
Multicomponent DNA Nanomachines for Amplification-Free Viral RNA Detection
 ,
,
Abstract
1. Introduction
2. Results
2.1. Selection of RNA Fragments
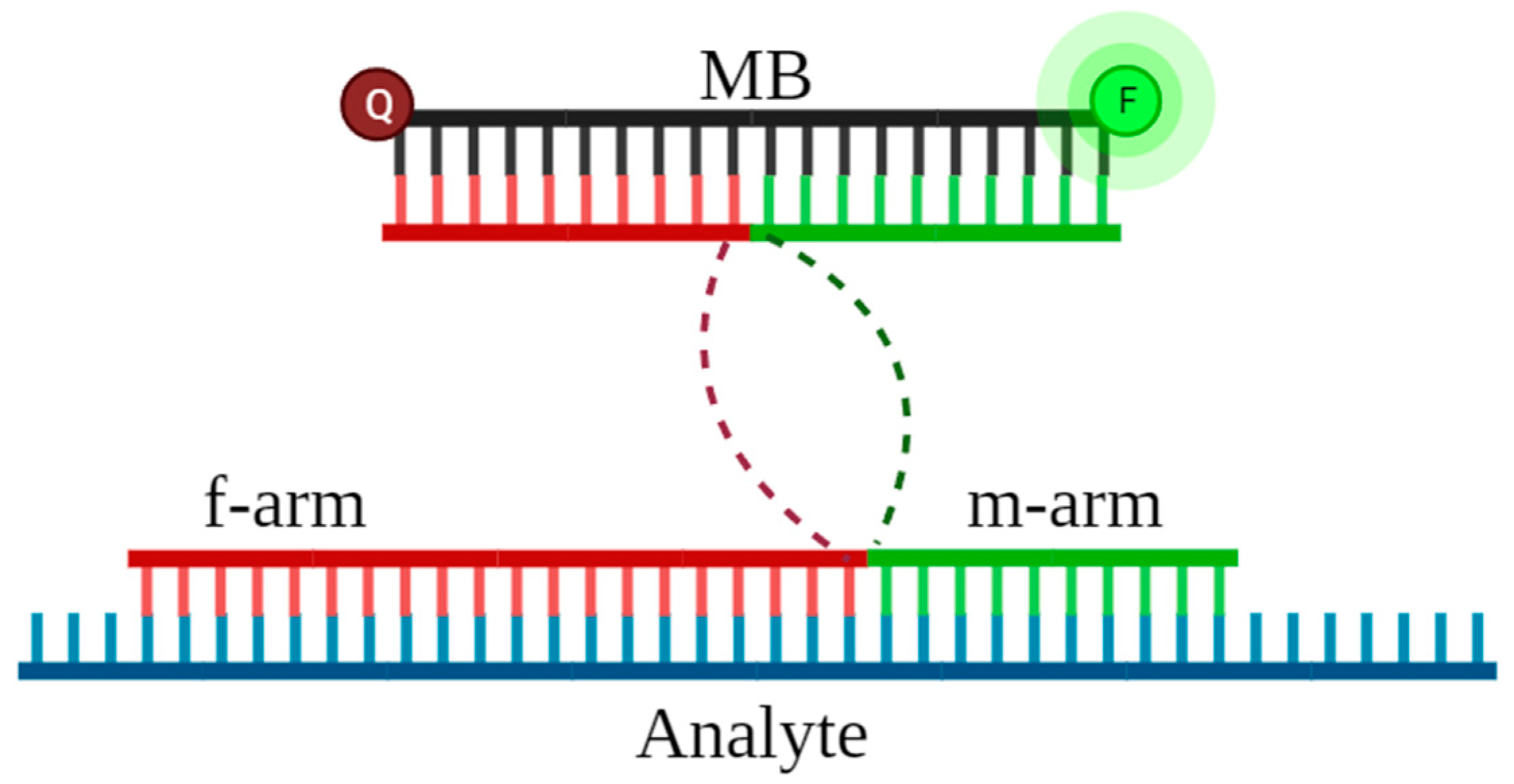
2.2. Design of DNA Nanosensors
2.3. Detection by MB-DNSs
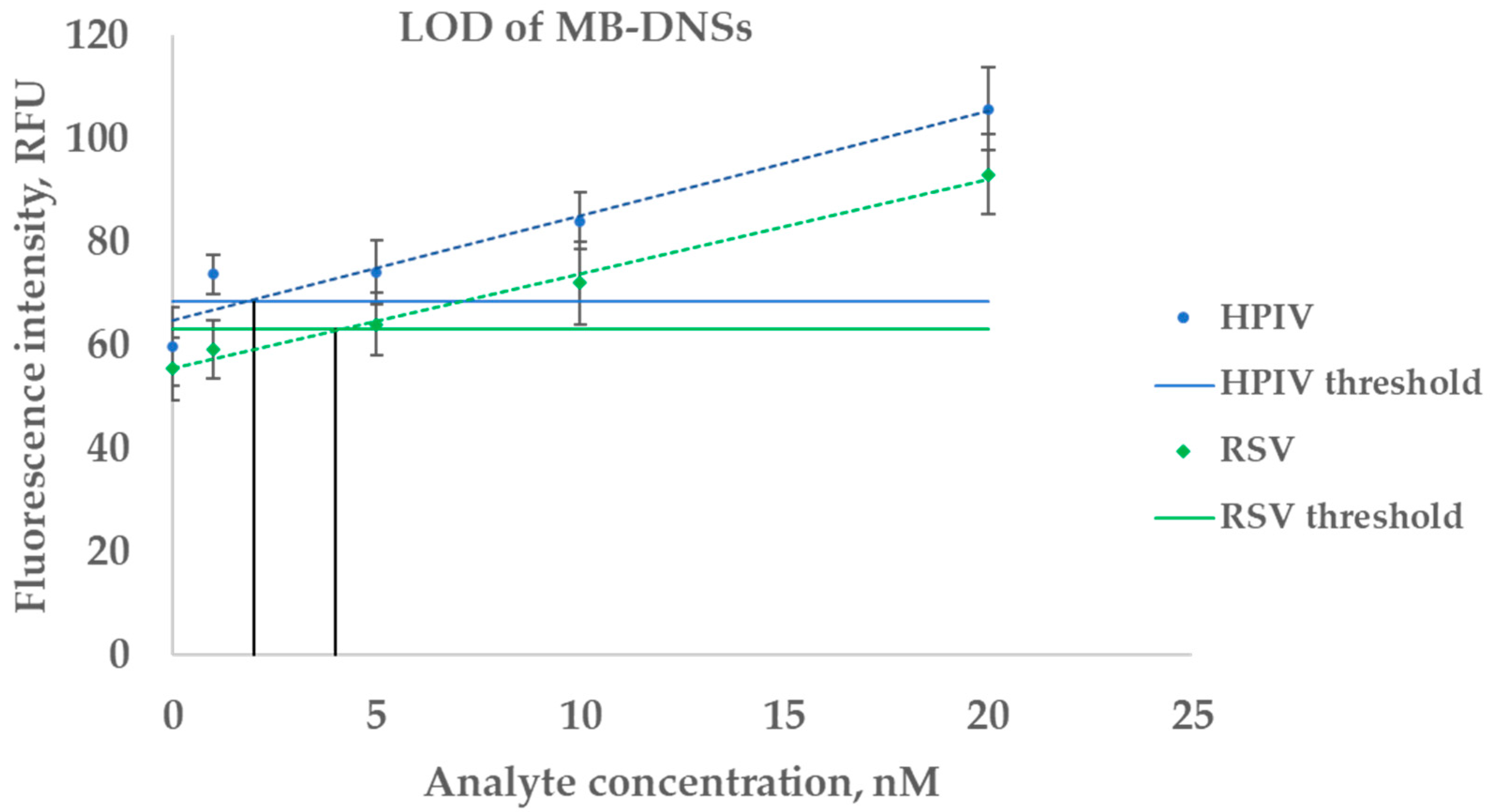
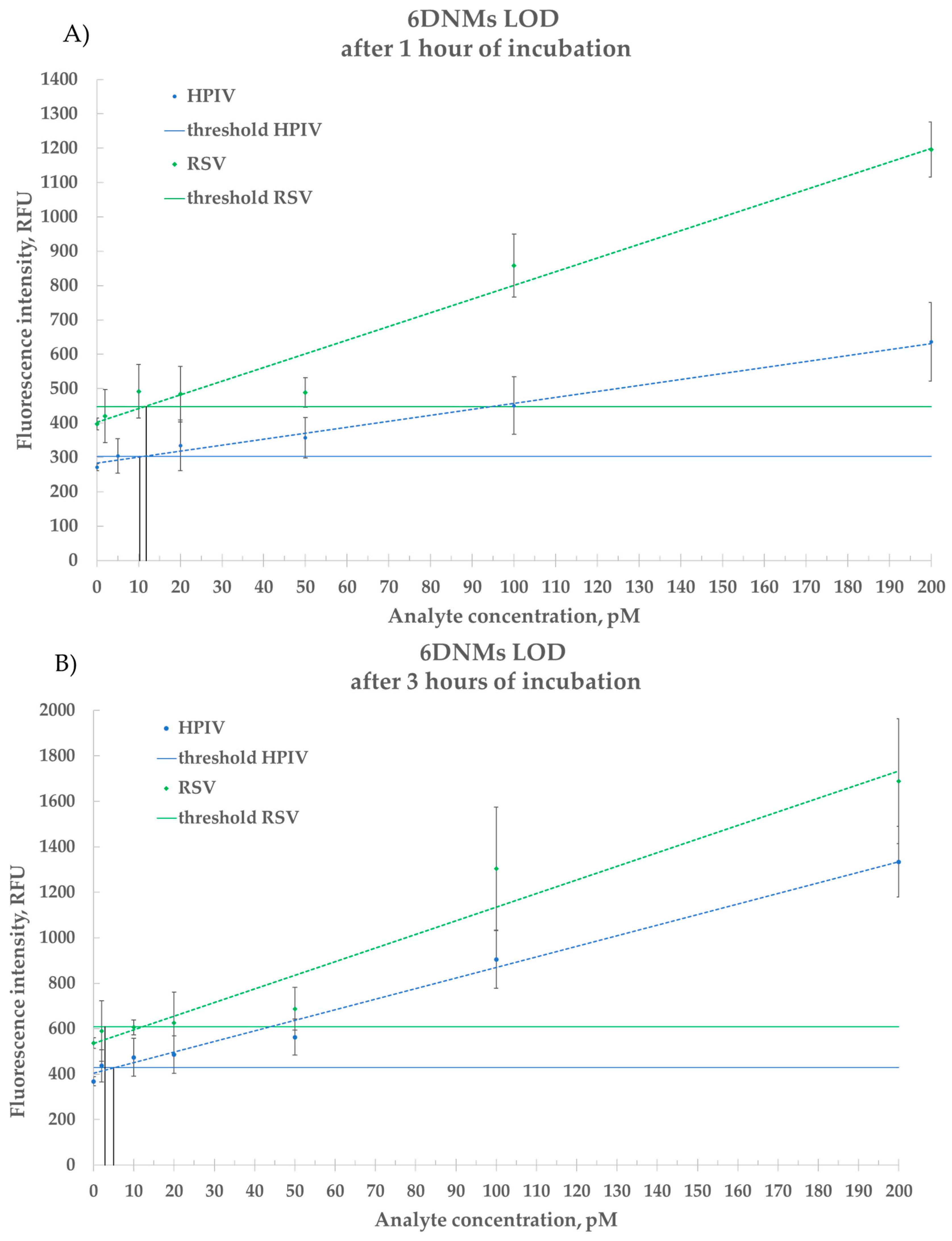
2.3.1. Estimation of LOD on a Synthetic Analyte
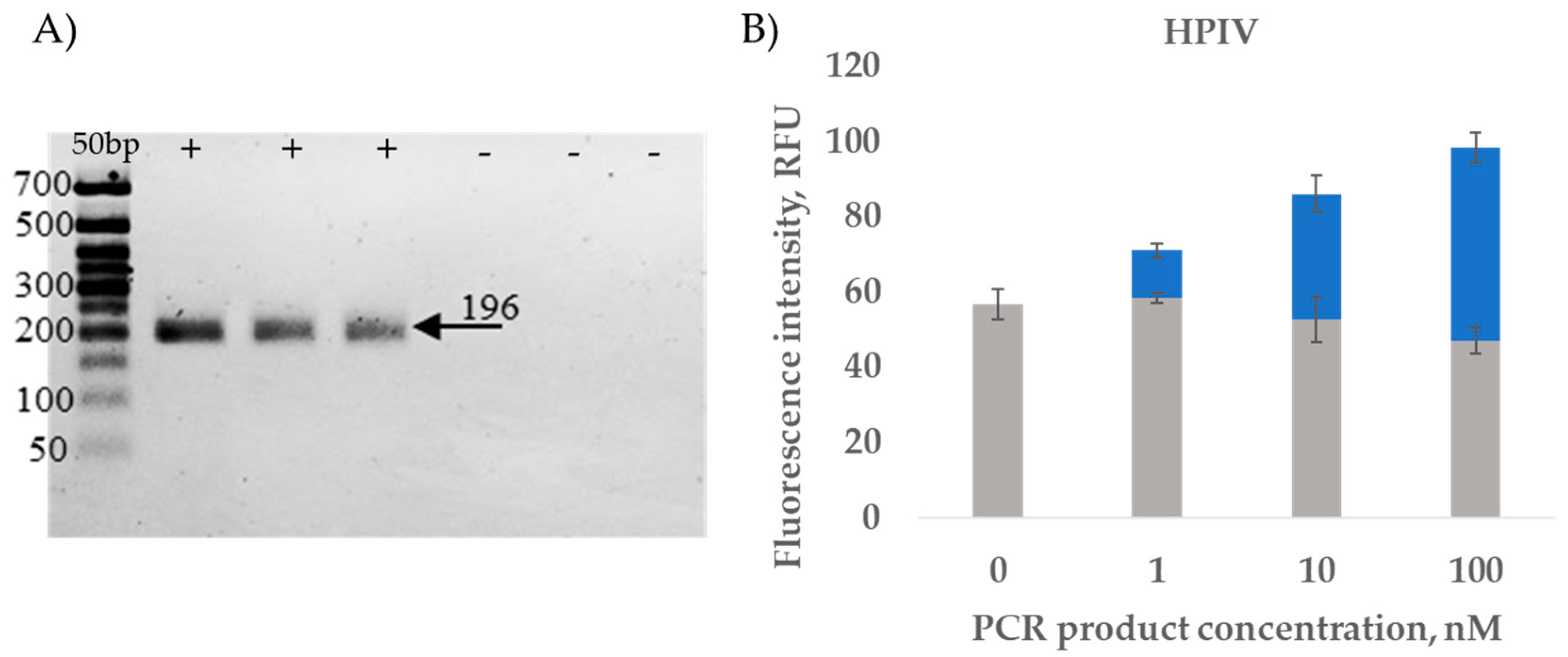
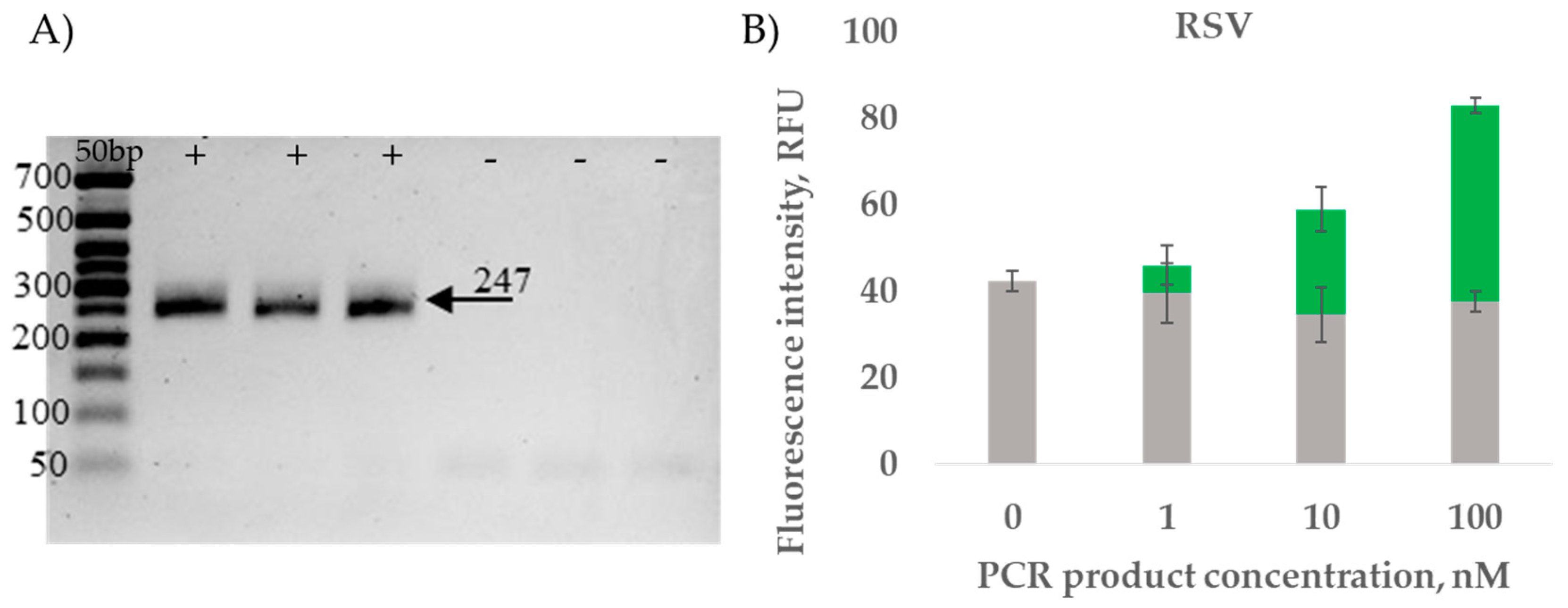
2.3.2. RT-PCR Amplicon Detection Results
2.3.3. Clinical Sample Detection Results
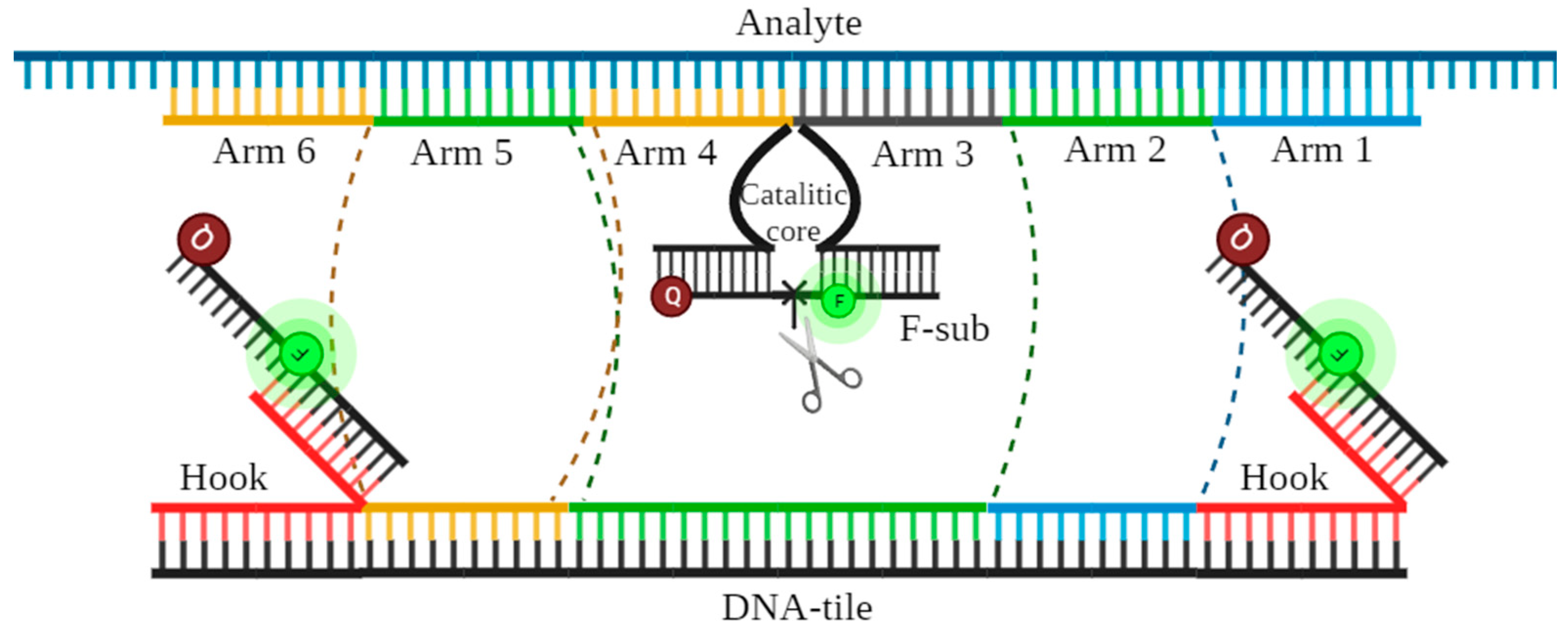
2.4. Detection by DNMs
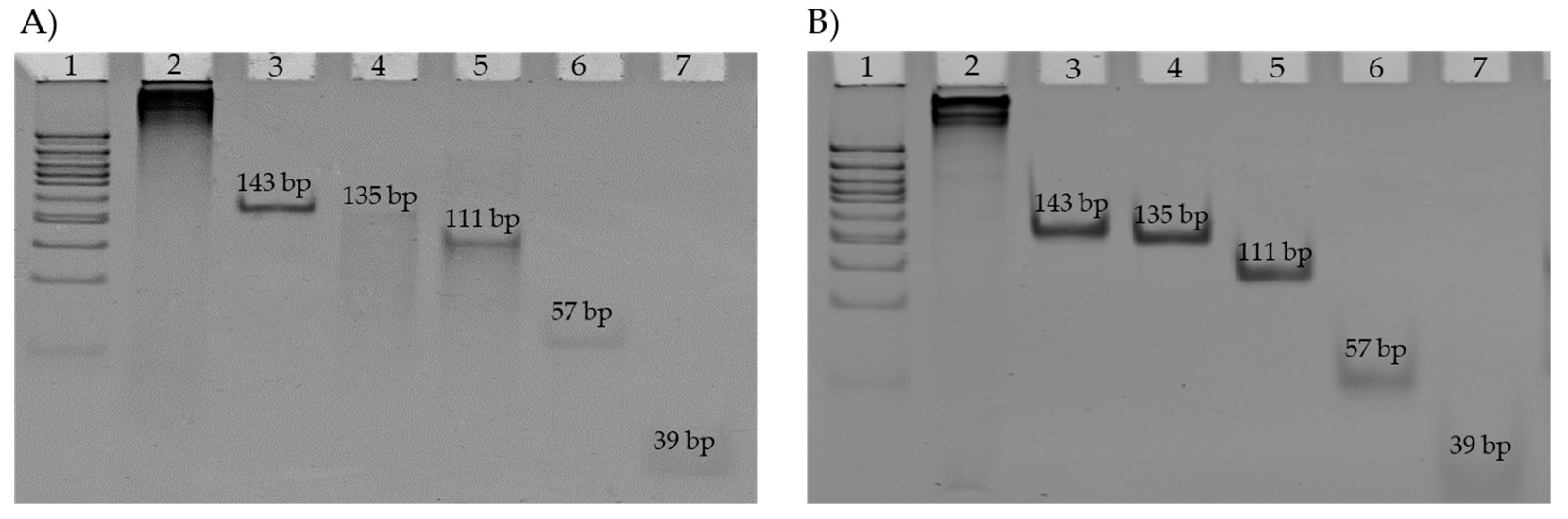
2.4.1. DNM Assembly Visualization
2.4.2. Estimation of LOD for Synthetic DNA
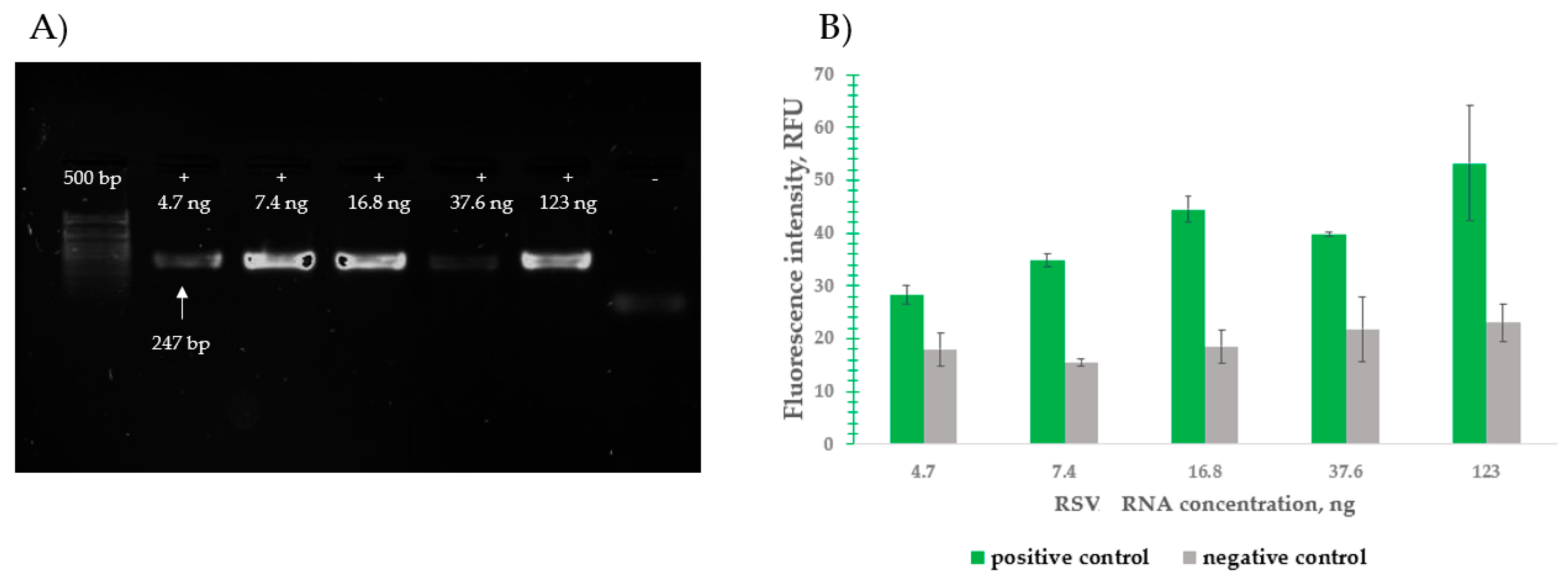
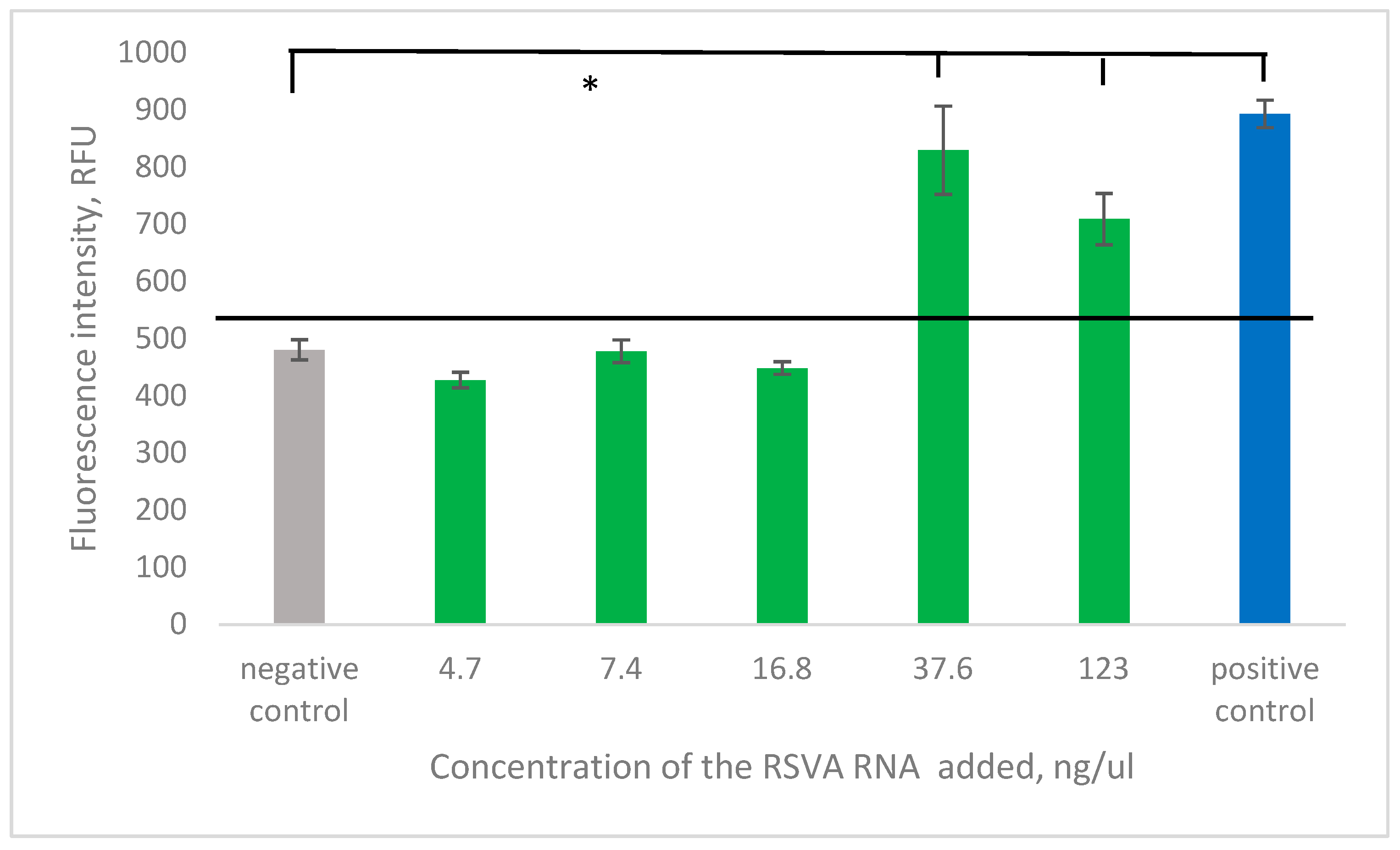
2.4.3. Detection of Total Genomic RNA
2.4.4. Detection of RNA Extracted from Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Detection Assay
4.1.2. RNA Extraction
4.1.3. RT-PCR
4.1.4. Electrophoresis
4.1.5. Purification of PCR Products
4.2. Methods
4.2.1. Objects of the Research
4.2.2. Design of MB-DNSs and DNMs
4.2.3. DNA Nanomachine Assembly
4.2.4. Polyacrylamide Gel Electrophoresis
4.2.5. RT-PCR
4.2.6. Agarose Gel Electrophoresis
4.2.7. Purification of Double-Stranded DNA Fragments
4.2.8. Fluorescent Detection Method: Detection by MB-DNSs
4.2.9. Fluorescent Detection Method: Detection by Multicomponent DNMs
4.2.10. Limit of Detection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patchett, D.; Yang, J.; Northern, J.; Salinas, M.; Bauer, B.A. Viral Respiratory Infections: An Ounce of Prevention Is Worth a Pound of Cure. Mayo Clin. Proc. Innov. Qual. Outcomes 2021, 5, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, H.R.; Yu, H. Viral Respiratory Infections. In Hunter’s Tropical Medicine and Emerging Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2020; pp. 284–288. [Google Scholar] [CrossRef]
- Karron, R.A. Respiratory Syncytial Virus and Parainfluenza Virus Vaccines. In Vaccines, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 1146–1153. [Google Scholar] [CrossRef]
- Wetzke, M.; Schwerk, N. Respiratory Syncytial Virus Infection. Pneumologe 2022, 16, 232–241. [Google Scholar] [CrossRef]
- Dronina, J.; Samukaite-Bubniene, U.; Ramanavicius, A. Advances and Insights in the Diagnosis of Viral Infections. J. Nanobiotechnol. 2021, 19, 348. [Google Scholar] [CrossRef]
- Reta, D.H. Molecular and Immunological Diagnostic Techniques of Medical Viruses. Int. J. Microbiol. 2020, 2020, 8832728. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Zhang, L.; Lu, Y.; Wang, X.; Shen, M.; Li, N.; Feng, L.; Jing, J.; Cao, B.; et al. Sensitive and rapid diagnosis of respiratory virus coinfection using a microfluidic chip-powered CRISPR/Cas12a system. Small 2022, 18, 2200854. [Google Scholar] [CrossRef]
- Ngoc, L.T.N.; Lee, Y.-C. Current Trends in RNA Virus Detection via Nucleic Acid Isothermal Amplification-Based Platforms. Biosensors 2024, 14, 97. [Google Scholar] [CrossRef]
- Kolpashchikov, D.M. Evolution of Hybridization Probes to DNA Machines and Robots. Acc. Chem. Res. 2019, 52, 1949–1956. [Google Scholar] [CrossRef]
- Gerasimova, Y.V.; Kolpashchikov, D.M. Detection of Bacterial 16S rRNA Using a Molecular Beacon-Based X Sensor. Biosens. Bioelectron. 2013, 41, 386–390. [Google Scholar] [CrossRef]
- Gerasimova, Y.V.; Cornett, E.; Kolpashchikov, D.M. RNA-Cleaving Deoxyribozyme Sensor for Nucleic Acid Analysis: The Limit of Detection. ChemBioChem 2010, 11, 811–817. [Google Scholar] [CrossRef]
- Silverman, S.K. Catalytic DNA: Scope, Applications, and Biochemistry of Deoxyribozymes. Trends Biochem. Sci. 2016, 41, 595–609. [Google Scholar] [CrossRef]
- Gorbenko, D.A.; Shkodenko, L.A.; Rubel, M.S.; Slita, A.V.; Nikitina, E.V.; Martens, E.A.; Kolpashchikov, D.M. DNA Nanomachine for Visual Detection of Structured RNA and Double-Stranded DNA. Chem. Commun. 2022, 58, 5395–5398. [Google Scholar] [CrossRef] [PubMed]
- Maltzeva, Y.I.; Gorbenko, D.A.; Nikitina, E.V.; Rubel, M.S.; Kolpashchikov, D.M. Visual Detection of Stem-Loop Primer Amplification (SPA) Products without Denaturation Using Peroxidase-like DNA Machines (PxDM). Int. J. Mol. Sci. 2023, 24, 7812. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.J.; Bengtson, H.N.; Gerasimova, Y.V.; Rohde, K.H.; Kolpashchikov, D.M. DNA Antenna Tile-Associated Deoxyribozyme Sensor with Improved Sensitivity. ChemBioChem 2016, 17, 2038–2041. [Google Scholar] [CrossRef]
- Ateiah, M.; Gandalipov, E.R.; Rubel, A.A.; Rubel, M.S.; Kolpashchikov, D.M. DNA Nanomachine (DNM) Biplex Assay for Differentiating Bacillus cereus Species. Int. J. Mol. Sci. 2023, 24, 4473. [Google Scholar] [CrossRef]
- Hussein, Z.; Nour, M.A.; Kozlova, A.V.; Kolpashchikov, D.M.; Komissarov, A.B.; El-Deeb, A.A. DNAzyme Nanomachine with Fluorogenic Substrate Delivery Function: Advancing Sensitivity in Nucleic Acid Detection. Anal. Chem. 2023, 95, 18667–18672. [Google Scholar] [CrossRef]
- Lyalina, T.A.; Goncharova, E.A.; Prokofeva, N.Y.; Voroshilina, E.S.; Kolpashchikov, D.M. A DNA Minimachine for Selective and Sensitive Detection of DNA. Analyst 2019, 144, 416–420. [Google Scholar] [CrossRef]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Kolpashchikov, D.M. A Binary Deoxyribozyme for Nucleic Acid Analysis. ChemBioChem 2007, 8, 2039–2042. [Google Scholar] [CrossRef]
- Kolpashchikov, D.M. Binary Probes for Nucleic Acid Analysis. Chem. Rev. 2010, 110, 4709–4723. [Google Scholar] [CrossRef]
- Gerasimova, Y.V.; Peck, S.H.; Kolpashchikov, D.M. Enzyme-Assisted Binary Probe for Sensitive Detection of RNA and DNA. Chem. Commun. 2010, 46, 8761–8763. [Google Scholar] [CrossRef]
- Rubel, M.S.; Shkodenko, L.A.; Gorbenko, D.A.; Solyanikova, V.V.; Maltzeva, Y.I.; Rubel, A.A.; Koshel, E.I.; Kolpashchikov, D.M. Detection of Multiplex NASBA RNA Products Using Colorimetric Split G Quadruplex Probes. In RNA Nanostructures; Methods in Molecular Biology; Humana: New York, NY, USA, 2023; Volume 2709, pp. 325–336. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, P.; Ahmed, R.; Wang, J.; Akin, D.; Soto, F.; Liu, B.F.; Li, P.; Demirci, U. Advanced point-of-care testing technologies for human acute respiratory virus detection. Adv. Mater. 2022, 34, 2103646. [Google Scholar] [CrossRef] [PubMed]
- MacDougall, D.; Crummett, W.B. Guidelines for data acquisition and data quality evaluation in environmental chemistry. Anal. Chem. 1980, 52, 2242–2249. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | MB-Based DNA Nanosensors | Deoxyribozyme DNA Nanomachines | RT-qPCR |
|---|---|---|---|
| Detection speed | 15–30 min | 30–180 min | 90–120 min |
| Ease of development | Computer tools are used for designing Based on the principle of complementarity | Computer tools are used for designing Based on the principle of complementarity | Computer tools are used to select primers based on the principle of complementarity The amplification and visualization protocols are additionally selected, often empirically |
| Sensitivity | Approximately 1–3 nM | Approximately 5–10 pM | Approximately 3 attoM |
| Specificity | Specific | Specific | Specific |
| Complexity | Does not require assembly step, easy to design | The assembly is necessary, the reaction is simple, require addition equipment to incubate samples, easy to design | Method requires specific expensive equipment, reagents and enzymes, laboratory condition, highly qualified personnel |
| Cost of the analysis | USD 0.35 per reaction | USD 0.6 per reaction | USD 1.7 per reaction |
| Competitiveness with existing methods | Cannot achieve the same sensitivity as PCR; works in combination as an alternative to visualization and recognition of specific PCR product | Cannot achieve the same sensitivity as PCR but able to achieve amplification-free detection of fM concentrations of ssRNA | Remains the gold standard for accurate quantification of nucleic acids. The success of detection strongly depends on the stages of sample preparation and amplification |
| Feasibility of viral RNA detection | Effectively detects short ssDNA, PCR products | Detects short DNA analyte as well as long viral RNA samples | Detects DNA/RNA after amplification |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solyanikova, V.V.; Gorbenko, D.A.; Zryacheva, V.V.; Shtro, A.A.; Rubel, M.S. Multicomponent DNA Nanomachines for Amplification-Free Viral RNA Detection. Int. J. Mol. Sci. 2025, 26, 3652. https://doi.org/10.3390/ijms26083652
Solyanikova VV, Gorbenko DA, Zryacheva VV, Shtro AA, Rubel MS. Multicomponent DNA Nanomachines for Amplification-Free Viral RNA Detection. International Journal of Molecular Sciences. 2025; 26(8):3652. https://doi.org/10.3390/ijms26083652
Chicago/Turabian StyleSolyanikova, Valeria V., Daria A. Gorbenko, Valeriya V. Zryacheva, Anna A. Shtro, and Maria S. Rubel. 2025. "Multicomponent DNA Nanomachines for Amplification-Free Viral RNA Detection" International Journal of Molecular Sciences 26, no. 8: 3652. https://doi.org/10.3390/ijms26083652
APA StyleSolyanikova, V. V., Gorbenko, D. A., Zryacheva, V. V., Shtro, A. A., & Rubel, M. S. (2025). Multicomponent DNA Nanomachines for Amplification-Free Viral RNA Detection. International Journal of Molecular Sciences, 26(8), 3652. https://doi.org/10.3390/ijms26083652