Maloberti, T.; Gruppioni, E.; Altimari, A.; Solaroli, E.; Lodi Rizzini, E.; et al. Clinical Relevance of Gene Mutations and Rearrangements in Advanced Differentiated Thyroid Cancer. ESMO Open 2023, 8, 102039. [Google Scholar] [CrossRef] [PubMed]
- Sahakian, N.; Castinetti, F.; Romanet, P. Molecular Basis and Natural History of Medullary Thyroid Cancer: It Is (Almost) All in the RET. Cancers 2023, 15, 4865. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Qu, N.; Chen, D.; Ma, B.; Zhang, L.; Wang, Q.; Wang, Y.; Wang, H.; Ni, Z.; Wang, W.; Liao, T.; et al. Integrated Proteogenomic and Metabolomic Characterization of Papillary Thyroid Cancer with Different Recurrence Risks. Nat. Commun. 2024, 15, 3175. [Google Scholar] [CrossRef]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on Fundamental Mechanisms of Thyroid Cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Shong, Y.K.; Kim, T.Y.; Viola, D.; Elisei, R.; Bendlová, B.; Yip, L.; Mian, C.; et al. Association Between BRAF V600E Mutation and Recurrence of Papillary Thyroid Cancer. J. Clin. Oncol. 2015, 33, 42–50. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone Deacetylation of NIS Promoter Underlies BRAF V600E-Promoted NIS Silencing in Thyroid Cancer. Endocr.-Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef]
- Riccio, I.; Laforteza, A.; Landau, M.B.; Hussein, M.H.; Linhuber, J.; Staav, J.; Issa, P.P.; Toraih, E.A.; Kandil, E. Decoding RAS Mutations in Thyroid Cancer: A Meta-Analysis Unveils Specific Links to Distant Metastasis and Increased Mortality. Am. J. Otolaryngol. 2025, 46, 104570. [Google Scholar] [CrossRef]
- Howell, G.M.; Hodak, S.P.; Yip, L. RAS Mutations in Thyroid Cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef]
- Bikas, A.; Ahmadi, S.; Pappa, T.; Marqusee, E.; Wong, K.; Nehs, M.A.; Cho, N.L.; Haase, J.; Doherty, G.M.; Sehgal, K.; et al. Additional Oncogenic Alterations in RAS-Driven Differentiated Thyroid Cancers Associate with Worse Clinicopathologic Outcomes. Clin. Cancer Res. 2023, 29, 2678–2685. [Google Scholar] [CrossRef]
- Lodish, M.B.; Stratakis, C.A. RET Oncogene in MEN2, MEN2B, MTC, and Other Forms of Thyroid Cancer: Molecular Genetics and Therapeutic Advances. Expert. Rev. Anticancer Ther. 2008, 8, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Moccia, M.; Federico, G.; Carlomagno, F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes 2020, 11, 424. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, J.S.; Effraimidis, G.; Rossing, M.; Rasmussen, Å.K.; Hoejberg, L.; Bastholt, L.; Godballe, C.; Oturai, P.; Feldt-Rasmussen, U. Multiple Endocrine Neoplasia Type 2: A Review. Semin. Cancer Biol. 2022, 79, 163–179. [Google Scholar] [CrossRef]
- Kim, M.; Kim, B.H. Current Guidelines for Management of Medullary Thyroid Carcinoma. Endocrinol. Metab. 2021, 36, 514–524. [Google Scholar] [CrossRef]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT Promoter Mutations Are a Major Indicator of Poor Outcome in Differentiated Thyroid Carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xing, M. TERT Promoter Mutations in Thyroid Cancer. Endocr.-Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly Prevalent TERT Promoter Mutations in Aggressive Thyroid Cancers. Endocr.-Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, X.; Jiang, Y.; Guo, M.; Zhang, S.; Li, J.; He, J.; Liu, J.; Wang, J.; Ouyang, L. Recent Advances in the Development of Dual VEGFR and C-Met Small Molecule Inhibitors as Anticancer Drugs. Eur. J. Med. Chem. 2016, 108, 495–504. [Google Scholar] [CrossRef]
- Bandargal, S.; Chen, T.; Pusztaszeri, M.P.; Forest, V.-I.; da Silva, S.D.; Payne, R.J. Prognostic Indicators of EIF1AX-Mutated Thyroid Tumor Malignancy and Cancer Aggressiveness. Cancers 2022, 14, 6097. [Google Scholar] [CrossRef]
- Karunamurthy, A.; Panebianco, F.; Hsiao, S.J.; Vorhauer, J.; Nikiforova, M.N.; Chiosea, S.; Nikiforov, Y.E. Prevalence and Phenotypic Correlations of EIF1AX Mutations in Thyroid Nodules. Endocr.-Relat. Cancer 2016, 23, 295–301. [Google Scholar] [CrossRef]
- Kozak, M. Initiation of Translation in Prokaryotes and Eukaryotes. Gene 1999, 234, 187–208. [Google Scholar] [CrossRef]
- Krishnamoorthy, G.P.; Davidson, N.R.; Leach, S.D.; Zhao, Z.; Lowe, S.W.; Lee, G.; Landa, I.; Nagarajah, J.; Saqcena, M.; Singh, K.; et al. EIF1AX and RAS Mutations Cooperate to Drive Thyroid Tumorigenesis through ATF4 and C-MYC. Cancer Discov. 2019, 9, 264–281. [Google Scholar] [CrossRef]
- Leandro-García, L.J.; Landa, I. Mechanistic Insights of Thyroid Cancer Progression. Endocrinology 2023, 164, bqad118. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Bullock, M.; Robinson, B.G.; Clifton-Bligh, R. Multikinase Inhibitors: A New Option for the Treatment of Thyroid Cancer. Nat. Rev. Endocrinol. 2011, 7, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 Inhibition of Oncogenic RET Mutants. JNCI J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef]
- Yu, C.; Bruzek, L.M.; Meng, X.W.; Gores, G.J.; Carter, C.A.; Kaufmann, S.H.; Adjei, A.A. The Role of Mcl-1 Downregulation in the Proapoptotic Activity of the Multikinase Inhibitor BAY 43-9006. Oncogene 2005, 24, 6861–6869. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Jean, G.W.; Mani, R.M.; Jaffry, A.; Khan, S.A. Toxic Effects of Sorafenib in Patients With Differentiated Thyroid Carcinoma Compared With Other Cancers. JAMA Oncol. 2016, 2, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase. Clin. Cancer Res. 2008, 14, 5459–5465. [Google Scholar] [CrossRef]
- Xue, L.; Gong, Z.; Vlantis, A.C.; Chan, J.Y.; Meehan, K.; van Hasselt, C.A.; Li, D.; Zeng, X.; Wei, M.; Tong, M.C.; et al. Autophagy Regulates Anti-Angiogenic Property of Lenvatinib in Thyroid Cancer. Am. J. Cancer Res. 2023, 13, 1457–1470. [Google Scholar]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an Orally Available Inhibitor of KDR Tyrosine Kinase Activity, Efficiently Blocks Oncogenic RET Kinases1. Cancer Res. 2002, 62, 7284–7290. [Google Scholar]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 Inhibits Vascular Endothelial Growth Factor Signaling, Angiogenesis, and Tumor Growth Following Oral Administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients With Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. JCO 2012, 30, 134–141. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Naylor, R.M.; Tabruyn, S.P.; You, W.; Aftab, D.T.; McDonald, D.M. Abstract A13: Reduction of Tumor Invasiveness and Metastasis and Prolongation of Survival of RIP-Tag2 Mice after Inhibition of VEGFR plus c-Met by XL184. Mol. Cancer Ther. 2009, 8 (Suppl. 12), A13. [Google Scholar] [CrossRef]
- Schlumberger, M.; Elisei, R.; Müller, S.; Schöffski, P.; Brose, M.; Shah, M.; Licitra, L.; Krajewska, J.; Kreissl, M.C.; Niederle, B.; et al. Overall Survival Analysis of EXAM, a Phase III Trial of Cabozantinib in Patients with Radiographically Progressive Medullary Thyroid Carcinoma. Ann. Oncol. 2017, 28, 2813–2819. [Google Scholar] [CrossRef]
- Brose, M.S.; Robinson, B.; Sherman, S.I.; Krajewska, J.; Lin, C.-C.; Vaisman, F.; Hoff, A.O.; Hitre, E.; Bowles, D.W.; Hernando, J.; et al. Cabozantinib for Radioiodine-Refractory Differentiated Thyroid Cancer (COSMIC-311): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Robinson, B.G.; Sherman, S.I.; Jarzab, B.; Lin, C.-C.; Vaisman, F.; Hoff, A.O.; Hitre, E.; Bowles, D.W.; Sen, S.; et al. Cabozantinib for Previously Treated Radioiodine-Refractory Differentiated Thyroid Cancer: Updated Results from the Phase 3 COSMIC-311 Trial. Cancer 2022, 128, 4203–4212. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-Enhanced Radioiodine Uptake in Advanced Thyroid Cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.A.; Sherman, E.J.; Baxi, S.S.; Tchekmedyian, V.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J. Clin. Endocrinol. Metab. 2019, 104, 1417–1428. [Google Scholar] [CrossRef]
- Hofmann, M.-C.; Kunnimalaiyaan, M.; Wang, J.R.; Busaidy, N.L.; Sherman, S.I.; Lai, S.Y.; Zafereo, M.; Cabanillas, M.E. Molecular Mechanisms of Resistance to Kinase Inhibitors and Redifferentiation in Thyroid Cancers. Endocr.-Relat. Cancer 2022, 1, R173–R190. [Google Scholar] [CrossRef]
- Laquerre, S.; Arnone, M.; Moss, K.; Yang, J.; Fisher, K.; Kane-Carson, L.S.; Smitheman, K.; Ward, J.; Heidrich, B.; Rheault, T.; et al. Abstract B88: A Selective Raf Kinase Inhibitor Induces Cell Death and Tumor Regression of Human Cancer Cell Lines Encoding B-RafV600E Mutation. Mol. Cancer Ther. 2009, 8 (Suppl. 12), B88. [Google Scholar] [CrossRef]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in Patients with Melanoma, Untreated Brain Metastases, and Other Solid Tumours: A Phase 1 Dose-Escalation Trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Zeiser, R.; Andrlová, H.; Meiss, F. Trametinib (GSK1120212). In Small Molecules in Oncology; Martens, U.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 91–100. [Google Scholar] [CrossRef]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus Trametinib in Patients with BRAF V600E-Mutant Anaplastic Thyroid Cancer: Updated Analysis from the Phase II ROAR Basket Study. Ann. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef]
- Busaidy, N.L.; Konda, B.; Wei, L.; Wirth, L.J.; Devine, C.; Daniels, G.A.; DeSouza, J.A.; Poi, M.; Seligson, N.D.; Cabanillas, M.E.; et al. Dabrafenib Versus Dabrafenib + Trametinib in BRAF-Mutated Radioactive Iodine Refractory Differentiated Thyroid Cancer: Results of a Randomized, Phase 2, Open-Label Multicenter Trial. Thyroid 2022, 32, 1184–1192. [Google Scholar] [CrossRef]
- Leboulleux, S.; Do Cao, C.; Zerdoud, S.; Attard, M.; Bournaud, C.; Lacroix, L.; Benisvy, D.; Taïeb, D.; Bardet, S.; Terroir-Cassou-Mounat, M.; et al. A Phase II Redifferentiation Trial with Dabrafenib-Trametinib and 131I in Metastatic Radioactive Iodine Refractory BRAF p.V600E-Mutated Differentiated Thyroid Cancer. Clin. Cancer Res. 2023, 29, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, S.M.; McFadden, D.G.; Palmer, E.L.; Daniels, G.H.; Wirth, L.J. Redifferentiation of Iodine-Refractory BRAF V600E-Mutant Metastatic Papillary Thyroid Cancer with Dabrafenib. Clin. Cancer Res. 2015, 21, 1028–1035. [Google Scholar] [CrossRef]
- Leboulleux, S.; Cao, C.D.; Zerdoud, S.; Attard, M.; Bournaud, C.; Benisvy, D.; Taieb, D.; Bardet, S.; Terroir-Cassou-Mounat, M.; Betrian, S.; et al. MERAIODE: A Redifferentiation Phase II Trial With Trametinib and Dabrafenib Followed by Radioactive Iodine Administration for Metastatic Radioactive Iodine Refractory Differentiated Thyroid Cancer Patients With a BRAFV600E Mutation (NCT 03244956). J. Endocr. Soc. 2021, 5 (Suppl. 1), A876. [Google Scholar] [CrossRef]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.W.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in Patients with BRAFV600E-Positive Metastatic or Unresectable Papillary Thyroid Cancer Refractory to Radioactive Iodine: A Non-Randomised, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Garbe, C.; Eigentler, T.K. Vemurafenib. In Small Molecules in Oncology; Martens, U.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 77–89. [Google Scholar] [CrossRef]
- van Berge Henegouwen, J.M.; van der Wijngaart, H.; Zeverijn, L.J.; Hoes, L.R.; Meertens, M.; Huitema, A.D.R.; Devriese, L.A.; Labots, M.; Verheul, H.M.W.; Voest, E.E.; et al. Efficacy and Toxicity of Vemurafenib and Cobimetinib in Relation to Plasma Concentrations, after Administration via Feeding Tube in Patients with BRAF-Mutated Thyroid Cancer: A Case Series and Review of Literature. Cancer Chemother. Pharmacol. 2022, 90, 97–104. [Google Scholar] [CrossRef]
- Cancer Research UK. DETERMINE (Determining Extended Therapeutic Indications for Existing Drugs in Rare Molecularly Defined Indications Using a National Evaluation Platform Trial): An Umbrella-Basket Platform Trial to Evaluate the Efficacy of Targeted Therapies in Rare Adult, Paediatric and Teenage/Young Adult (TYA) Cancers With Actionable Genomic Alterations, Including Common Cancers With Rare Actionable Alterations Treatment Arm 05: Vemurafenib in Combination With Cobimetinib in Adult Patients With BRAF Positive Cancers.; Clinical Trial Registration NCT05768178. 2023. Available online: https://clinicaltrials.gov/study/NCT05768178 (accessed on 16 February 2025).
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Tian, M.; Hu, L.; Ruan, X.; Zhang, W.; Zheng, X.; Gao, M. Early Combined SHP2 Targeting Reverses the Therapeutic Resistance of Vemurafenib in Thyroid Cancer. J. Cancer 2023, 14, 1592–1604. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Levin, R.S.; Gordan, J.D.; Oses-Prieto, J.A.; Webber, J.T.; Salt, M.; Hann, B.; Burlingame, A.L.; McCormick, F.; Bandyopadhyay, S.; et al. Oncogene Mimicry as a Mechanism of Primary Resistance to BRAF Inhibitors. Cell Rep. 2014, 8, 1037–1048. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of Feedback Inhibition of HER3 Transcription by RAF and MEK Inhibitors Attenuates Their Antitumor Effects in BRAF-Mutant Thyroid Carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Garcia-Rendueles, M.E.R.; Krishnamoorthy, G.; Saqcena, M.; Acuña-Ruiz, A.; Revilla, G.; de Stanchina, E.; Knauf, J.A.; Lester, R.; Xu, B.; Ghossein, R.A.; et al. Yap Governs a Lineage-Specific Neuregulin1 Pathway-Driven Adaptive Resistance to RAF Kinase Inhibitors. Mol. Cancer 2022, 21, 213. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Stevens, S.E.; Lin, J.J.; Nagy, R.; Ferris, L.; Shaw, A.T.; Gainor, J.F. Emergence of a RET V804M Gatekeeper Mutation During Treatment With Vandetanib in RET-Rearranged NSCLC. J. Thorac. Oncol. 2018, 13, e226–e227. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Capdevila, J. The Right Compound for the Right Target: Tackling RET. Ann. Oncol. 2018, 29, 1623–1625. [Google Scholar] [CrossRef]
- Nakaoku, T.; Kohno, T.; Araki, M.; Niho, S.; Chauhan, R.; Knowles, P.P.; Tsuchihara, K.; Matsumoto, S.; Shimada, Y.; Mimaki, S.; et al. A Secondary RET Mutation in the Activation Loop Conferring Resistance to Vandetanib. Nat. Commun. 2018, 9, 625. [Google Scholar] [CrossRef]
- Gil-Bernabé, S.; García-DeLaFuente, L.; García-Álvarez, A.; García-Rostán, G.; Capdevila, J.; Hernando, J. Genomics Review of Selective RET Inhibitors Sensitivity in Thyroid Cancer Clinical Trials. Am. J. Med. Genet. Part C Semin. Med. Genet. 2025, e32127. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET -Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective RET Kinase Inhibition for Patients with RET-Altered Cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef]
- Brandhuber, B.; Haas, J.; Tuch, B.; Ebata, K.; Bouhana, K.; McFaddin, E.; Williams, L.; Winski, S.; Brown, E.; Burkhard, M.; et al. The Development of a Potent, KDR/VEGFR2-Sparing RET Kinase Inhibitor for Treating Patients with RET-Dependent Cancers. Eur. J. Cancer 2016, 69, S144. [Google Scholar] [CrossRef]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V.; et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-Agnostic Efficacy and Safety of Selpercatinib in Patients with RET Fusion-Positive Solid Tumours Other than Lung or Thyroid Tumours (LIBRETTO-001): A Phase 1/2, Open-Label, Basket Trial. Lancet Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef]
- Hadoux, J.; Elisei, R.; Brose, M.S.; Hoff, A.O.; Robinson, B.G.; Gao, M.; Jarzab, B.; Isaev, P.; Kopeckova, K.; Wadsley, J.; et al. Phase 3 Trial of Selpercatinib in Advanced RET-Mutant Medullary Thyroid Cancer. N. Engl. J. Med. 2023, 389, 1851–1861. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Shen, T.; Terzyan, S.S.; Liu, X.; Hu, X.; Patel, K.P.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural Basis of Acquired Resistance to Selpercatinib and Pralsetinib Mediated by Non-Gatekeeper RET Mutations. Ann. Oncol. 2021, 32, 261–268. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Curigliano, G.; Brose, M.S.; Zhu, V.W.; Leboulleux, S.; Bowles, D.W.; et al. Pralsetinib for Patients with Advanced or Metastatic RET-Altered Thyroid Cancer (ARROW): A Multi-Cohort, Open-Label, Registrational, Phase 1/2 Study. Lancet Diabetes Endocrinol. 2021, 9, 491–501. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.; Mansfield, A.S.; Taylor, M.H.; Schuler, M.; Zhu, V.W.; Hadoux, J.; Curigliano, G.; Wirth, L.; Gainor, J.F.; et al. Pralsetinib in Patients with Advanced/Metastatic Rearranged During Transfection (RET)-Altered Thyroid Cancer: Updated Efficacy and Safety Data from the ARROW Study. Thyroid 2024, 34, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.; Fisher, G.; Brook, S.; Patel, S.; Arkenau, H.-T. Selective RET Inhibitors (SRIs) in Cancer: A Journey from Multi-Kinase Inhibitors to the Next Generation of SRIs. Cancers 2023, 16, 31. [Google Scholar] [CrossRef]
- Schoffski, P.; Cho, B.C.; Italiano, A.; Loong, H.H.F.; Massard, C.; Medina Rodriguez, L.; Shih, J.-Y.; Subbiah, V.; Verlingue, L.; Andreas, K.; et al. BOS172738, a Highly Potent and Selective RET Inhibitor, for the Treatment of RET-Altered Tumors Including RET-Fusion+ NSCLC and RET-Mutant MTC: Phase 1 Study Results. J. Clin. Oncol. 2021, 39 (Suppl. 15), 3008. [Google Scholar] [CrossRef]
- Miyazaki, I.; Ishida, K.; Kato, M.; Suzuki, T.; Fujita, H.; Ohkubo, S.; Iwasawa, Y. Abstract P06-02: Discovery of TAS0953/HM06, a Novel next Generation RET-Specific Inhibitor Capable of Inhibiting RET Solvent Front Mutations. Mol. Cancer Ther. 2021, 20 (Suppl. 12), P06-02. [Google Scholar] [CrossRef]
- Miyazaki, I.; Odintsov, I.; Ishida, K.; Lui, A.J.W.; Kato, M.; Suzuki, T.; Zhang, T.; Wakayama, K.; Kurth, R.I.; Cheng, R.; et al. Vepafestinib Is a Pharmacologically Advanced RET-Selective Inhibitor with High CNS Penetration and Inhibitory Activity against RET Solvent Front Mutations. Nat. Cancer 2023, 4, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Shouyao Holdings (Beijing), Co. LTD. A Phase I/II, Open-Label, Single-Arm, Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Antineoplastic Activity of SY-5007 in Patients With RET-Altered Advanced Solid Tumor; Clinical Trial Registration NCT05278364. 2023. Available online: https://clinicaltrials.gov/study/NCT05278364 (accessed on 1 January 2024).
- Ellipses Pharma. A Modular, Open-Label, Phase I/II Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Efficacy of EP0031 in Patients With Advanced RET-Altered Malignancies; Clinical Trial Registration NCT05443126. 2024. Available online: https://clinicaltrials.gov/study/NCT05443126 (accessed on 18 February 2025).
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-Translational Modifications and Regulation of the RAS Superfamily of GTPases as Anticancer Targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef]
- Untch, B.R.; Dos Anjos, V.; Garcia-Rendueles, M.E.R.; Knauf, J.A.; Krishnamoorthy, G.P.; Saqcena, M.; Bhanot, U.K.; Socci, N.D.; Ho, A.L.; Ghossein, R.; et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res. 2018, 78, 4642–4657. [Google Scholar] [CrossRef]
- Hong, D.S.; Sebti, S.M.; Newman, R.A.; Blaskovich, M.A.; Ye, L.; Gagel, R.F.; Moulder, S.; Wheler, J.J.; Naing, A.; Tannir, N.M.; et al. Phase I Trial of a Combination of the Multikinase Inhibitor Sorafenib and the Farnesyltransferase Inhibitor Tipifarnib in Advanced Malignancies. Clin. Cancer Res. 2009, 15, 7061–7068. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ventura, S.; Pojo, M.; Matias, A.T.; Moura, M.M.; Marques, I.J.; Leite, V.; Cavaco, B.M. The Efficacy of HRAS and CDK4/6 Inhibitors in Anaplastic Thyroid Cancer Cell Lines. J. Endocrinol. Invest. 2019, 42, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.M.; Alkhilaiwi, F.A.; Khan, N.U.; Tasleem, M. In Silico Screening and Validation of Achyranthes Aspera as a Potential Inhibitor of BRAF and NRAS in Controlling Thyroid Cancer. Anticancer Agents Med. Chem. 2023, 23, 2111–2126. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Hamidi, S.; Ouni, R.; Rico, R.; Henderson, Y.C.; Puche, M.; Alekseev, S.; Colunga-Minutti, J.G.; Zafereo, M.E.; Lai, S.Y.; et al. Emerging Therapeutic Options for Follicular-Derived Thyroid Cancer in the Era of Immunotherapy. Front. Immunol. 2024, 15, 1369780. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Turkmen, E.; Sogutlu, F.; Erdogan, M.; Biray Avci, C. Evaluation of the Anticancer Effect of Telomerase Inhibitor BIBR1532 in Anaplastic Thyroid Cancer in Terms of Apoptosis, Migration and Cell Cycle. Med. Oncol. 2023, 40, 196. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Mousa, M.H.A.; Sharaky, M.; Mourad, M.A.E.; El-Dessouki, A.M.; Hamouda, A.O.; Alnajjar, R.; Ayed, A.A.; Shaldam, M.A.; Tawfik, H.O. Lead Optimization of BIBR1591 To Improve Its Telomerase Inhibitory Activity: Design and Synthesis of Novel Four Chemical Series with In Silico, In Vitro, and In Vivo Preclinical Assessments. J. Med. Chem. 2024, 67, 492–512. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of Telomeres and Telomerase in Cancer, and Advances in Telomerase-Targeted Therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Z.; Liu, Y.; Wang, P.; Fang, X.; Sun, M.; Ma, K.; Wang, B.; Han, W. Design and Synthesis of Novel Structures with Anti-Tumor Effects: Targeting Telomere G-Quadruplex and hTERT. Bioorganic Med. Chem. Lett. 2025, 118, 130083. [Google Scholar] [CrossRef]
- Long, W.; Zeng, Y.-X.; Zheng, B.-X.; Li, Y.-B.; Wang, Y.-K.; Chan, K.-H.; She, M.-T.; Lu, Y.-J.; Cao, C.; Wong, W.-L. Targeting hTERT Promoter G-Quadruplex DNA Structures with Small-Molecule Ligand to Downregulate hTERT Expression for Triple-Negative Breast Cancer Therapy. J. Med. Chem. 2024, 67, 13363–13382. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Chu, J.; Meinguet, C.; Kiss, R.; Vandenbussche, G.; Masereel, B.; Wouters, J.; Kornienko, A.; Pelletier, J.; Mathieu, V. A Harmine-Derived Beta-Carboline Displays Anti-Cancer Effects in Vitro by Targeting Protein Synthesis. Eur. J. Pharmacol. 2017, 805, 25–35. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.-C.; Prawira, A.; de Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and Antitumor Activity of the Anti–PD-1 Antibody Pembrolizumab in Patients with Advanced, PD-L1–Positive Papillary or Follicular Thyroid Cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Algazi, A.; Capdevila, J.; Longo, F.; Miller, W., Jr.; Chun Bing, J.T.; Bonilla, C.E.; Chung, H.C.; Guren, T.K.; Lin, C.-C.; et al. Efficacy and Safety of Pembrolizumab Monotherapy in Patients with Advanced Thyroid Cancer in the Phase 2 KEYNOTE-158 Study. Cancer 2023, 129, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Seufert, J.; Aumann, K.; Ruf, J.; Klein, C.; Kiefer, S.; Rassner, M.; Boerries, M.; Zielke, A.; la Rosee, P.; et al. Combination of Lenvatinib and Pembrolizumab Is an Effective Treatment Option for Anaplastic and Poorly Differentiated Thyroid Carcinoma. Thyroid 2021, 31, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- French, J.D.; Haugen, B.R.; Worden, F.P.; Bowles, D.W.; Gianoukakis, A.G.; Konda, B.; Dadu, R.; Sherman, E.J.; McCue, S.; Foster, N.R.; et al. Combination Targeted Therapy with Pembrolizumab and Lenvatinib in Progressive, Radioiodine-Refractory Differentiated Thyroid Cancers. Clin. Cancer Res. 2024, 30, 3757–3767. [Google Scholar] [CrossRef]
- Hamidi, S.; Iyer, P.C.; Dadu, R.; Gule-Monroe, M.K.; Maniakas, A.; Zafereo, M.E.; Wang, J.R.; Busaidy, N.L.; Cabanillas, M.E. Checkpoint Inhibition in Addition to Dabrafenib/Trametinib for BRAFV600E-Mutated Anaplastic Thyroid Carcinoma. Thyroid 2024, 34, 336–346. [Google Scholar] [CrossRef]
- Naing, A.; Gainor, J.F.; Gelderblom, H.; Forde, P.M.; Butler, M.O.; Lin, C.-C.; Sharma, S.; de Olza, M.O.; Varga, A.; Taylor, M.; et al. A First-in-Human Phase 1 Dose Escalation Study of Spartalizumab (PDR001), an Anti–PD-1 Antibody, in Patients with Advanced Solid Tumors. J. Immunother. Cancer 2020, 8, e000530. [Google Scholar] [CrossRef]
- Capdevila, J.; Wirth, L.J.; Ernst, T.; Ponce Aix, S.; Lin, C.-C.; Ramlau, R.; Butler, M.O.; Delord, J.-P.; Gelderblom, H.; Ascierto, P.A.; et al. PD-1 Blockade in Anaplastic Thyroid Carcinoma. J. Clin. Oncol 2020, 38, 2620–2627. [Google Scholar] [CrossRef]
- Deng, R.; Bumbaca, D.; Pastuskovas, C.V.; Boswell, C.A.; West, D.; Cowan, K.J.; Chiu, H.; McBride, J.; Johnson, C.; Xin, Y.; et al. Preclinical Pharmacokinetics, Pharmacodynamics, Tissue Distribution, and Tumor Penetration of Anti-PD-L1 Monoclonal Antibody, an Immune Checkpoint Inhibitor. mAbs 2016, 8, 593–603. [Google Scholar] [CrossRef]
- Inman, B.A.; Longo, T.A.; Ramalingam, S.; Harrison, M.R. Atezolizumab: A PD-L1–Blocking Antibody for Bladder Cancer. Clin. Cancer Res. 2017, 23, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; Dadu, R.; Ferrarotto, R.; Gule-Monroe, M.; Liu, S.; Fellman, B.; Williams, M.D.; Zafereo, M.; Wang, J.R.; Lu, C.; et al. Anti–Programmed Death Ligand 1 Plus Targeted Therapy in Anaplastic Thyroid Carcinoma: A Nonrandomized Clinical Trial. JAMA Oncol. 2024, 10, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Ding, H.-L.; Li, X.-L.; Wang, W.-Y.; Wang, X.-L.; Gu, J. Comparison of Prognosis between Oncocytic Thyroid Carcinoma and Follicular Thyroid Carcinoma: A Population-Based Propensity Score Matching Analysis. Eur. Arch. Otorhinolaryngol. 2025, 282, 993–1003. [Google Scholar] [CrossRef]
- Gulwani, D.; Upadhyay, P.; Goel, R.; Sarangthem, V.; Singh, T.D. Nanomedicine Mediated Thyroid Cancer Diagnosis and Treatment: An Approach from Generalized to Personalized Medicine. Discov. Oncol. 2024, 15, 789. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Coperchini, F.; Croce, L.; Magri, F.; Teliti, M.; Rotondi, M. Drug Repositioning in Thyroid Cancer Treatment: The Intriguing Case of Anti-Diabetic Drugs. Front. Pharmacol. 2023, 14, 1303844. [Google Scholar] [CrossRef]

{kind=link}
| Drug | Target | Indication | Clinical Trial | Efficacy | Common Adverse Effects | |
|---|---|---|---|---|---|---|
| PFS | Response Rate | |||||
| Sorafenib | VEGFR-1, VEGFR-2, VEGFR-3, RET, BRAF, KIT, and PDGFR | RAIR DTC | DECISION Phase III | 10.8 months | 12.2% | Secondary malignancy, dyspnoea, and pleural effusion |
| Lenvatinib | VEGFR-1, VEGFR-2, VEGFR-3, RET, BRAF, KIT, PDGFR, and FGFR | RAIR DTC | SELECT Phase III | 18.3 months | 64.8% | Diarrhoea, hypertension, proteinuria, and decreased appetite |
| Vandetanib | VEGFR, PDGFR, EGFR, and RET | MTC | NCT00410761 Phase III | 30.5 months | 45% | Diarrhoea, rash, nausea, hypertension, and headache |
| Cabozantinib | MET, RET, KIT, and VEGFR | MTC | EXAM Phase III | 11.2 months | 28% | Diarrhoea, skin reaction, fatigue, and hypertension |
| Second-line RAIR DTC | Cosmic-311 Phase III | 11 months | 15% | |||
| Dabrafenib | BRAF V600E mutation | RAIR DTC with BRAF mutations | NCT01723202 Phase II | 10.7 months | 35% | Skin and subcutaneous tissue disorders, fever, and hyperglycemia |
| Dabrafenib + trametinib | BRAF V600E mutation + MEK1 and MEK2 | BRAF-mutant ATC | ROAR Phase II | 6.7 months | 56% | Pyrexia, anaemia, decreased appetite, and fatigue |
| RAIR DTC with BRAF mutations | NCT01723202 Phase II | 15.1 months | 30% | Fever, nausea, chills, and fatigue | ||
| Vemurafenib | BRAF | RAIR DTC | NCT01286753 Phase II | 18.2 months | 38.5% | Rash, fatigue, or weight loss |
| Selpercatinib | RET | RET-altered TC | LIBRETTO Phase I/II | 1-year PFS rate 64–92% | 69–79% | Hypertension and increased alanine and aspartate aminotransferase levels |
| RET-altered MTC | LIBRETTO-531 Phase III | 1-year PFS rate 86.8% | 69.4% | |||
| Pralsetinib | RET | RET-mutant MTC | ARROW Phase I/II | 1-year PFS rate 75–81% | 60–71% | Elevated aspartate aminotransferase, anaemia, and hypertension |
| RET fusion-positive TC | ARROW Phase I/II | 1-year PFS rate 81% | 89% | |||
| Pembrolizumab | PD-1 | Papillary or follicular TC | KEYNOTE-158 Phase II | 4.2 months | 6.8% | Fatigue, pruritus, and rash |
| Spartalizumab | PD-1 | ATC | NCT02404441 Phase I/II | 1-year PFS 52.1% of PD-L1 + patients | 19% | Diarrhea, pruritus, fatigue, and pyrexia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil-Bernabé, S.; García-DeLaFuente, L.; García-Rostán, G. The Revolution of Targeted Therapies in Thyroid Cancer Treatment: Present and Future Promising Anti-Cancer Drugs. Int. J. Mol. Sci. 2025, 26, 3663. https://doi.org/10.3390/ijms26083663
Gil-Bernabé S, García-DeLaFuente L, García-Rostán G. The Revolution of Targeted Therapies in Thyroid Cancer Treatment: Present and Future Promising Anti-Cancer Drugs. International Journal of Molecular Sciences. 2025; 26(8):3663. https://doi.org/10.3390/ijms26083663
Chicago/Turabian StyleGil-Bernabé, Sara, Lucía García-DeLaFuente, and Ginesa García-Rostán. 2025. "The Revolution of Targeted Therapies in Thyroid Cancer Treatment: Present and Future Promising Anti-Cancer Drugs" International Journal of Molecular Sciences 26, no. 8: 3663. https://doi.org/10.3390/ijms26083663
APA StyleGil-Bernabé, S., García-DeLaFuente, L., & García-Rostán, G. (2025). The Revolution of Targeted Therapies in Thyroid Cancer Treatment: Present and Future Promising Anti-Cancer Drugs. International Journal of Molecular Sciences, 26(8), 3663. https://doi.org/10.3390/ijms26083663