

Acquired Hemophilia Associated with Rheumatoid Arthritis: A Case Report and Review of the Literature

 , , and
, , and
Abstract
:1. Introduction
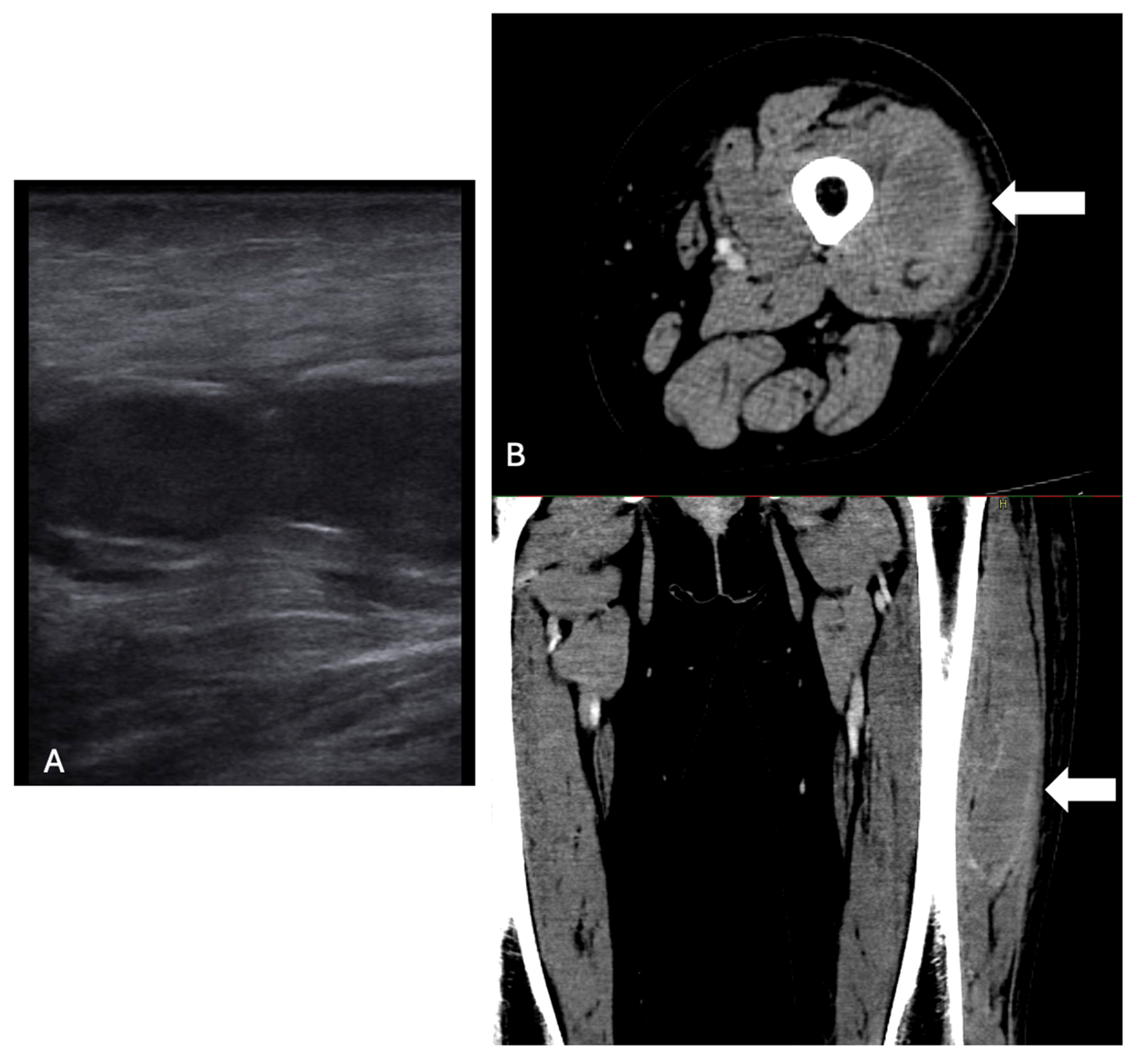
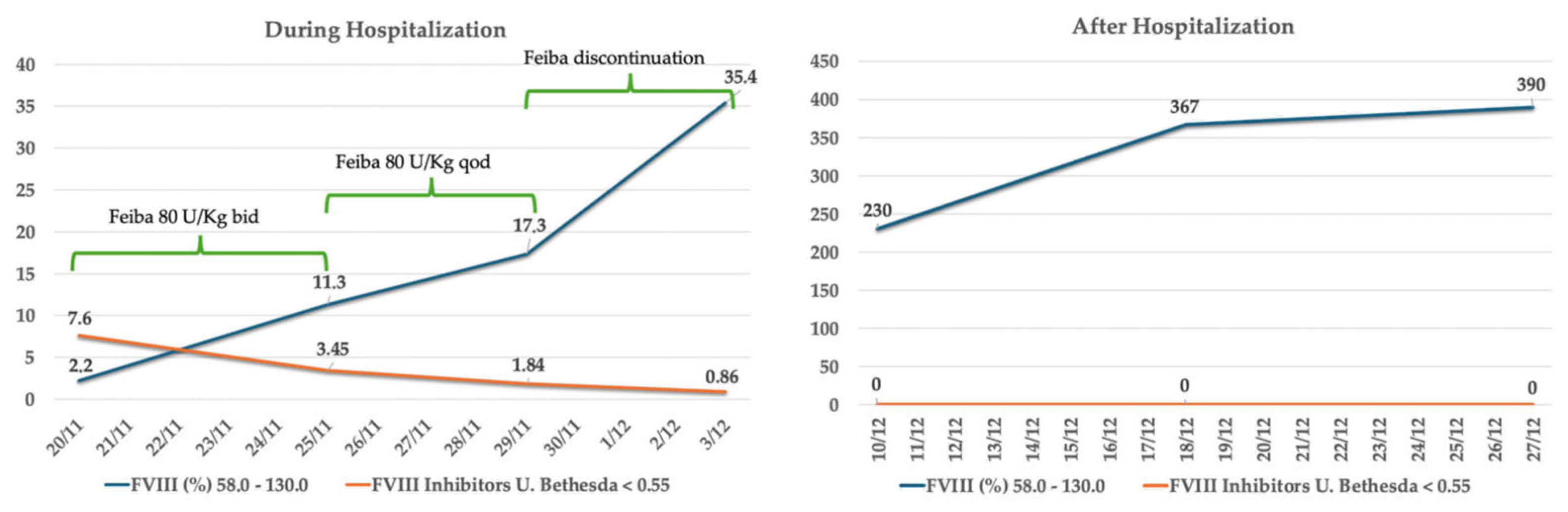
2. Case Description
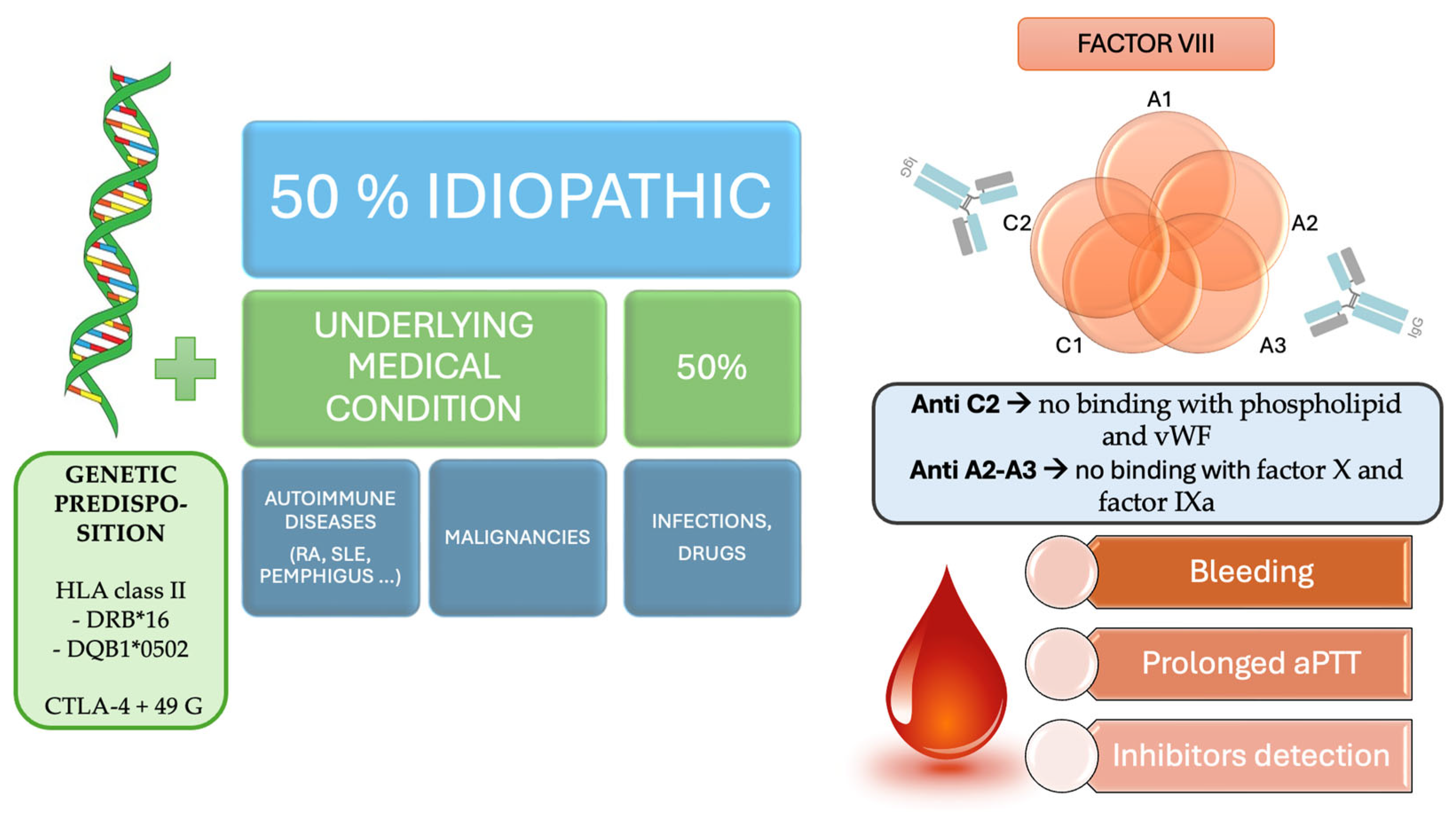
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huth-Kühne, A.; Baudo, F.; Collins, P.; Ingerslev, J.; Kessler, C.M.; Lévesque, H.; Castellano, M.E.; Shima, M.; St-Louis, J. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica 2009, 94, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Jayakar, J.P.; O’Neill, N.; Yan, M.; Nisenbaum, R.; Garvey, M.B.; Teitel, J.; Sholzberg, M. Retrospective review of Acquired Haemophilia A from the largest Canadian Haemophilia treatment centre. Haemophilia 2018, 24, e383–e387. [Google Scholar] [CrossRef]
- Collins, P.W.; Hirsch, S.; Baglin, T.P.; Dolan, G.; Hanley, J.; Makris, M.; Keeling, D.M.; Liesner, R.; Brown, S.A.; Hay, C.R.; et al. Acquired hemophilia A in the United Kingdom: A 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007, 109, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Knoebl, P.; Marco, P.; Baudo, F.; Collins, P.; Huth-Kühne, A.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Lévesque, H.; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J. Thromb. Haemost. 2012, 10, 622–631. [Google Scholar] [CrossRef]
- Tiede, A.; Klamroth, R.; Scharf, R.E.; Trappe, R.U.; Holstein, K.; Huth-Kühne, A.; Gottstein, S.; Geisen, U.; Schenk, J.; Scholz, U.; et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): Results from the GTH-AH 01/2010 study. Blood 2015, 125, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Asahina, A.; Nakatani, E.; Imaichi, Y.; Ohata, E.; Oshima, M.; Miyakoshi, A.; Miyake, H.; Ichikawa, Y.; Dote, H.; Ubukata, N.; et al. Risk factors, treatment and survival rates of late-onset acquired haemophilia A: A cohort study from the Shizuoka Kokuho Database. Haemophilia 2023, 29, 799–808. [Google Scholar] [CrossRef]
- Napolitano, M.; Siragusa, S.; Mancuso, S.; Kessler, C.M. Acquired haemophilia in cancer: A systematic and critical literature review. Haemophilia 2018, 24, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Tiede, A.; Collins, P.; Knoebl, P.; Teitel, J.; Kessler, C.; Shima, M.; Di Minno, G.; d’Oiron, R.; Salaj, P.; Jiménez-Yuste, V.; et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica 2020, 105, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Lechner, K. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb. Haemost. 1981, 45, 200–203. [Google Scholar] [CrossRef]
- Michiels, J.J. Acquired hemophilia A in women postpartum: Clinical manifestations, diagnosis, and treatment. Clin. Appl. Thromb. Hemost. 2000, 6, 82–86. [Google Scholar] [CrossRef]
- Franchini, M.; Zaffanello, M.; Lippi, G. Acquired hemophilia in pediatrics: A systematic review. Pediatr. Blood Cancer 2010, 55, 606–611. [Google Scholar] [CrossRef]
- Huang, S.Y.; Tsay, W.; Lin, S.Y.; Hsu, S.C.; Hung, M.H.; Shen, M.C. A study of 65 patients with acquired hemophilia A in Taiwan. J. Formos. Med. Assoc. 2015, 114, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Gheisari, R.; Bomke, B.; Hoffmann, T.; Scharf, R.E. Clinical features and outcome of acquired haemophilia A. Interim analysis of the Düsseldorf study. Hamostaseologie 2010, 30, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kruse-Jarres, R.; Kempton, C.L.; Baudo, F.; Collins, P.W.; Knoebl, P.; Leissinger, C.A.; Tiede, A.; Kessler, C.M. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am. J. Hematol. 2017, 92, 695–705. [Google Scholar] [CrossRef]
- Lavigne-Lissalde, G.; Schved, J.F.; Granier, C.; Villard, S. Anti-factor VIII antibodies: A 2005 update. Thromb. Haemost. 2005, 94, 760–769. [Google Scholar] [CrossRef]
- Scandella, D.; Mattingly, M.; de Graaf, S.; Fulcher, C.A. Localization of epitopes for human factor VIII inhibitor antibodies by immunoblotting and antibody neutralization. Blood 1989, 74, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.D.; Carrizosa, D. Acquired factor VIII inhibitors: Pathophysiology and treatment. Hematol. Am. Soc. Hematol. Educ. Program. 2006, 2006, 432–437. [Google Scholar] [CrossRef]
- Whelan, S.F.; Hofbauer, C.J.; Horling, F.M.; Allacher, P.; Wolfsegger, M.J.; Oldenburg, J.; Male, C.; Windyga, J.; Tiede, A.; Schwarz, H.P.; et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood 2013, 12, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- van Helden, P.M.; van den Berg, H.M.; Gouw, S.C.; Kaijen, P.H.; Zuurveld, M.G.; Mauser-Bunschoten, E.P.; Aalberse, R.C.; Vidarsson, G.; Voorberg, J. IgG subclasses of anti-FVIII antibodies during immune tolerance induction in patients with hemophilia A. Br. J. Haematol. 2008, 142, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Lippi, G. Acquired factor VIII inhibitors. Blood 2008, 112, 250–255. [Google Scholar] [CrossRef]
- Lehoczki, A.; Fekete, M.; Mikala, G.; Bodó, I. Acquired hemophilia A as a disease of the elderly: A comprehensive review of epidemiology, pathogenesis, and novel therapy. Geroscience 2024, 47, 503–514. [Google Scholar] [CrossRef]
- Collins, P.; Baudo, F.; Huth-Kühne, A.; Ingerslev, J.; Kessler, C.M.; Castellano, M.E.; Shima, M.; St-Louis, J.; Lévesque, H. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res. Notes 2010, 3, 161. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Mannucci, P.M. Acquired haemophilia A: A 2013 update. Thromb. Haemost. 2013, 110, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Zeitler, H.; Pavlova, A. Genetic markers in acquired haemophilia. Haemophilia 2010, 16, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wu, B.; Mo, L.; Chen, H.; Zhao, Y.; Tan, T.; Chen, L.; Li, Y.; Yao, P.; Tang, Y. Associations between biological ageing and the risk of, genetic susceptibility to, and life expectancy associated with rheumatoid arthritis: A secondary analysis of two observational studies. Lancet Healthy Longev. 2024, 5, e45–e55. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S. From aging to long COVID: Exploring the convergence of immunosenescence, inflammaging, and autoimmunity. Front. Immunol. 2023, 14, 1298004. [Google Scholar] [CrossRef] [PubMed]
- Larsen, O.F.A. Nurturing by nutrition: On the future of gut microbiota management strategies for autoimmune disease. Front. Nutr. 2023, 9, 1107016. [Google Scholar] [CrossRef]
- Banse, C.; Benhamou, Y.; Lequerré, T.; Le Cam-Duchez, V.; Lévesque, H.; Vittecoq, O. Acquired hemophilia possibly induced by etanercept in a patient with rheumatoid arthritis. Jt. Bone Spine 2015, 82, 200–202. [Google Scholar] [CrossRef]
- Brinster, A.; Bertrand, M.A.; Guillot, X.; Sondag, M.; Wendling, D. Acquired hemophilia, rheumatoid arthritis, and TNFalpha antagonists: Comment on the article “Acquired hemophilia possibly induced by etanercept in a patient with rheumatoid arthritis” by Banse et al., Joint Bone Spine 2015;82:200-2. Jt. Bone Spine 2015, 82, 384–385. [Google Scholar] [CrossRef]
- Cegledi, A.; Batai, A.; Dolgos, J.; Fekete, M.; Gopcsa, L.; Kiraly, V.; Lakatos, G.; Nagy, G.; Szemlaky, Z.; Varkonyi, A.; et al. Case Report: Effective management of adalimumab-induced acquired hemophilia A with the CyDRI protocol. Pathol. Oncol. Res. 2024, 30, 1611720. [Google Scholar] [CrossRef] [PubMed]
- Doshi, B.S.; Rana, J.; Castaman, G.; Shaheen, M.A.; Kaczmarek, R.; Butterfield, J.S.; Meeks, S.L.; Leissinger, C.; Biswas, M.; Arruda, V.R. B cell-activating factor modulates the factor VIII immune response in hemophilia A. J. Clin. Investig. 2021, 131, 142906. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. B Cells in Rheumatoid Arthritis: Pathogenic Mechanisms and Treatment Prospects. Front. Immunol. 2021, 12, 750753. [Google Scholar]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Melchiorre, D.; Morfini, M.; Linari, S.; Zignego, A.L.; Innocenti, M.; Matucci Cerinic, M. Anti-TNF-alpha therapy prevents the recurrence of joint bleeding in haemophilia and arthritis. Rheumatology 2014, 53, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Caliogna, L.; Berni, M.; Torriani, C.; Mancuso, M.E.; Di Minno, M.N.D.; Brancato, A.M.; Jannelli, E.; Mosconi, M.; Pasta, G. Pathogenesis of osteoarthritis, rheumatoid arthritis, and hemophilic arthropathy: The role of angiogenesis. Haemophilia 2024, 30, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Sanges, S.; Jeanpierre, E.; Lopez, B.; Russick, J.; Delignat, S.; Carpentier, B.; Dubois, R.; Dubucquoi, S.; Guerrier, T.; Hachulla, É.; et al. Acquired Hemophilia A in IgG4-Related Disease: Case Report, Immunopathogenic Study, and Review of the Literature. Front. Immunol. 2020, 11, 558811. [Google Scholar] [CrossRef]
- Mai Thanh, C.; Nguyen Thi, K.; Nguyen Canh, H.; Nguyen Thi Dieu, T. Pediatric IgG4-related dacryoadenitis and sialadenitis (Mikulicz’s disease) with acquired hemophilia A: A case report and review of literature. Int. J. Immunopathol. Pharmacol. 2024, 38, 3946320241301734. [Google Scholar] [CrossRef]
- Aljasser, M.I.; Sladden, C.; Crawford, R.I.; Au, S. Bullous pemphigoid associated with acquired hemophilia a: A rare association of autoimmune disease. J. Cutan. Med. Surg. 2014, 18, 123–126. [Google Scholar] [CrossRef]
- Pathirana, U.G.; Gunawardena, N.; Abeysinghe, H.; Copley, H.C.; Somarathne, M.G. Acquired haemophilia A associated with autoimmune thyroiditis: A case report. J. Med. Case Rep. 2014, 29, 469. [Google Scholar] [CrossRef]
- Marques Dias, J.I.; Ferreira, M.A.; Grilo, A.; Reis, A.I. Acquired haemophilia A associated to autoimmune thyroiditis and pangastritis. BMJ Case Rep. 2022, 15, 248701. [Google Scholar] [CrossRef]
- Nishiura, N.; Ujimoto, D.; Fujita, J.; Maeda, T.; Nakagawa, Y.; Kashiwagi, H.; Oritani, K.; Tomiyama, Y.; Kanakura, Y. Autoimmune bullous disease and Hashimoto’s disease complicated by acquired hemophilia A. Rinsho Ketsueki 2017, 58, 233–238. [Google Scholar]
- Amisha, F.; Saluja, P.; Malik, P.; Van Rhee, F. Acquired hemophilia A (AHA) due to anti-SARS-CoV-2 vaccination: A systematic review. EJHaem 2023, 4, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, H.; Tane, M.; Kosako, H.; Ibe, M.; Takeyama, M.; Murata, S.; Mushino, T.; Sonoki, T. Acute-type acquired hemophilia A after COVID-19 mRNA vaccine administration: A new disease entity? J. Autoimmun. 2022, 133, 102915. [Google Scholar] [CrossRef] [PubMed]
- Moulis, G.; Pugnet, G.; Bagheri, H.; Courtellemont, C.; Huart, A.; Chauveau, D.; Pourrat, J.; Montastruc, J.L. Acquired factor VIII haemophilia following influenza vaccination. Eur. J. Clin. Pharmacol. 2010, 66, 1069–1070. [Google Scholar] [CrossRef] [PubMed]
- Tengborn, L.; Baudo, F.; Huth-Kühne, A.; Knoebl, P.; Lévesque, H.; Marco, P.; Pellegrini, F.; Nemes, L.; Collins, P.; EACH2 registry contributors. Pregnancy-associated acquired haemophilia A: Results from the European Acquired Haemophilia (EACH2) registry. BJOG 2012, 119, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Borg, J.Y.; Guillet, B.; Le Cam-Duchez, V.; Goudemand, J.; Lévesque, H.; SACHA Study Group. Outcome of acquired haemophilia in France: The prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registry. Haemophilia 2013, 19, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Werwitzke, S.; Geisen, U.; Nowak-Göttl, U.; Eichler, H.; Stephan, B.; Scholz, U.; Holstein, K.; Klamroth, R.; Knöbl, P.; Huth-Kühne, A.; et al. Diagnostic and prognostic value of factor VIII binding antibodies in acquired hemophilia A: Data from the GTH-AH 01/2010 study. J. Thromb. Haemost. 2016, 14, 940–947. [Google Scholar] [CrossRef]
- Ma, A.D.; Kessler, C.M.; Al-Mondhiry, H.A.; Gut, R.Z.; Cooper, D.L. Use of recombinant activated factor VII for acute bleeding episodes in acquired hemophilia: Final analysis from the Hemostasis and Thrombosis Research Society Registry acquired hemophilia study. Blood Coagul. Fibrinolysis 2016, 27, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Nemes, L.; Pitlik, E. New protocol for immune tolerance induction in acquired hemophilia. Haematologica 2000, 85, 64–68. [Google Scholar]
- Amano, K.; Seita, I.; Higasa, S.; Sawada, A.; Kuwahara, M.; Shima, M. Treatment of acute bleeding in acquired haemophilia A with recombinant activated factor VII: Analysis of 10-year Japanese postmarketing surveillance data. Haemophilia 2017, 23, 50–58. [Google Scholar] [CrossRef]
- Dimichele, D.; Négrier, C. A retrospective postlicensure survey of FEIBA efficacy and safety. Haemophilia 2006, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Leissinger, C.; Gringeri, A.; Antmen, B.; Berntorp, E.; Biasoli, C.; Carpenter, S.; Cortesi, P.; Jo, H.; Kavakli, K.; Lassila, R.; et al. Anti-inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N. Engl. J. Med. 2011, 365, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.J.; Henzl, M.J.; Gomperts, E.D. Safety of factor VIII inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic adverse events. Haemophilia 2002, 8, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Giangrande, P.L. Porcine factor VIII. Haemophilia 2012, 18, 305–309. [Google Scholar] [CrossRef]
- Tang, Q.; Liao, J.; Xie, X. Acquired Hemophilia Associated with Rheumatic Diseases: A Case-Based Systematic Review. J. Inflamm. Res. 2022, 15, 4385–4393. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | In Medicine Department | Reference Range |
|---|---|---|
| Creatinine (mg/dL) | 0.62 | 0.50–0.9 |
| Na+ (mmol/L) | 140 | 136–145 |
| K+ (mmol/L) | 4.5 | 3.4–5.5 |
| Ca+ (mmol/L) | 2.11 | 2.10–2.50 |
| Proteins (g/L) | 59 | 60–82 |
| Albumin (g/L) | 38 | 35–55 |
| Amylase (U/L) | 68 | 28–100 |
| Lipase (U/L) | 40 | 13–60 |
| AST (U/L) | 8 | 9–45 |
| ALT (U/L) | 14 | 10–40 |
| Total Bilirubin (mg/dL) | 1.45 | 0.30–1.20 |
| Direct Bilirubine (mg/dL) | 0.51 | <0.20 |
| CPK (U/L) | 693 | 20–200 |
| LDH (U/L) | 365 | 135–225 |
| Haptoglobin (mg/L) | 240 | 300–2000 |
| Folate (ng/mL) | >20 | 3.8–16.0 |
| Vitamin B12 (pg/mL) | 215 | 191–663 |
| Ferritin (µg/L) | 73 | 30–400 |
| Reticulocytes (%) | 5.99 | 0.50–2.50 |
| CRP (µg/L) | 22,800 | 100–6000 |
| ESR (mm/h) | 47 | 0–35 |
| Hb (g/dL) | 8.2 | 12.2–15.3 |
| WBC (×109/L) | 10.72 | 4.40–11.30 |
| N (×109/L) | 7.39 | 1.80–7.70 |
| L (×109/L) | 2.18 | 1.80–4.80 |
| PLT (×109/L) | 469 | 150–450 |
| Fibrinogen (g/L) | 5.50 | 1.50–4.00 |
| INR | 0.92 | 0.81–1.20 |
| aPTT ratio | 1.91 | 0.8–1.30 |
| C3 (mg/dL) | 136.00 | 90.00–180.00 |
| C4 (mg/dL) | 32.30 | 10.00–40.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gioia, C.; Paroli, M.; Morace, V.; Nardacci, L.; Ruffo, S.M.; Rossi, E.; Pignatelli, P.; Accapezzato, D. Acquired Hemophilia Associated with Rheumatoid Arthritis: A Case Report and Review of the Literature. Int. J. Mol. Sci. 2025, 26, 3628. https://doi.org/10.3390/ijms26083628
Gioia C, Paroli M, Morace V, Nardacci L, Ruffo SM, Rossi E, Pignatelli P, Accapezzato D. Acquired Hemophilia Associated with Rheumatoid Arthritis: A Case Report and Review of the Literature. International Journal of Molecular Sciences. 2025; 26(8):3628. https://doi.org/10.3390/ijms26083628
Chicago/Turabian StyleGioia, Chiara, Marino Paroli, Valentina Morace, Lucrezia Nardacci, Sara Martina Ruffo, Elisabetta Rossi, Pasquale Pignatelli, and Daniele Accapezzato. 2025. "Acquired Hemophilia Associated with Rheumatoid Arthritis: A Case Report and Review of the Literature" International Journal of Molecular Sciences 26, no. 8: 3628. https://doi.org/10.3390/ijms26083628
APA StyleGioia, C., Paroli, M., Morace, V., Nardacci, L., Ruffo, S. M., Rossi, E., Pignatelli, P., & Accapezzato, D. (2025). Acquired Hemophilia Associated with Rheumatoid Arthritis: A Case Report and Review of the Literature. International Journal of Molecular Sciences, 26(8), 3628. https://doi.org/10.3390/ijms26083628