
The Role of N6-Methyladenosine in Mitochondrial Dysfunction and Pathology


Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
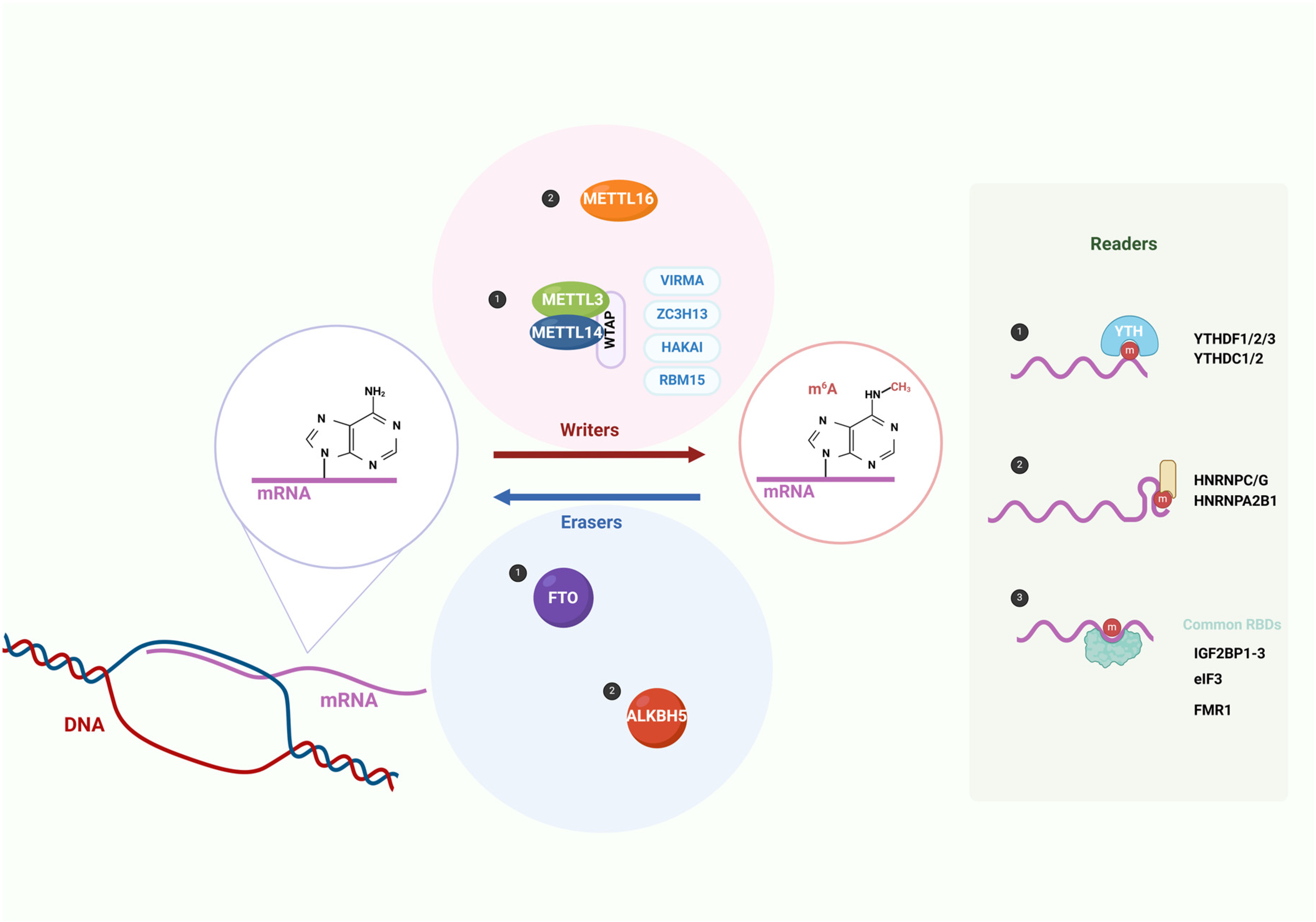
| Category | Factor | Full Name | Function | Reference |
|---|---|---|---|---|
| Writers | METTL3 | Methyltransferase-like 3 | As the catalytic core of the m6A methyltransferase complex, it catalyzes m6A modification. | [28] |
| METTL14 | Methyltransferase-like 14 | Acts as an RNA binding platform within the m6A methyltransferase complex and forms a heterodimer with METTL3 to catalyze m6A modification. | [5,28] | |
| WTAP | Wilms tumor 1- associated protein | As a regulatory subunit in the m6A methyltransferase complex, it interacts with METTL3 and METTL14 and directs them to nuclear speckles. | [29,30] | |
| METTL16 | Methyltransferase-like 16 | A single-component methyltransferase that can methylate U6-snRNA, the MALAT1 long non-coding RNA, and the MAT2A pre-mRNA. | [31,32] | |
| Writers | VIRMA (KIAA1429) | Vir-like m6A methyltransferase-associated | Recruits the core components of the methyltransferase METTL3/METTL14/WTAP to specific regions for selective methylation. | [33] |
| RBM15/ RBM15B | RNA binding motif protein 15/ RNA binding motif protein 15B | Binds to the m6A methyltransferase complex and guides it to specific RNA sites. | [34] | |
| ZC3H13 | Zinc finger CCCH-type containing 13 | Controls the nuclear localization of the Zc3h13–WTAP–Virilizer–Hakai complex and promotes m6A methylation. | [35] | |
| HAKAI (CBLL1) | Cbl Proto-Oncogene Like 1 | Maintains the stability of the m6A methyltransferase complex. | [36] | |
| Erasers | FTO | Fat mass and obesity-associated | Removes m6A modification and promotes mRNA splicing. | [37] |
| ALKBH5 | AlkB homolog 5 | Removes m6A modification on nuclear RNA, thereby regulating nuclear mRNA export. | [14] | |
| Readers | YTHDF1 | YTH N6-methyladenosine RNA-binding protein 1 | Promotes the translation initiation of m6A-modified mRNA. | [18] |
| YTHDF2 | YTH N6-methyladenosine RNA-binding protein 2 | Promotes the degradation of m6A-modified mRNA. | [17] | |
| YTHDF3 | YTH N6-methyladenosine RNA-binding protein 3 | As a partner of YTHDF1 and YTHDF2, it interacts with YTHDF1/YTHDF2 to promote mRNA translation or degradation. | [19] | |
| YTHDC1 | YTH domain containing 1 | Promotes mRNA splicing. | [20] | |
| YTHDC2 | YTH domain containing 2 | Increases the translation efficiency of the target mRNA. | [21,22] | |
| IGF2BP1/2/3 | Insulin-like growth factor 2 mRNA-binding protein 1/2/3 | Promotes the stability of the target mRNA and facilitates its translation. | [24] | |
| HNRNPC | Heterogeneous nuclear ribonucleoprotein C | Affects the stability, alternative splicing, and translation of pre-mRNA. | [25] | |
| HNRNPA2B1 | Heterogeneous nuclear ribonucleoprotein A2/B1 | Promotes the processing of nuclear pri-miRNA and mRNA splicing. | [26] | |
| eIF3 | Eukaryotic initiation factor 3 | Promotes m6A-mediated cap-independent translation. | [27] |
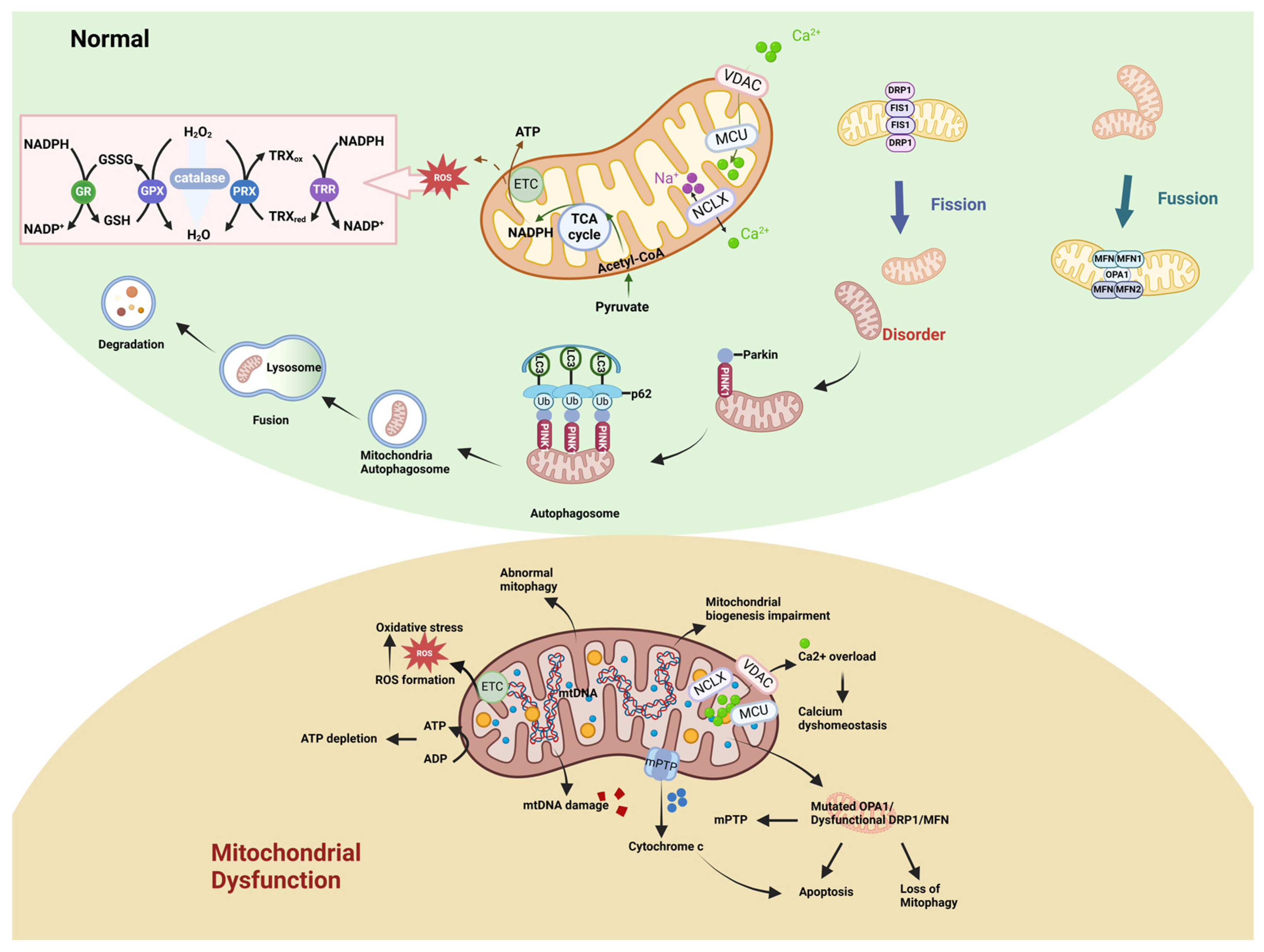
2. Mitochondrial Function and Dysregulation
2.1. Mitochondrial Structure and Function
2.2. Manifestations of Mitochondrial Dysfunction
2.2.1. Abnormal Mitochondrial Energy Metabolism and Impaired Biogenesis
2.2.2. Alterations in Mitochondrial Dynamics
2.2.3. Abnormal Mitophagy
2.2.4. Imbalance Between Generation and Clearance of Mitochondrial ROS
2.2.5. Imbalance of Calcium Homeostasis
2.2.6. Cell Death
2.2.7. Mitochondria-Targeted Therapy in Diseases
| Category | Factor | Full Name | Function | Reference |
|---|---|---|---|---|
| Mitochondrial Biogenesis | PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1 alpha | Regulates processes such as mitochondrial biogenesis, fission, fusion, and mitophagy by modulating coactivators and downstream effector factors. | [126] |
| PGC-1β | Peroxisome proliferator-activated receptor gamma coactivator 1 beta | Promotes mitochondrial biogenesis and is essential for normal OXPHOS and mitochondrial function. | [126] | |
| PRC | PGC-1-related coactivator | Activates the transcription factors NRF1 and NRF2, which are associated with the expression of the respiratory chain. | [127] | |
| TFAM | Mitochondrial transcription factor A | Essential for the transcription and replication of mtDNA. | [58] | |
| Mitochondrial Dynamics | DRP1 (DLP1/ DNM1L) | The GTPase dynamin-related protein 1 | Recruited from the cytoplasm to the outer mitochondrial membrane and mediate the process of mitochondrial fission. | [64,65] |
| MID49 | Mitochondrial dynamics proteins of 49 | Recruits DRP1 and promotes mitochondrial fission. | [128] | |
| MID51 | Mitochondrial dynamics proteins of 51 | Recruits DRP1 and promotes mitochondrial fission. | [128] | |
| MFF | Mitochondrial fission factor | Acts as a DRP1 receptor on the mitochondrial membrane, recruits DRP1, and promotes mitochondrial fission. | [129] | |
| FIS1 | Fission protein 1 | Recruits DRP1 and facilitates mitochondrial fission. | [128] | |
| MTP18 (MTFP1) | Mitochondrial fission process 1 | Maintains the mitochondrial morphology by regulating mitochondrial fission. | [130] | |
| MFN1 | Mitofusin 1 | Together with MFN2, mediates the fusion of the OMM in a GTP-dependent manner, coordinating the sequential fusion of the OMM and the IMM with the IMM fusion regulator OPA1. | [129] | |
| MFN2 | Mitofusin 2 | Together with MFN1, it mediates the fusion of the OMM in a GTP-dependent manner, mediates mitochondria–ER tethering, and transfers phosphatidylserine from ER to mitochondria. | [129] | |
| OPA1 | Optic atrophy protein 1 | Controls the fusion of the IMM and participates in processes such as regulating the shape of cristae, the arrangement of ETC supercomplexes, and the control of Cyt c release. | [73] | |
| Mitophagy | PINK1 | Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 | Recruits Parkin to initiate mitophagy and eliminate damaged mitochondria. | [130] |
| Parkin | Parkin | E3 ubiquitin ligase, ubiquitinates multiple OMM proteins, recruits autophagy receptors to damaged mitochondria, and promotes mitophagy. | [130] | |
| BNIP3L (NIX) | BCL2-interacting protein 3 like | Participates in the process of mitophagy as a mitophagy receptor and promotes the formation of autophagosomes. | [131] | |
| BNIP3 | BCL2-interacting protein 3 | Induces mitophagy by binding to LC3 through the BH3 domain. | [132] | |
| FUNDC1 | FUN14 domain containing 1 | A ubiquitin-independent mitophagy receptor that can directly bind to LC3 to initiate mitophagy. | [133] | |
| Mitochondrial Oxidative Stress | SOD | Superoxide dismutase | An antioxidant enzyme that converts superoxide anion radicals into hydrogen peroxide and oxygen. | [84] |
| GPX | Glutathione peroxidase | An antioxidant enzyme family that utilizes reduced glutathione as an electron donor to catalyze the reduction of hydrogen peroxide or organic hydroperoxides to water or the corresponding alcohols. | [134] | |
| TRX2 | Thioredoxin-2 | Clears ROS in the cell through the TRX2/PRX system and regulates the apoptotic signaling pathway by inhibiting oxidative stress. | [135] | |
| PRX | Peroxiredoxin | A multifunctional enzyme that reduces peroxides through the cysteine residues at the active center and also acts as a redox signaling regulator, chaperone, and pro-inflammatory factor. | [136] | |
| Calcium Homeostasis | MCU | Mitochondrial calcium uniporter | Forms a pore through which calcium ions enter the mitochondria and regulates the concentration of Ca2+ in the mitochondria. | [137,138] |
| MICU1 | Mitochondrial calcium uptake 1 | A key regulatory factor for mitochondrial Ca2+ uptake. When the intracellular Ca2+ level is high, it promotes the influx of calcium ions into the mitochondria. | [139,140] | |
| MICU2 | Mitochondrial calcium uptake 2 | When the level of calcium ions outside the mitochondria is low, it turns off the activity of the MCU. | [138] | |
| MCUR1 | MCU regulator 1 | Acts as a scaffolding factor to bind the MCU and EMRE. | [141] | |
| MCUb | MCU dominant-negative β-subunit | An inhibitory subunit of the MCU complex that forms a multimer with the MCU to inhibit the influx of Ca2+. | [142] | |
| SCL25A23 | Solute carrier 25A23 | Participates in mitochondrial Ca2+ uptake and interacts with the MCU and MICU1 to enhance the activity of the MCU channel. | [143] | |
| EMRE | Essential MCU regulator | Activates the function of the MCU, increases the uptake of Ca2+, and maintains the MICU regulation of the MCU pore. | [137] | |
| NCLX | Mitochondrial Na+/Ca2+/Li+ exchanger | Mediates the efflux of mitochondrial Ca2+ using the entry of sodium ions into the mitochondria along their concentration gradient as the driving force; transports calcium ions out of the mitochondria to maintain mitochondrial calcium homeostasis. | [98] | |
| Cell Death | BAK | BCL2-antagonist/killer | Together with BAX, mediates the permeabilization of the OMM in the mitochondrial pathway and promotes apoptosis. | [144] |
| BAX | BCL2 Associated X Protein | Together with BAK, mediates the permeabilization of the OMM in the mitochondrial pathway and promotes apoptosis. | [144] | |
| RIPK1 | Receptor-interacting protein kinase 1 | A key mediator of the apoptotic, necroptotic, and inflammatory pathways that mediate necroptosis. | [145] | |
| RIPK3 | Receptor-interacting protein kinase 3 | Acts as a downstream mediator of RIPK1 to trigger necroptosis. | [146] | |
| GPX4 | Glutathione-dependent peroxidase 4 | A form of glutathione peroxidase that specifically catalyzes the conversion of lipid hydroperoxides into non-toxic lipid alcohols to alleviate ferroptosis. | [147] |
3. M6A and Mitochondrial Dysfunction
3.1. Role of m6A in Influencing Mitochondrial Function in Disease
3.2. Role of m6A in Influencing Mitochondrial Function in Cancer
| Factor | Full Name | Function | Reference |
|---|---|---|---|
| TRAF6 | Tumor necrosis factor receptor-associated factor 6 | Belonging to the TRAFs family, an adaptor protein is recruited to the intracellular region when activated and executes a variety of physiological functions through the TLR4 signaling pathway. | [157] |
| ECSIT | Evolutionarily conserved signaling intermediate in Toll pathways | A multifunctional protein partially located in the IMM, which participates in the assembly of oxidative phosphorylation complex I. | [157] |
| LONP1 | Lon peptidase 1 | A protein quality control protease that plays an important role in regulating mitochondrial protein homeostasis and maintains the integrity of mtDNA by selectively degrading abnormal and oxidatively damaged proteins. | [158] |
| MTHFD2 | Methylenetetrahydrofolate dehydrogenase 2 | A mitochondrial enzyme encoded by the nucleus, which participates in folate metabolism and one-carbon metabolism in mitochondria and maintains intracellular redox balance. | [230] |
| NDUFA4 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 4 | Encodes a subunit in the electron transport chain complex of the mitochondrial respiratory chain to generate ATP. | [219] |
| PDK4 | Pyruvate dehydrogenase kinase 4 | Promotes the transition from mitochondrial oxidative phosphorylation to glycolysis and regulates glucose metabolism by phosphorylating pyruvate dehydrogenase. | [231] |
| RRM2B | Ribonucleotide reductase regulatory TP53 inducible subunit M2B | A key subunit of ribonucleotide reductase (RR) plays important roles in DNA repair, replication, oxidative stress, and mtDNA synthesis. | [232] |
| RR | Ribonucleotide reductase | Catalyzes ribonucleoside diphosphates to deoxyribonucleoside diphosphates and plays an important role in DNA synthesis and repair. | [232] |
| Caveolin-1 | Caveolin-1 | A membrane protein that is essential for maintaining the structure and function of mitochondria and is associated with the number of mitochondria and their bioenergetic functions. | [222] |
| APOE | Apolipoprotein E | A glycoprotein that functions as a lipid transport protein. | [233] |
| DCP2 | Decapping MRNA 2 | A major decapping enzyme during 5′ to 3′ mRNA decay, controlling the expression of PINK1 and Parkin to regulate mitophagy and the level of mitochondrial damage. | [226] |
| OLA1 | Obg-like ATPase 1 | An ATP hydrolase that mediates mitochondrial energy metabolism, including ATP hydrolysis and glycolysis. | [227] |
3.3. Small Molecule Drug Therapy
4. Summary and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, Y.; Xu, C.; An, P.; Luo, Y.; Jiao, L.; Luo, J.; Li, Y. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. IJMS 2022, 23, 16053. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.-L.; Cheng, S.S.-Y.; Cheung, Y.-T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.-S.; Lee, S.M.-Y.; Chang, R.C.-C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, D.F. The regulation of tumor cell physiology by mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2018, 500, 9–16. [Google Scholar] [CrossRef]
- An, Y. The Role of m6A RNA methylation in cancer metabolism. Mol. Cancer 2022, 21, 14. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 30 UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Liu, Q.; Gregory, R.I. RNAmod: An Integrated System for the Annotation of mRNA Modifications. Nucleic Acids Res. 2019, 47, W548–W555. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Höbartner, C.; Sloan, K.E.; Bohnsack, M.T. Human METTL16 is a n6-methyladenosine (m6a) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985.e5. [Google Scholar] [CrossRef]
- Zheng, G. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Qu, J.; Yan, H.; Hou, Y.; Cao, W.; Liu, Y.; Zhang, E.; He, J.; Cai, Z. RNA Demethylase ALKBH5 in cancer: From mechanisms to therapeutic potential. J. Hematol. Oncol. 2022, 15, 8. [Google Scholar] [CrossRef]
- Chen, X.-Y. The Role of m6A RNA Methylation in Human Cancer. Mol. Cancer 2019, 18, 1–9. [Google Scholar] [CrossRef]
- Wang, X. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Wang, X. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Shi, H. YTHDF3 facilitates translation and decay of N6-methyladenosine-Modified RNA. Cell Res. 2017, 27. [Google Scholar] [CrossRef]
- Xiang, Y.; Laurent, B.; Hsu, C.-H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6A Methylation Regulates the Ultraviolet-Induced DNA Damage Response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Hsu, P.J. Ythdc2 Is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef]
- Jain, D.; Puno, M.R.; Meydan, C.; Lailler, N.; Mason, C.E.; Lima, C.D.; Anderson, K.V.; Keeney, S. Ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. eLife 2018, 7, e30919. [Google Scholar] [CrossRef]
- Liao, S.; Sun, H.; Xu, C. YTH domain: A family of N. 6 -methyladenosine (m 6 A) readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a mediator of m6A-dependent nuclear RNA processing events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m6A promotes cap-independent translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Lu, F.; Zhang, Y. Cell totipotency: Molecular features, induction, and maintenance. Natl. Sci. Rev. 2015, 2, 217–225. [Google Scholar] [CrossRef]
- Schöller, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m6 a generating METTL3–METTL14–WTAP complex. RNA 2018, 24, 499–512. [Google Scholar] [CrossRef]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, J.; Lin, X.; Yang, L.; Zhou, Q.; Mi, X.; Li, Q.; Wang, S.; Li, D.; Liu, X.-M.; et al. METTL16 Promotes translation and lung tumorigenesis by sequestering cytoplasmic eIF4E2. Cell Rep. 2023, 42, 112150. [Google Scholar] [CrossRef]
- Satterwhite, E.R.; Mansfield, K.D. RNA methyltransferase METTL16: Targets and function. WIREs RNA 2022, 13, e1681. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′utr and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 2018, 69, 1028–1038.e6. [Google Scholar] [CrossRef] [PubMed]
- Bawankar, P.; Lence, T.; Paolantoni, C.; Haussmann, I.U.; Kazlauskiene, M.; Jacob, D.; Heidelberger, J.B.; Richter, F.M.; Nallasivan, M.P.; Morin, V.; et al. Hakai is required for stabilization of core components of the m6A mRNA methylation machinery. Nat. Commun. 2021, 12, 3778. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B.-F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.-J.; Ping, X.-L.; Chen, Y.-S.; Wang, W.-J.; et al. FTO-dependent demethylation of n6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Walsh, C.T.; Tu, B.P.; Tang, Y. Eight kinetically stable but thermodynamically activated molecules that power cell metabolism. Chem. Rev. 2018, 118, 1460–1494. [Google Scholar] [CrossRef]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.W.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA. 2010, 107, 16823–16827. [Google Scholar] [CrossRef]
- Sun, F.; Zhou, Q.; Pang, X.; Xu, Y.; Rao, Z. Revealing various coupling of electron transfer and proton pumping in mitochondrial respiratory chain. Curr. Opin. Struct. Biol. 2013, 23, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Et. Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Doke, T.; Susztak, K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. 2022, 32, 841–853. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Y.; Pi, C.; Yu, X.; Gao, X.; Zhang, C.; Sun, H.; Zhang, H.; Shi, Y.; He, X. Nicotinamide mononucleotide supplementation improves mitochondrial dysfunction and rescues cellular senescence by NAD+/Sirt3 pathway in mesenchymal stem cells. IJMS 2022, 23, 14739. [Google Scholar] [CrossRef]
- Mito, T.; Vincent, A.E.; Faitg, J.; Taylor, R.W.; Khan, N.A.; McWilliams, T.G.; Suomalainen, A. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab. 2022, 34, 197–208.e5. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, X.; Zhang, N.; Yang, Y.; Yi, Y.; Shao, Q.; Liu, M.; Zhang, X. Mitochondrial dysfunction as a therapeutic strategy for neurodegenerative diseases: Current insights and future directions. Ageing Res. Rev. 2024, 102, 102577. [Google Scholar] [CrossRef]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial diseases: Hope for the future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Hoogenraad, N.J.; Ryan, M.T. Translocation of proteins into mitochondria. IUBMB Life 2001, 51, 345–350. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309. [Google Scholar] [CrossRef] [PubMed]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Medi 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Kozhukhar, N.; Alexeyev, M.F. Limited predictive value of TFAM in mitochondrial biogenesis. Mitochondrion 2019, 49, 156–165. [Google Scholar] [CrossRef]
- McQuate, A.; Raible, D.W. Finding the balance: The elusive mechanisms underlying auditory hair cell mitochondrial biogenesis and mitophagy. Hear. Res. 2023, 428, 108664. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.; Zhao, G. The mitophagy pathway and its implications in human diseases. Sig. Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Sig. Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Et. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.B.; Sánchez-Guerrero, Á.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef]
- Chang, C.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein drp1. Ann. New York Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef]
- Adaniya, S.M.; O-Uchi, J.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. -Cell Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Galloway, C.A.; Lee, H.; Yoon, Y. Mitochondrial morphology—emerging role in bioenergetics. Free Radic. Biol. Med. 2012, 53, 2218–2228. [Google Scholar] [CrossRef]
- Labbé, K.; Murley, A.; Nunnari, J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391. [Google Scholar] [CrossRef]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 Requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef]
- Zheng, J.; Croteau, D.L.; Bohr, V.A.; Akbari, M. Diminished OPA1 expression and impaired mitochondrial morphology and homeostasis in aprataxin-deficient cells. Nucleic Acids Res. 2019, 47, 4086–4110. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Scorrano, L. Determinants and outcomes of mitochondrial dynamics. Mol. Cell 2023, 83, 857–876. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Et. Biophys. Acta (BBA)—Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef]
- Uoselis, L.; Nguyen, T.N.; Lazarou, M. Mitochondrial degradation: Mitophagy and beyond. Mol. Cell 2023, 83, 3404–3420. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Lizama, B.N.; Chu, C.T. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol. Asp. Med. 2021, 82, 100972. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Kuksal, N.; Chalker, J.; Mailloux, R.J. Progress in understanding the molecular oxygen paradox—Function of mitochondrial reactive oxygen species in cell signaling. Biol. Chem. 2017, 398, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Gill, R.; Mailloux, R.J. Protein S-Glutathionylation: The linchpin for the transmission of regulatory information on redox buffering capacity in mitochondria. Chem. -Biol. Interact. 2019, 299, 151–162. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Slade, L.; Chalker, J.; Kuksal, N.; Young, A.; Gardiner, D.; Mailloux, R.J. Examination of the superoxide/hydrogen peroxide forming and quenching potential of mouse liver mitochondria. Biochim. Et. Biophys. Acta (BBA) - Gen. Subj. 2017, 1861, 1960–1969. [Google Scholar] [CrossRef]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Moraes, C.T. Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol. 2001, 12, 449–457. [Google Scholar] [CrossRef]
- Wang, R.; Wang, M.; He, S.; Sun, G.; Sun, X. Targeting calcium homeostasis in myocardial ischemia/reperfusion injury: An overview of regulatory mechanisms and therapeutic reagents. Front. Pharmacol. 2020, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Lederer, W.J. How the mitochondrial calcium uniporter complex (MCUcx) works. proc. Natl. Acad. Sci. USA 2020, 117, 22634–22636. [Google Scholar] [CrossRef]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.R.; Vangheluwe, P. Primary active Ca2+ transport systems in health and disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035113. [Google Scholar] [CrossRef] [PubMed]
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. -Cell Physiol. 2021, 320, C465–C482. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, F.; Li, P.; Gao, Y. Mitochondrial Ca2+ homeostasis: Emerging roles and clinical significance in cardiac remodeling. IJMS 2022, 23, 3025. [Google Scholar] [CrossRef]
- Strubbe-Rivera, J.O.; Chen, J.; West, B.A.; Parent, K.N.; Wei, G.-W.; Bazil, J.N. Modeling the effects of calcium overload on mitochondrial ultrastructural remodeling. Appl. Sci. 2021, 11, 2071. [Google Scholar] [CrossRef]
- Walkon, L.L.; Strubbe-Rivera, J.O.; Bazil, J.N. Calcium overload and mitochondrial metabolism. Biomolecules 2022, 12, 1891. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Cecchi, C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. IJMS 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- Saqirile; Deng, Y.; Li, K.; Yan, W.; Li, K.; Wang, C. Gene expression regulation and the signal transduction of programmed cell death. CIMB 2024, 46, 10264–10298. [Google Scholar] [CrossRef] [PubMed]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and cell death. Nat. Cell Biol. 2024, 26, 1434–1446. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Wei, S.; Nguyen, T.H.; Jo, Y.; Zhang, Y.; Park, W.; Gariani, K.; Oh, C.-M.; Kim, H.H.; Ha, K.-T.; et al. Mitochondria-associated programmed cell death as a therapeutic target for age-related disease. Exp. Mol. Med. 2023, 55, 1595–1619. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Yang, C.; Yang, X.; Harrington, A.; Potts, C.; Kaija, A.; Ryzhova, L.; Liaw, L. Notch signaling regulates mouse perivascular adipose tissue function via mitochondrial pathways. Genes. 2023, 14, 1964. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial Regulation of Ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Mohammed, J.N.; Gelles, J.D.; Chen, Y. Mechanistic connections between mitochondrial biology and regulated cell death. Dev. Cell 2021, 56, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Ding, H.; Li, Y.; Li, X.; Liu, X.; Wang, K.; Wen, M.; Jiang, W.; Zeng, H. Hypercapnia promotes microglial pyroptosis via inhibiting mitophagy in hypoxemic adult rats. CNS Neurosci. Ther. 2020, 26, 1134–1146. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Chen, G.; Chen, Q. Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis. J. Biomed. Sci. 2023, 30, 86. [Google Scholar] [CrossRef] [PubMed]
- Wongrakpanich, A.; Geary, S.M.; Joiner, M.A.; Anderson, M.E.; Salem, A.K. Mitochondria-targeting particles. Nanomedicine 2014, 9, 2531–2543. [Google Scholar] [CrossRef] [PubMed]
- Batheja, S.; Gupta, S.; Tejavath, K.K.; Gupta, U. TPP-based conjugates: Potential targeting ligands. Drug Discov. Today 2024, 29, 103983. [Google Scholar] [CrossRef]
- Nam, H.Y.; Hong, J.-A.; Choi, J.; Shin, S.; Cho, S.K.; Seo, J.; Lee, J. Mitochondria-targeting peptoids. Bioconjugate Chem. 2018, 29, 1669–1676. [Google Scholar] [CrossRef]
- Xu, R.; Huang, L.; Liu, J.; Zhang, Y.; Xu, Y.; Li, R.; Su, S.; Xu, X. Remodeling of mitochondrial metabolism by a mitochondria-targeted RNAi nanoplatform for effective cancer therapy. Small 2024, 20. [Google Scholar] [CrossRef]
- Liao, J.; He, W.; Zhang, S.; Wang, J.; Cheng, H.; Gong, L.; Li, Y.; Sun, Y.; Lu, D.; Zhang, C.; et al. Magnetic field driven ceria nanosystems for mitochondria targeted therapy of ischemic stroke. Adv. Funct. Mater. 2025, 2423291. [Google Scholar] [CrossRef]
- Wen, C.; Tang, J.; Wu, M.; Liu, H.; Lin, X.; Fan, M.; Liu, G.; Zhang, J.; Liang, L.; Liu, X.; et al. Preparation, characterization, and stability of pectin-whey protein isolate-based nanoparticles with mitochondrial targeting ability. Int. J. Biol. Macromol. 2025, 301, 140383. [Google Scholar] [CrossRef]
- Duan, Z.; Li, L.; Zhan, Q.; Chen, J.; Li, Q.; Liu, R.; Tu, Y. Mitochondria-targeting type-i photodynamic therapy based on phenothiazine for realizing enhanced immunogenic cancer cell death via mitochondrial oxidative stress. Int. J. Nurs. 2025, 20, 125–139. [Google Scholar] [CrossRef]
- Wang, W.; Yao, S.-Y.; Luo, J.; Ding, C.; Huang, Q.; Yang, Y.; Shi, Z.; Lin, J.; Pan, Y.-C.; Zeng, X.; et al. Engineered hypoxia-responsive albumin nanoparticles mediating mitophagy regulation for cancer therapy. Nat. Commun. 2025, 16, 596. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Song, B.; Yang, G.; Wang, Y.; Wu, Z.; Si, M.; Yang, Z.; Chen, H.; Liu, C.; Li, M.; et al. In situ transformable nanoparticle effectively suppresses bladder cancer by damaging mitochondria and blocking mitochondrial autophagy flux. Adv. Sci. 2025, 12, 2409425. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, Y.; Xie, Y.; Liu, Y.; Hu, H.; Xie, C.; Xu, S.; Zhang, Z.; Zhang, J.; Shen, Y.; et al. Mitochondria-targeted icaritin nanoparticles induce immunogenic cell death in hepatocellular carcinoma. ACS Appl. Mater. Interfaces 2025, 17, 2899–2910. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome Proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Sig. Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y. Acad. Sci. 2008, 1147, 321–334. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Tábara, L.-C.; Segawa, M.; Prudent, J. Molecular mechanisms of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2025, 26, 123–146. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Behera, B.P.; Patra, S.; Patil, S.; Patro, B.S.; Bhutia, S.K. The inner mitochondrial membrane fission protein MTP18 Serves as a mitophagy receptor to prevent apoptosis in oral cancer. J. Cell Sci. 2023, 136, jcs259986. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, W.; Lu, Y.; Zheng, Y.; Pan, L.; Wu, X.; Yuan, Y.; Shen, Z.; Ma, S.; Zhang, X.; et al. BNIP3L/NIX-mediated mitophagy: Molecular mechanisms and implications for human disease. Cell Death Dis. 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Niemi, N.M.; Friedman, J.R. Coordinating BNIP3/NIX-mediated mitophagy in space and time. Biochem. Soc. Trans. 2024, 52, 1969–1979. [Google Scholar] [CrossRef]
- Bi, Y.; Liu, S.; Qin, X.; Abudureyimu, M.; Wang, L.; Zou, R.; Ajoolabady, A.; Zhang, W.; Peng, H.; Ren, J.; et al. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J. Adv. Res. 2024, 55, 45–60. [Google Scholar] [CrossRef]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef]
- Tanaka, T. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef]
- Wu, M.; Deng, C.; Lo, T.-H.; Chan, K.-Y.; Li, X.; Wong, C.-M. Peroxiredoxin, senescence, and cancer. Cells 2022, 11, 1772. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, J.; Hu, H.; Chen, L. MCU complex: Exploring emerging targets and mechanisms of mitochondrial physiology and pathology. J. Adv. Res. 2025, 68, 271–298. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial Ef hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Prasad, V.; Grimes, K.M.; Vanhoutte, D.; Blair, N.S.; Lin, S.-C.; Bround, M.J.; Bers, D.M.; Molkentin, J.D. MCUb is an inducible regulator of calcium-dependent mitochondrial metabolism and substrate utilization in muscle. Cell Reports 2023, 42, 113465. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress–mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Peña-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Liu, S.; Joshi, K.; Denning, M.F.; Zhang, J. RIPK3 signaling and its role in the pathogenesis of cancers. Cell. Mol. Life Sci. 2021, 78, 7199–7217. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and epigenetics—crosstalk in homeostasis and stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
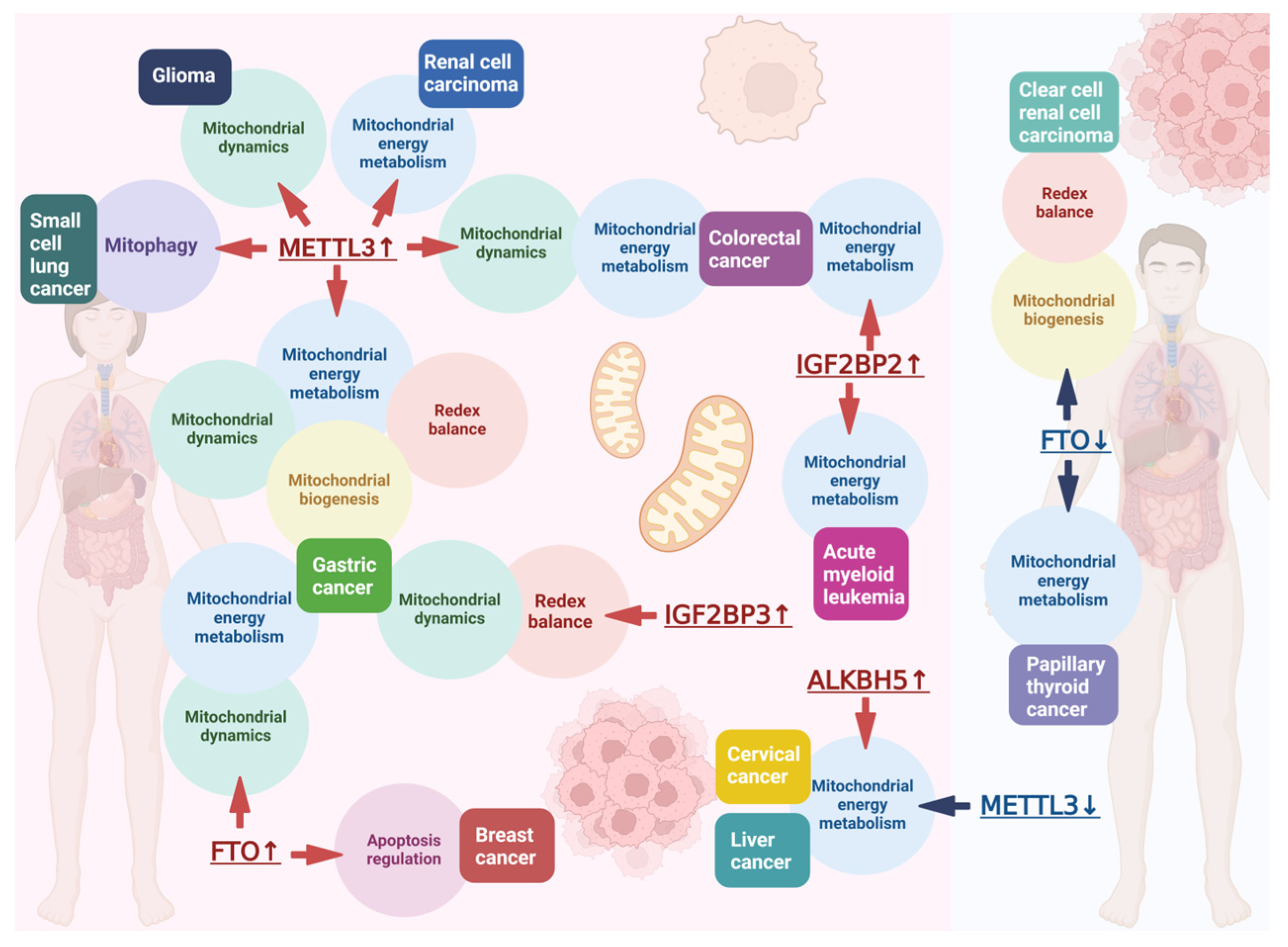
- Kahl, M.; Xu, Z.; Arumugam, S.; Edens, B.M.; Fischietti, M.; Zhu, A.C.; Platanias, L.C.; He, C.; Zhuang, X.; Ma, Y.C. m6A RNA methylation regulates mitochondrial function. Hum. Mol. Genet. 2024, 33, 969–980. [Google Scholar] [CrossRef]
- Wang, X.; Huang, N.; Yang, M.; Wei, D.; Tai, H.; Han, X.; Gong, H.; Zhou, J.; Qin, J.; Wei, X.; et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1α pathway-mediated mitochondria biogenesis. Cell Death Dis. 2017, 8, e2702. [Google Scholar] [CrossRef]
- Kang, H.; Zhang, Z.; Yu, L.; Li, Y.; Liang, M.; Zhou, L. FTO Reduces mitochondria and promotes hepatic fat accumulation through rna demethylation. J. Cell. Biochem. 2018, 119, 5676–5685. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.D.; Guo, W.Y.; Han, C.H.; Wang, Y.; Chen, X.S.; Li, D.W.; Liu, J.L.; Zhang, M.; Zhu, N.; Wang, X. N6-Methyladenosine demethylase FTO impairs hepatic ischemia–reperfusion injury via inhibiting Drp1-mediated mitochondrial fragmentation. Cell Death Dis. 2021, 12, 442. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Zhang, W.; Gao, Y.; Chen, S.; Jiang, M.; He, R.; Chen, W.; Wei, X.; Hu, B.; Xu, H.; et al. N6-methyladenosine demethyltransferase FTO alleviates sepsis by upregulating BNIP3 to induce mitophagy. J. Cell. Physiol. 2024, 239, e31448. [Google Scholar] [CrossRef]
- Hou, L.; Li, S.; Li, S.; Wang, R.; Zhao, M.; Liu, X. FTO inhibits oxidative stress by mediating m6A demethylation of Nrf2 to alleviate cerebral ischemia/reperfusion injury. J. Physiol. Biochem. 2023, 79, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Chang, J.; Li, Y.; Gao, Z.; Qiu, Q.; Wang, Q.; Han, G.; Chai, J.; Feng, M.; Wang, P.; et al. Differential m6A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell 2022, 29, 149–159.e7. [Google Scholar] [CrossRef]
- Chen, H.; Xing, H.; Zhong, C.; Lin, X.; Chen, R.; Luo, N.; Chen, L.; Huang, Y. METTL3 confers protection against mitochondrial dysfunction and cognitive impairment in an alzheimer disease mouse model by upregulating Mfn2 via N6-methyladenosine modification. J. Neuropathol. Exp. Neurol. 2024, 83, 606–614. [Google Scholar] [CrossRef]
- Yang, P.; Wang, Y.; Ge, W.; Jing, Y.; Hu, H.; Yin, J.; Xue, M.; Wang, Y.; Li, X.; Li, X.; et al. m6A Methyltransferase METTL3 contributes to sympathetic hyperactivity post-Mi via promoting TRAF6-dependent mitochondrial ROS production. Free Radic. Biol. Med. 2023, 209, 342–354. [Google Scholar] [CrossRef]
- Shetty, R.; Noland, R.; Nandi, G.; Suzuki, C.K. Powering down the mitochondrial lonp1 protease: A novel strategy for anticancer therapeutics. Expert. Opin. Ther. Targets 2024, 28, 9–15. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, S.; Xu, L.; Li, M.; Tian, B.; Yao, L.; Zhu, X. LONP1 Alleviates ageing-related renal fibrosis by maintaining mitochondrial homeostasis. J. Cell. Mol. Medi. 2024, 28, e70090. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, D.; Wang, Y.; Guo, K.; Spencer, C.B.; Ortoga, L.; Qu, M.; Shi, Y.; Shao, Y.; Wang, Z.; et al. METTL3-mediated N6-methyladenosine exacerbates ferroptosis via m6A-IGF2BP2-dependent mitochondrial metabolic reprogramming in sepsis-induced acute lung injury. Clin. Transl. Med. 2023, 13, e1389. [Google Scholar] [CrossRef]
- Tu, B.; Song, K.; Zhou, Y.; Sun, H.; Liu, Z.-Y.; Lin, L.-C.; Ding, J.-F.; Sha, J.-M.; Shi, Y.; Yang, J.-J.; et al. METTL3 boosts mitochondrial fission and induces cardiac fibrosis by enhancing LncRNA GAS5 methylation. Pharmacol. Res. 2023, 194, 106840. [Google Scholar] [CrossRef] [PubMed]
- Ke, M.-Y.; Fang, Y.; Cai, H.; Lu, J.-W.; Yang, L.; Wang, Y.; Wu, R.-Q.; Zhang, X.-F.; Lv, Y.; Dong, J. The m6 a reader YTHDF1 attenuates fulminant hepatitis via MFG-E8 translation in an m6 a dependent manner. Int. J. Biol. Sci. 2023, 19, 3987–4003. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, W.; Wang, Z.; Liu, Z.; Yi, X.; Wu, J. Mettl3-m6A-YTHDF1 axis promotion of mitochondrial dysfunction in metabolic dysfunction–associated steatotic liver disease. Cell. Signal. 2024, 121, 111303. [Google Scholar] [CrossRef]
- Moqbel, S.A.A.; Zeng, R.; Ma, D.; Xu, L.; Lin, C.; He, Y.; Ma, C.; Xu, K.; Ran, J.; Jiang, L.; et al. The effect of mitochondrial fusion on chondrogenic differentiation of cartilage progenitor/stem cells via Notch2 signal pathway. Stem Cell Res. Ther. 2022, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Lin, L.-C.; Liu, Z.-Y.; Song, K.; Tu, B.; Sun, H.; Zhou, Y.; Mao, S.; Zhang, Y.; Li, R.; et al. N6-methyladenosine-mediated phase separation suppresses NOTCH1 expression and promotes mitochondrial fission in diabetic cardiac fibrosis. Cardiovasc. Diabetol. 2024, 23, 347. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Y.; Sun, F.; Luo, Y.; Yang, Y.; Li, J.; Hu, W.; Tao, H.; Lu, C.; Yang, J.-J. ALKBH5 Attenuates mitochondrial fission and ameliorates liver fibrosis by reducing Drp1 methylation. Pharmacol. Res. 2023, 187, 106608. [Google Scholar] [CrossRef]
- Liu, L.; Wu, J.; Lu, C.; Ma, Y.; Wang, J.; Xu, J.; Yang, X.; Zhang, X.; Wang, H.; Xu, J.; et al. WTAP-mediated m6A modification of lncRNA Snhg1 improves myocardial ischemia-reperfusion injury via miR-361-5p/OPA1-dependent mitochondrial fusion. J. Transl. Med. 2024, 22, 499. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The glycolytic switch in tumors: How many players are involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Bueno, M.J.; Ruiz-Sepulveda, J.L.; Quintela-Fandino, M. Mitochondrial inhibition: A treatment strategy in cancer? Curr. Oncol. Rep. 2021, 23, 49. [Google Scholar] [CrossRef]
- Miller, J.J.; Gonzalez Castro, L.N.; McBrayer, S.; Weller, M.; Cloughesy, T.; Portnow, J.; Andronesi, O.; Barnholtz-Sloan, J.S.; Baumert, B.G.; Berger, M.S.; et al. Isocitrate dehydrogenase (IDH) mutant gliomas: A society for neuro-oncology (SNO) consensus review on diagnosis, management, and future directions. Neuro-Oncol. 2023, 25, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.J.; Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef]
- Li, J.; Yu, T.; Zeng, P.; Tian, J.; Liu, P.; Qiao, S.; Wen, S.; Hu, Y.; Liu, Q.; Lu, W.; et al. Wild-type IDH2 Is a therapeutic target for triple-negative breast cancer. Nat. Commun. 2024, 15, 3445. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Tan, Z.; Lu, J.; Li, L.; Song, X.; Shi, F.; Xie, L.; You, S.; Luo, X.; et al. Wild-Type IDH2 promotes the warburg effect and tumor growth through HIF1α in lung cancer. Theranostics 2018, 8, 4050–4061. [Google Scholar] [CrossRef]
- Chen, X.; Xu, W.; Wang, C.; Guan, S.; Sun, Y.; Wang, X.; An, D.; Wen, Z.; Chen, P.; Cheng, Y. The clinical significance of isocitrate dehydrogenase 2 in esophageal squamous cell carcinoma. Science 2017, 7, 700. [Google Scholar]
- Fan, S.; Yan, X.; Hu, X.; Liu, X.; Zhao, S.; Zhang, Y.; Zhou, X.; Shen, X.; Qi, Q.; Chen, Y. Shikonin Blocks CAF-induced TNBC metastasis by suppressing mitochondrial biogenesis through GSK-3β/NEDD4-1 mediated phosphorylation-dependent degradation of PGC-1α. J. Exp. Clin. Cancer Res. 2024, 43, 180. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Taddei, M.L.; Sacco, E.; Navari, N.; Correnti, M.; Piombanti, B.; Pastore, M.; Campani, C.; Pranzini, E.; Iorio, J.; et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J. Hepatol. 2021, 74, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Sig. Transduct. Target. Ther. 2020, 5, 59. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca2+ Inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC Cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The Role of the Mitochondrial Calcium Uniporter (MCU) Complex in Cancer. Pflug. Arch.—Eur. J. Physiol. 2018, 470, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corrà, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef]
- Gil-Hernández, A.; Arroyo-Campuzano, M.; Simoni-Nieves, A.; Zazueta, C.; Gomez-Quiroz, L.E.; Silva-Palacios, A. Relevance of membrane contact sites in cancer progression. Front. Cell Dev. Biol. 2021, 8, 622215. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Z.; Hu, S.; Chen, L.; Li, W.; Yang, Z. miRNA-874-3p inhibits the migration, invasion and proliferation of breast cancer cells by targeting VDAC1. Aging 2023, 15, 705–717. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Yang, Q.; Wan, L.; Zhao, J.; Wu, Y.; Wang, J.; Yang, Y.; Niu, M.; Liu, H.; et al. Increased Drp1 Promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J. Exp. Clin. Cancer Res. 2022, 41, 76. [Google Scholar] [CrossRef]
- Liang, J.; Yang, Y.; Bai, L.; Li, F.; Li, E. DRP1 Upregulation promotes pancreatic cancer growth and metastasis through increased aerobic glycolysis. J. Gastro Hepatol. 2020, 35, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chang, C.; Chien, C.; Huang, G.; Chen, Y.; Su, L.; Tsai, H.; Lin, Y.; Fang, F.; Chen, C. DRP1 contributes to head and neck cancer progression and induces glycolysis through modulated FOXM1/MMP12 Axis. Mol. Oncol. 2022, 16, 2585–2606. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Liu, H.; Feng, L.; Xue, Y.; Tang, H.; Xu, P. How mitochondrial dynamics imbalance affects the progression of breast cancer:A mini review. Med. Oncol. 2024, 41, 238. [Google Scholar] [CrossRef]
- Zhan, L.; Cao, H.; Wang, G.; Lyu, Y.; Sun, X.; An, J.; Wu, Z.; Huang, Q.; Liu, B.; Xing, J. Drp1-Mediated mitochondrial fission promotes cell proliferation through crosstalk of P53 and NF-κB pathways in hepatocellular carcinoma. Oncotarget 2016, 7, 65001–65011. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef]
- Li, S.; Han, S.; Zhang, Q.; Zhu, Y.; Zhang, H.; Wang, J.; Zhao, Y.; Zhao, J.; Su, L.; Li, L.; et al. FUNDC2 Promotes Liver tumorigenesis by inhibiting MFN1-mediated mitochondrial fusion. Nat. Commun. 2022, 13, 3486. [Google Scholar] [CrossRef]
- Yeon, S.Y.; Jo, Y.S.; Choi, E.J.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations in repeat sequences of ANK3, HACD4, TCP10L, TP53BP1, MFN1, LCMT2, RNMT, TRMT6, METTL8 and METTL16 genes in colon cancers. Pathol. Oncol. Res. 2018, 24, 617–622. [Google Scholar] [CrossRef]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Pang, G.; Xie, Q.; Yao, J. Mitofusin 2 inhibits bladder cancer cell proliferation and invasion via the Wnt/Β-catenin pathway. Oncol. Lett. 2019, 18, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Jiang, X.; Gu, Y.; Zheng, H.; Wang, X.; Zhang, H.; Wu, J.; Cheng, Y. OPA1 supports mitochondrial dynamics and immune evasion to CD8+ T cell in lung adenocarcinoma. PeerJ 2022, 10, e14543. [Google Scholar] [CrossRef]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef]
- Soares, C.D.; Morais, T.M.D.L.; Carlos, R.; De Almeida, O.P.; Mariano, F.V.; Altemani, A.; De Carvalho, M.G.F.; Corrêa, M.B.; Dos Reis, R.R.D.; Amorim, L.S.; et al. Prognostic importance of mitochondrial markers in mucosal and cutaneous head and neck melanomas. Hum. Pathol. 2019, 85, 279–289. [Google Scholar] [CrossRef]
- Abo Elwafa, R.; Gamaleldin, M.; Ghallab, O. The clinical and prognostic significance of FIS1, SPI1, PDCD7 and Ang2 expression levels in acute myeloid leukemia. Cancer Genet. 2019, 233–234, 84–95. [Google Scholar] [CrossRef]
- Li, Q.; Chu, Y.; Li, S.; Yu, L.; Deng, H.; Liao, C.; Liao, X.; Yang, C.; Qi, M.; Cheng, J.; et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell Death Dis. 2022, 13, 899. [Google Scholar] [CrossRef]
- Abdel-Moety, A.; Baddour, N.; Salem, P.; El-Tobgy, H.; El-Shendidi, A. SQSTM1 expression in hepatocellular carcinoma and relation to tumor recurrence after radiofrequency ablation. J. Clin. Exp. Hepatol. 2022, 12, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Miyata, H.; Makino, T.; Masuike, Y.; Furukawa, H.; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. High expression of the mitophagy-related protein Pink1 is associated with a poor response to chemotherapy and a poor prognosis for patients treated with neoadjuvant chemotherapy for esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2017, 24, 4025–4032. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, Q.-X.; Zhang, J.; Zhou, D.; Yang, G.-X.; Li, M.-Y.; Qiu, Y.; Chen, Q.; Zheng, H.; Dai, J.-G. PINK1 Overexpression promotes cell migration and proliferation via regulation of autophagy and predicts a poor prognosis in lung cancer cases. CMAR 2020, 12, 7703–7714. [Google Scholar] [CrossRef]
- Claudia, C.V.D.S.; Rosimeri, K.S.B.; Nayanne, L.C.P.; Amanda, P.A.; Marcia, O.; Lúcia, D.N.; Vanessa, S.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61. [Google Scholar] [CrossRef]
- Toma, M.I.; Wuttig, D.; Kaiser, S.; Herr, A.; Weber, T.; Zastrow, S.; Koch, R.; Meinhardt, M.; Baretton, G.B.; Wirth, M.P.; et al. PARK2 and PACRG are commonly downregulated in clear-cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes. Chromosomes Cancer 2013, 52, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Kasperbauer, J.L.; Tombers, N.M.; Wang, V.; Mayer, K.; Smith, D.I. A selected group of large common fragile site genes have decreased expression in oropharyngeal squamous cell carcinomas. Genes. Chromosomes Cancer 2014, 53, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev. Cell 2018, 46, 441–455.e8. [Google Scholar] [CrossRef]
- Macher-Goeppinger, S.; Keith, M.; Hatiboglu, G.; Hohenfellner, M.; Schirmacher, P.; Roth, W.; Tagscherer, K.E. Expression and functional characterization of the BNIP3 protein in renal cell carcinomas. Transl. Oncol. 2017, 10, 869–875. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, D.; Zhou, L.; Pei, Y.; Zhuang, Y.; Cui, W.; Chen, J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 2019, 41, 384–394. [Google Scholar] [CrossRef]
- Hou, H.; Er, P.; Cheng, J.; Chen, X.; Ding, X.; Wang, Y.; Chen, X.; Yuan, Z.; Pang, Q.; Wang, P.; et al. High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 2017, 6, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Green, N.H.; Galvan, D.L.; Badal, S.S.; Chang, B.H.; LeBleu, V.S.; Long, J.; Jonasch, E.; Danesh, F.R. MTHFD2 Links RNA Methylation to metabolic reprogramming in renal cell carcinoma. Oncogene 2019, 38, 6211–6225. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhuang, C.; Luo, X.; Huang, X.; Yao, L.; Li, J.; Li, Y.; Xiong, T.; Ye, J.; Zhang, F.; et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO - PGC -1α signalling axis. J. Cell. Mol. Medi. 2019, 23, 2163–2173. [Google Scholar] [CrossRef]
- Xu, W.; Lai, Y.; Pan, Y.; Tan, M.; Ma, Y.; Sheng, H.; Wang, J. m6A RNA methylation-mediated NDUFA4 promotes cell proliferation and metabolism in gastric cancer. Cell Death Dis. 2022, 13, 715. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Y.; Li, J.; Chen, Z.; Chen, F.; Tu, J.; Lin, S.; Wang, H. N6-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020, 11, 2578. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, H.; Zhong, K.; Tao, L.; Lin, Y.; Xie, G.; Tan, Y.; Wu, Y.; Lu, Y.; Chen, Z.; et al. N6-methyladenosine facilitates mitochondrial fusion of colorectal cancer cells via induction of GSH synthesis and stabilization of OPA1 mRNA. Natl. Sci. Rev. 2024, 11, nwae039. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Q.; Deng, H.; Xu, B.; Zhou, Y.; Liu, J.; Liu, Y.; Shi, Y.; Zheng, X.; Jiang, J. N6-methyladenosine demethylase FTO promotes growth and metastasis of gastric cancer via m6A modification of caveolin-1 and metabolic regulation of mitochondrial dynamics. Cell Death Dis. 2022, 13, 72. [Google Scholar] [CrossRef]
- Yan, Y.; Luo, A.; Liu, S.; Cai, M.; Liu, X.; Zhang, X.; Zhang, S.; Liu, Y.; Zeng, J.; Xu, X.; et al. METTL3-mediated LINC00475 alternative splicing promotes glioma progression by inducing mitochondrial fission. Research 2024, 7, 0324. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef]
- Huang, J.; Sun, W.; Wang, Z.; Lv, C.; Zhang, T.; Zhang, D.; Dong, W.; Shao, L.; He, L.; Ji, X.; et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J. Exp. Clin. Cancer Res. 2022, 41, 42. [Google Scholar] [CrossRef]
- Sun, Y.; Shen, W.; Hu, S.; Lyu, Q.; Wang, Q.; Wei, T.; Zhu, W.; Zhang, J. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J. Exp. Clin. Cancer Res. 2023, 42, 65. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Han, L.; Hu, X.; Sun, T.; Xu, D.; Li, Y.; Chen, Q.; Yao, W.; He, M.; Wang, Z.; et al. N6-methyladenosine reader IMP2 stabilizes the ZFAS1/OLA1 axis and activates the warburg effect: Implication in colorectal cancer. J. Hematol. Oncol. 2021, 14, 188. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, X.; Xie, K.; Luo, J.; Zhang, Y.; Huang, X.; Luo, J.; Tan, S. Mitochondrial dysfunction induced by HIF-1α under hypoxia contributes to the development of gastric mucosal lesions. Clin. Amp. Transl. Med. 2024, 14, e1653. [Google Scholar] [CrossRef]
- Weng, H.; Huang, F.; Yu, Z.; Chen, Z.; Prince, E.; Kang, Y.; Zhou, K.; Li, W.; Hu, J.; Fu, C.; et al. The m6A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell 2022, 40, 1566–1582.e10. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, S.; Zhou, H.; Li, R.; Xia, X.; Xiong, H. Identification of MTHFD2 as a Prognostic biomarker and ferroptosis regulator in triple-negative breast cancer. Front. Oncol. 2023, 13, 1098357. [Google Scholar] [CrossRef]
- Dou, X.; Fu, Q.; Long, Q.; Liu, S.; Zou, Y.; Fu, D.; Xu, Q.; Jiang, Z.; Ren, X.; Zhang, G.; et al. PDK4-dependent hypercatabolism and lactate production of senescent cells promotes cancer malignancy. Nat. Metab. 2023, 5, 1887–1910. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.-C.; Kuo, M.-L.; Cheng, J.; Cheng, Y.-C.; Hsieh, Y.-C.; Liu, Y.-R.; Hsieh, R.-H.; Yen, Y. RRM2B-mediated regulation of mitochondrial activity and inflammation under oxidative stress. Mediat. Inflamm. 2015, 2015, 287345. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.S.; Garner, B.; Ooi, L. Understanding the role of ApoE fragments in Alzheimer’s disease. Neurochem. Res. 2019, 44, 1297–1305. [Google Scholar] [CrossRef]
- Xia, Y.; Wang, Y.; Chen, K.; Zhang, M.; Jiang, Q.; Xu, T. Quercetin attenuated necroptosis and apoptosis caused by LPS-Induced mitochondrial function dysfunction through the METTL3-mediated PTEN m6A methylation/PI3K/AKT signaling in broiler livers. Phytomedicine 2025, 139, 156551. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Williams, A.; Wei, N. Quercetin ameliorated insulin resistance via regulating METTL3-mediated N6-methyladenosine modification of PRKD2 mRNA in skeletal muscle and C2C12 myocyte cell line. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 2655–2668. [Google Scholar] [CrossRef]
| Mitochondrial Function | Abnormalities of Functional Units | Cancer Type | Reference |
|---|---|---|---|
| Mitochondrial energy metabolism | Isocitrate dehydrogenase (IDH) mutant | Gliomas, acute myeloid leukemia, cholangiocarcinoma, chondrosarcoma | [176,177] |
| Upregulation of wild-type IDH2 | Triple-negative breast cancer, lung cancer, esophageal squamous cell carcinoma | [178,179,180] | |
| Upregulation of PGC-1α | Triple-negative breast cancer, cholangiocarcinoma | [181,182] | |
| Mitochondrial calcium homeostasis | Upregulation of MCU and downregulation of MICU1 | Colorectal cancer, hepatocellular carcinoma, breast cancer | [183,184,185] |
| Upregulation of MCUR1 | Hepatocellular carcinoma | [186] | |
| Downregulation of MCU | Prostate cancer, colon cancer | [187] | |
| Upregulation of VDAC | Breast cancer, head and neck cancer, lung adenocarcinoma | [188] | |
| Upregulation of VDAC1 | Breast cancer | [189] | |
| Mitochondrial dynamics | Upregulation of DRP1 | Esophageal squamous cell carcinoma, pancreatic cancer, head and neck cancer, breast cancer, hepatocellular carcinoma | [190,191,192,193,194] |
| Upregulation of DRP1 and downregulation of MFN1 | Hepatocellular carcinoma, breast cancer | [195,196] | |
| Upregulation of DRP1 and downregulation of MFN2 | Lung cancer | [197] | |
| The mitochondrial protein FUNDC2 inhibits MFN1 | Hepatocellular carcinoma | [198] | |
| MFN1 frameshift mutations | Colorectal cancer | [199] | |
| Downregulation of MFN2 | Breast cancer, lung cancer, bladder cancer | [200,201] | |
| Upregulation of OPA1 and MFN1 | Lung adenocarcinoma | [202] | |
| Upregulation of fission factor | Hepatocellular carcinoma | [203] | |
| Upregulation of FIS1 | Oral melanoma, acute myeloid leukemia | [204,205] | |
| Mitophagy | High expression of mucin 1 (MUC1) protects PINK1 from cleavage, thereby increasing mitochondrial autophagy | Breast cancer | [206] |
| Upregulation of sequestosome 1 (SQSTM1/p62) | Hepatocellular carcinoma | [207] | |
| Upregulation of PINK1 | Esophageal squamous cell carcinoma, non-small cell lung cancer | [208,209] | |
| Downregulation of Pankin | Colorectal cancer, clear-cell renal cell carcinoma, oropharyngeal squamous cell carcinoma, pancreatic ductal adenocarcinoma | [210,211,212,213] | |
| Upregulation of BNIP3 | Renal cell carcinoma | [214] | |
| Upregulation of FUNDC1 | Breast cancer, cervical cancer | [215,216] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, W.; Saqirile; Li, K.; Li, K.; Wang, C. The Role of N6-Methyladenosine in Mitochondrial Dysfunction and Pathology. Int. J. Mol. Sci. 2025, 26, 3624. https://doi.org/10.3390/ijms26083624
Yan W, Saqirile, Li K, Li K, Wang C. The Role of N6-Methyladenosine in Mitochondrial Dysfunction and Pathology. International Journal of Molecular Sciences. 2025; 26(8):3624. https://doi.org/10.3390/ijms26083624
Chicago/Turabian StyleYan, Wenxin, Saqirile, Ke Li, Kexin Li, and Changshan Wang. 2025. "The Role of N6-Methyladenosine in Mitochondrial Dysfunction and Pathology" International Journal of Molecular Sciences 26, no. 8: 3624. https://doi.org/10.3390/ijms26083624
APA StyleYan, W., Saqirile, Li, K., Li, K., & Wang, C. (2025). The Role of N6-Methyladenosine in Mitochondrial Dysfunction and Pathology. International Journal of Molecular Sciences, 26(8), 3624. https://doi.org/10.3390/ijms26083624