
Disruption of Man-6-P-Dependent Sorting to Lysosomes Confers IGF1R-Mediated Apoptosis Resistance

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
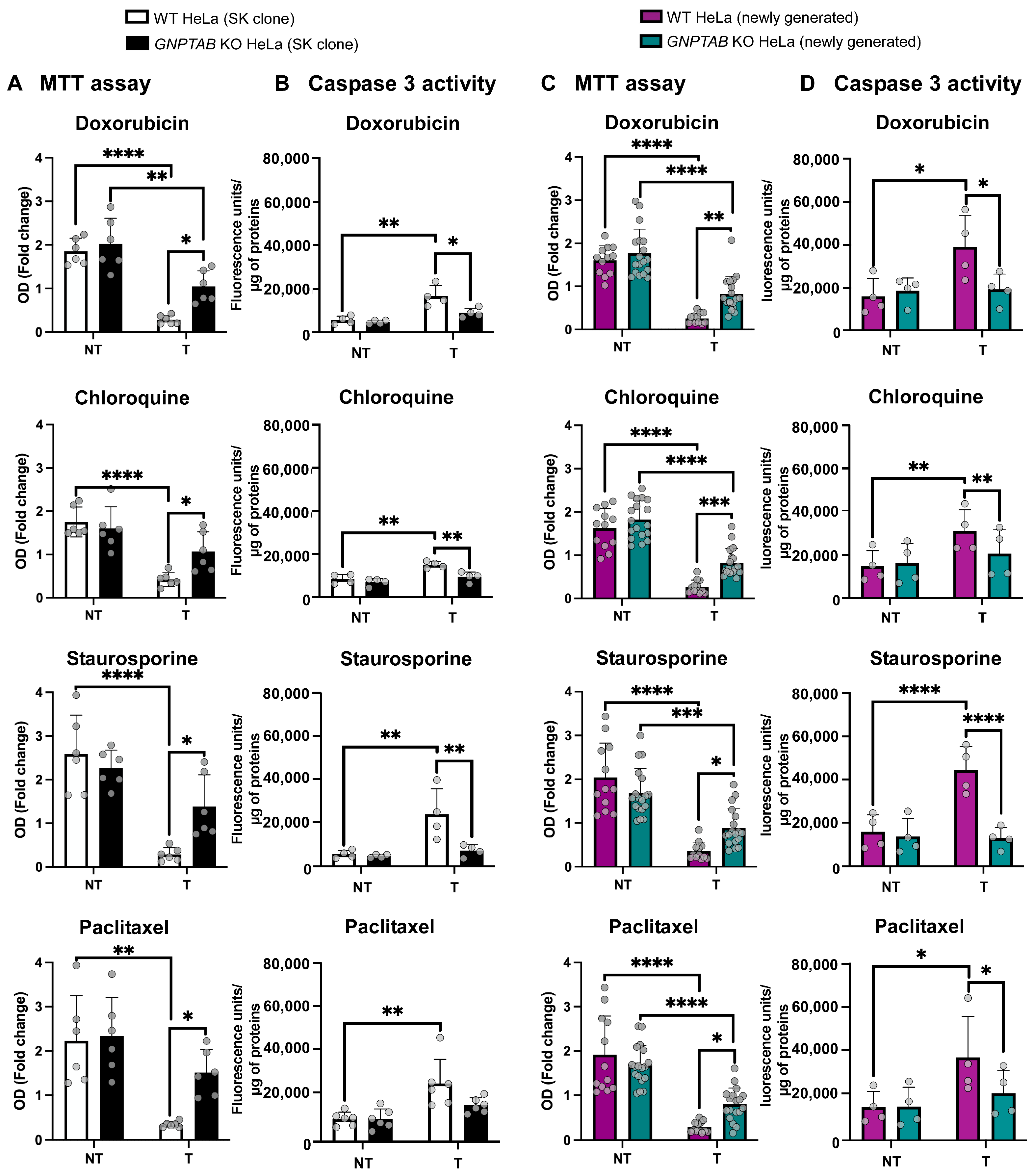
2.1. GNPTAB Knockout HeLa Cells Exhibit Resistance Against Several Cytotoxic Molecules
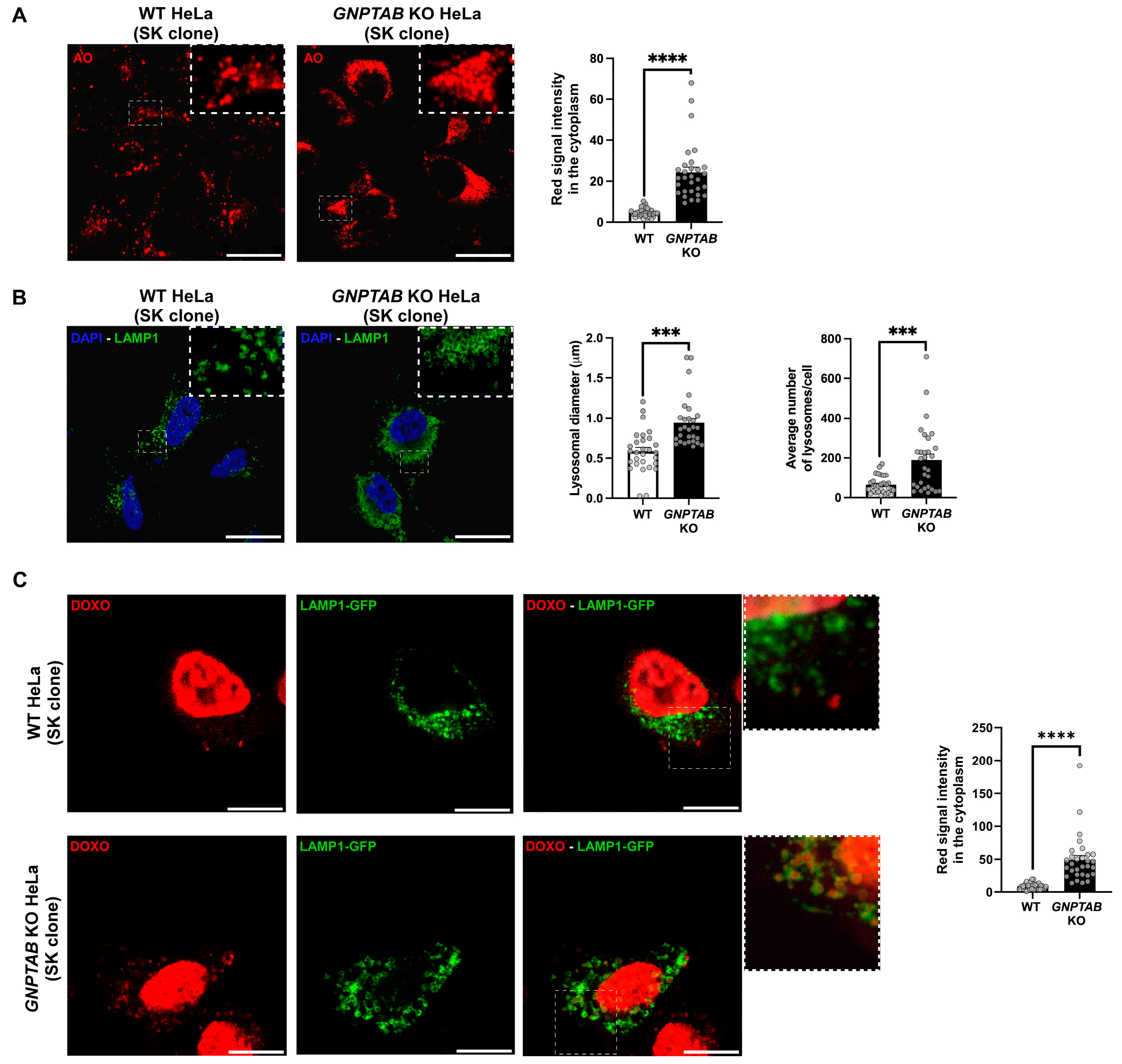
2.2. Investigation of Putative Resistance Mechanism(s)
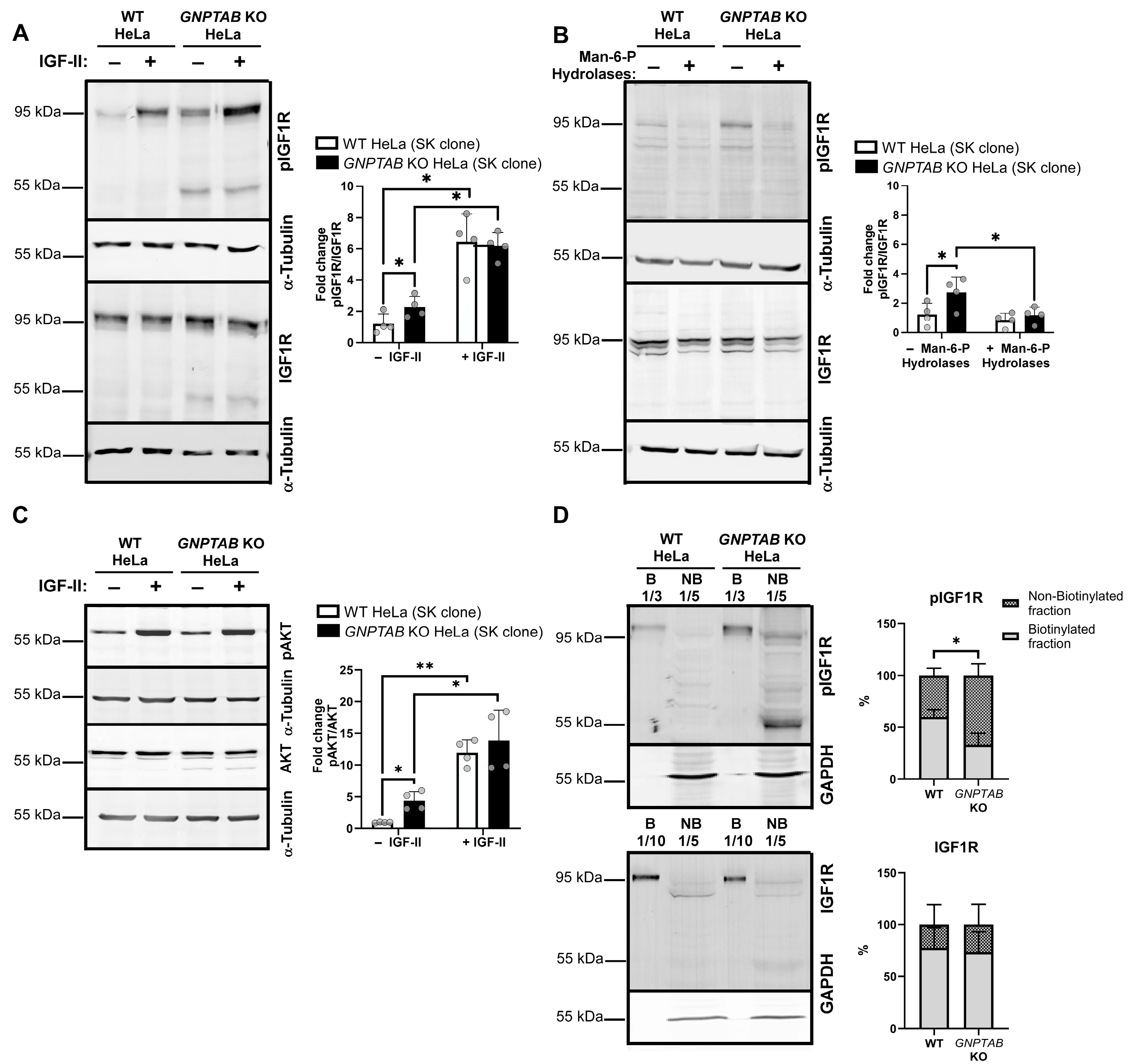
2.3. The IGF1R Pathway Is Hyperactivated in GNPTAB Knockout HeLa Cells
2.4. The IGF1R Signaling Pathway Is Involved in the Resistance of GNPTAB Knockout Cells to Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Treatments and Transient Transfections
4.2. MTT Assay
4.3. Caspase 3/7 Assay
4.4. Engineering of Additional GNPTAB KO HeLa Clones
4.5. Western Blotting
4.6. Fluorescence Microscopy Analyses
4.7. Cell-Surface Biotinylation Assay
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kornfeld, S. Trafficking of lysosomal enzymes. Faseb J. 1987, 1, 462–468. [Google Scholar] [CrossRef] [PubMed]
- von Figura, K.; Hasilik, A. Lysosomal enzymes and their receptors. Annu. Rev. Biochem. 1986, 55, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, S.; Mellman, I. The biogenesis of lysosomes. Annu. Rev. Cell Biol. 1989, 5, 483–525. [Google Scholar] [CrossRef] [PubMed]
- Braulke, T.; Carette, J.E.; Palm, W. Lysosomal enzyme trafficking: From molecular mechanisms to human diseases. Trends Cell Biol. 2024, 34, 198–210. [Google Scholar] [CrossRef]
- Reitman, M.L.; Kornfeld, S. UDP-N-acetylglucosamine:glycoprotein N-acetylglucosamine-1-phosphotransferase. Proposed enzyme for the phosphorylation of the high mannose oligosaccharide units of lysosomal enzymes. J. Biol. Chem. 1981, 256, 4275–4281. [Google Scholar] [CrossRef]
- Pohlmann, R.; Waheed, A.; Hasilik, A.; von Figura, K. Synthesis of phosphorylated recognition marker in lysosomal enzymes is located in the cis part of Golgi apparatus. J. Biol. Chem. 1982, 257, 5323–5325. [Google Scholar] [CrossRef]
- Varki, A.; Kornfeld, S. Identification of a rat liver alpha-N-acetylglucosaminyl phosphodiesterase capable of removing "blocking" alpha-N-acetylglucosamine residues from phosphorylated high mannose oligosaccharides of lysosomal enzymes. J. Biol. Chem. 1980, 255, 8398–8401. [Google Scholar] [CrossRef]
- Qian, Y.; Lee, I.; Lee, W.S.; Qian, M.; Kudo, M.; Canfield, W.M.; Lobel, P.; Kornfeld, S. Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J. Biol. Chem. 2010, 285, 3360–3370. [Google Scholar] [CrossRef]
- Kornfeld, S.S.W. I-Cell Disease and Pseudo-Hurler Polydystrophy: Disorders of Lysosomal Enzyme Phosphorylation and Localization in The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; National Institute of Health: Bethesda, MD, USA, 2001; pp. 3421–3452. [Google Scholar]
- Braulke, T.; Raas-Rothschild, A.; Kornfeld, S. I-Cell Disease and Pseudo-Hurler Polydystrophy: Disorders of Lysosomal Enzyme Phosphorylation and Localization. In The Online Metabolic and Molecular Bases of Inherited Diseas; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Reitman, M.L.; Varki, A.; Kornfeld, S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5′-diphosphate-N-acetylglucosamine: Glycoprotein N-acetylglucosaminylphosphotransferase activity. J. Clin. Investig. 1981, 67, 1574–1579. [Google Scholar] [CrossRef]
- Bach, G.; Bargal, R.; Cantz, M. I-cell disease: Deficiency of extracellular hydrolase phosphorylation. Biochem. Biophys. Res. Commun. 1979, 91, 976–981. [Google Scholar] [CrossRef]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Neuzil, J.; Kågedal, K.; Öllinger, K.; Brunk, U.T. Decreased Apoptotic Response of Inclusion-Cell Disease Fibroblasts: A Consequence of Lysosomal Enzyme Missorting? Exp. Cell Res. 2002, 274, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Tyynelä, J.; Hasilik, A.; Levade, T.; Andrieu-Abadie, N. Stress-induced apoptosis is impaired in cells with a lysosomal targeting defect but is not affected in cells synthesizing a catalytically inactive cathepsin D. Cell Death Differ. 2003, 10, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef]
- Kavčič, N.; Pegan, K.; Turk, B. Lysosomes in programmed cell death pathways: From initiators to amplifiers. Biol. Chem. 2017, 398, 289–301. [Google Scholar] [CrossRef]
- Hanewinkel, H.; Glössl, J.; Kresse, H. Biosynthesis of cathepsin B in cultured normal and I-cell fibroblasts. J. Biol. Chem. 1987, 262, 12351–12355. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D--many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2009, 1793, 605–614. [Google Scholar] [CrossRef]
- Staudt, C.; Puissant, E.; Boonen, M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci. 2017, 18, 47. [Google Scholar] [CrossRef]
- van Meel, E.; Lee, W.S.; Liu, L.; Qian, Y.; Doray, B.; Kornfeld, S. Multiple Domains of GlcNAc-1-phosphotransferase Mediate Recognition of Lysosomal Enzymes. J. Biol. Chem. 2016, 291, 8295–8307. [Google Scholar] [CrossRef]
- Goldman, S.D.; Funk, R.S.; Rajewski, R.A.; Krise, J.P. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis 2009, 1, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R.; Hongo, A.; Rubini, M.; Prisco, M.; Valentinis, B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim. Biophys. Acta 1997, 1332, F105–F126. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yin, Z.; Tao, K.; Wang, G.; Gao, J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol. Lett. 2018, 15, 41–47. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.G.; Pfeffer, S.R.; Coussens, L.; Tepper, M.A.; Brocklebank, C.M.; Mole, J.E.; Anderson, J.K.; Chen, E.; Czech, M.P.; Ullrich, A. A Single Receptor Binds Both Insulin-Like Growth Factor II and Mannose-6-Phosphate. Science 1988, 239, 1134–1137. [Google Scholar] [CrossRef]
- York, S.J.; Arneson, L.S.; Gregory, W.T.; Dahms, N.M.; Kornfeld, S. The rate of internalization of the mannose 6-phosphate/insulin-like growth factor II receptor is enhanced by multivalent ligand binding. J. Biol. Chem. 1999, 274, 1164–1171. [Google Scholar] [CrossRef]
- Osipo, C.; Dorman, S.; Frankfater, A. Loss of Insulin-like Growth Factor II Receptor Expression Promotes Growth in Cancer by Increasing Intracellular Signaling from both IGF-I and Insulin Receptors. Exp. Cell Res. 2001, 264, 388–396. [Google Scholar] [CrossRef]
- Zavorka, M.E.; Connelly, C.M.; Grosely, R.; MacDonald, R.G. Inhibition of insulin-like growth factor II (IGF-II)-dependent cell growth by multidentate pentamannosyl 6-phosphate-based ligands targeting the mannose 6-phosphate/IGF-II receptor. Oncotarget 2016, 7, 62386–62410. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.-I. 40 Years of IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef]
- Soori, M.; Lu, G.; Mason, R.W. Cathepsin Inhibition Prevents Autophagic Protein Turnover and Downregulates Insulin Growth Factor-1 Receptor-Mediated Signaling in Neuroblastoma. J. Pharmacol. Exp. Ther. 2016, 356, 375–386. [Google Scholar] [CrossRef]
- Sun, P.; Sleat, D.E.; Lecocq, M.; Hayman, A.R.; Jadot, M.; Lobel, P. Acid phosphatase 5 is responsible for removing the mannose 6-phosphate recognition marker from lysosomal proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 16590–16595. [Google Scholar] [CrossRef]
- García-Echeverría, C.; Pearson, M.A.; Marti, A.; Meyer, T.; Mestan, J.; Zimmermann, J.; Gao, J.; Brueggen, J.; Capraro, H.-G.; Cozens, R.; et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 2004, 5, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Warshamana-Greene, G.S.; Litz, J.; Buchdunger, E.; García-Echeverría, C.; Hofmann, F.; Krystal, G.W. The Insulin-Like Growth Factor-I Receptor Kinase Inhibitor, NVP-ADW742, Sensitizes Small Cell Lung Cancer Cell Lines to the Effects of Chemotherapy. Clin. Cancer Res. 2005, 11, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Scriver, C.R.; Beaudet, A.L.; Sly, W.S.; Valle, D.; Childs, B.; Kinzler, K.W.; Vogelstein, B. (Eds.) The Metabolic and Molecular Bases of Inherited Disease. Biochemistry 2001, 67, 611–612. [Google Scholar]
- Gelfman, C.M.; Vogel, P.; Issa, T.M.; Turner, C.A.; Lee, W.S.; Kornfeld, S.; Rice, D.S. Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5221–5228. [Google Scholar] [CrossRef]
- van Meel, E.; Boonen, M.; Zhao, H.; Oorschot, V.; Ross, F.P.; Kornfeld, S.; Klumperman, J. Disruption of the Man-6-P targeting pathway in mice impairs osteoclast secretory lysosome biogenesis. Traffic 2011, 12, 912–924. [Google Scholar] [CrossRef]
- Encarnação, M.; Kollmann, K.; Trusch, M.; Braulke, T.; Pohl, S. Post-translational modifications of the gamma-subunit affect intracellular trafficking and complex assembly of GlcNAc-1-phosphotransferase. J. Biol. Chem. 2011, 286, 5311–5318. [Google Scholar] [CrossRef]
- Kollmann, K.; Pestka, J.M.; Kühn, S.C.; Schöne, E.; Schweizer, M.; Karkmann, K.; Otomo, T.; Catala-Lehnen, P.; Failla, A.V.; Marshall, R.P.; et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol. Med. 2013, 5, 1871–1886. [Google Scholar] [CrossRef]
- Paton, L.; Bitoun, E.; Kenyon, J.; Priestman, D.A.; Oliver, P.L.; Edwards, B.; Platt, F.M.; Davies, K.E. A novel mouse model of a patient mucolipidosis II mutation recapitulates disease pathology. J. Biol. Chem. 2014, 289, 26709–26721. [Google Scholar] [CrossRef]
- Han, T.-U.; Root, J.; Reyes, L.D.; Huchinson, E.B.; Hoffmann, J.d.; Lee, W.-S.; Barnes, T.D.; Drayna, D. Human GNPTAB stuttering mutations engineered into mice cause vocalization deficits and astrocyte pathology in the corpus callosum. Proc. Natl. Acad. Sci. USA 2019, 116, 17515–17524. [Google Scholar] [CrossRef]
- Idol, R.A.; Wozniak, D.F.; Fujiwara, H.; Yuede, C.M.; Ory, D.S.; Kornfeld, S.; Vogel, P. Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ. PLoS ONE 2014, 9, e109768. [Google Scholar] [CrossRef] [PubMed]
- Westermann, L.M.; Fleischhauer, L.; Vogel, J.; Jenei-Lanzl, Z.; Ludwig, N.F.; Schau, L.; Morellini, F.; Baranowsky, A.; Yorgan, T.A.; Di Lorenzo, G.; et al. Imbalanced cellular metabolism compromises cartilage homeostasis and joint function in a mouse model of mucolipidosis type III gamma. Dis. Model. Mech. 2020, 13, dmm046425. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.M.; Stewart, C.E.; Liu, Z.; Bhatt, H.; Rotwein, P.; Stewart, C.L. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994, 8, 2953–2963. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Eggenschwiler, J.; Fisher, P.; D’Ercole, A.J.; Davenport, M.L.; Efstratiadis, A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev. Biol. 1996, 177, 517–535. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef]
- Antonsson, A.; Persson, J.L. Induction of apoptosis by staurosporine involves the inhibition of expression of the major cell cycle proteins at the G(2)/m checkpoint accompanied by alterations in Erk and Akt kinase activities. Anticancer. Res. 2009, 29, 2893–2898. [Google Scholar]
- Khing, T.M.; Choi, W.S.; Kim, D.M.; Po, W.W.; Thein, W.; Shin, C.Y.; Sohn, U.D. The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells. Sci. Rep. 2021, 11, 23490. [Google Scholar] [CrossRef]
- Repnik, U.; Česen, M.H.; Turk, B. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion 2014, 19, 49–57. [Google Scholar] [CrossRef]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Boonen, M.; Staudt, C.; Gilis, F.; Oorschot, V.; Klumperman, J.; Jadot, M. Cathepsin D and its newly identified transport receptor SEZ6L2 can modulate neurite outgrowth. J. Cell Sci. 2016, 129, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Doray, B. Elevated mRNA expression and defective processing of cathepsin D in HeLa cells lacking the mannose 6-phosphate pathway. FEBS Open Bio 2021, 11, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Autefage, H.; Garcia, V.; Levade, T.; Andrieu-Abadie, N. Mannose 6-phosphorylated proteins are required for tumor necrosis factor-induced apoptosis: Defective response in I-cell disease fibroblasts. J. Biol. Chem. 2004, 279, 52914–52923. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a008656. [Google Scholar] [CrossRef]
- Arends, M.J.; Wyllie, A.H. Apoptosis: Mechanisms and roles in pathology. Int. Rev. Exp. Pathol. 1991, 32, 223–254. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Hoes, M.F.; Tromp, J.; Ouwerkerk, W.; Bomer, N.; Oberdorf-Maass, S.U.; Samani, N.J.; Ng, L.L.; Lang, C.C.; van der Harst, P.; Hillege, H.; et al. The role of cathepsin D in the pathophysiology of heart failure and its potentially beneficial properties: A translational approach. Eur. J. Heart Fail. 2020, 22, 2102–2111. [Google Scholar] [CrossRef]
- Liu, L.; Chen, B.; Zhang, X.; Tan, L.; Wang, D.W. Increased Cathepsin D Correlates with Clinical Parameters in Newly Diagnosed Type 2 Diabetes. Dis. Markers 2017, 2017, 5286408. [Google Scholar] [CrossRef]
- Morena, F.; Argentati, C.; Trotta, R.; Crispoltoni, L.; Stabile, A.; Pistilli, A.; di Baldassarre, A.; Calafiore, R.; Montanucci, P.; Basta, G.; et al. A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches. Int. J. Mol. Sci. 2017, 18, 1806. [Google Scholar] [CrossRef]
- Baldwin, A.C.; Naatz, A.; Bohnsack, R.N.; Bartosiak, J.T.; Oleson, B.J.; Hansen, P.A.; Dahms, N.M.; Corbett, J.A. Cation-Independent Mannose 6-Phosphate Receptor Deficiency Enhances β-Cell Susceptibility to Palmitate. Mol. Cell Biol. 2018, 38, e00680-17. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, N.K.; Kingham, J.G.; Lloyd, B.; Lloyd, R.S.; Price, C.P.; Triger, D.R.; Wright, R. Acid hydrolases in monocytes from patients with inflammatory bowel disease, chronic liver disease, and rheumatoid arthritis. Lancet 1978, 1, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, R.J.; Niedbala, M.J.; Korytnyk, W. Glycosidases in cancer and invasion. Cancer Metastasis Rev. 1985, 4, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Wang, T.; Wiggins, K.; Lu, P.N.; Underwood, C.; Ochenkowska, K.; Samarut, E.; Pollard, L.M.; Flanagan-Steet, H.; Steet, R. Dysregulated lysosomal exocytosis drives protease-mediated cartilage pathogenesis in multiple lysosomal disorders. iScience 2024, 27, 109293. [Google Scholar] [CrossRef]
- Park, S.; Ahuja, M.; Kim, M.S.; Brailoiu, G.C.; Jha, A.; Zeng, M.; Baydyuk, M.; Wu, L.G.; Wassif, C.A.; Porter, F.D.; et al. Fusion of lysosomes with secretory organelles leads to uncontrolled exocytosis in the lysosomal storage disease mucolipidosis type IV. EMBO Rep. 2016, 17, 266–278. [Google Scholar] [CrossRef]
- Machado, E.R.; Annunziata, I.; van de Vlekkert, D.; Grosveld, G.C.; d’Azzo, A. Lysosomes and Cancer Progression: A Malignant Liaison. Front. Cell Dev. Biol. 2021, 9, 642494. [Google Scholar] [CrossRef]
- Makrypidi, G.; Damme, M.; Müller-Loennies, S.; Trusch, M.; Schmidt, B.; Schlüter, H.; Heeren, J.; Lübke, T.; Saftig, P.; Braulke, T. Mannose 6 dephosphorylation of lysosomal proteins mediated by acid phosphatases Acp2 and Acp5. Mol. Cell Biol. 2012, 32, 774–782. [Google Scholar] [CrossRef]
- Falcón-Pérez, J.M.; Nazarian, R.; Sabatti, C.; Dell’Angelica, E.C. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J. Cell Sci. 2005, 118 (Pt 22), 5243–5255. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aynaci, A.; Toussaint, M.; Gilis, F.; Albert, M.; Gaussin, J.-F.; Jadot, M.; Boonen, M. Disruption of Man-6-P-Dependent Sorting to Lysosomes Confers IGF1R-Mediated Apoptosis Resistance. Int. J. Mol. Sci. 2025, 26, 3586. https://doi.org/10.3390/ijms26083586
Aynaci A, Toussaint M, Gilis F, Albert M, Gaussin J-F, Jadot M, Boonen M. Disruption of Man-6-P-Dependent Sorting to Lysosomes Confers IGF1R-Mediated Apoptosis Resistance. International Journal of Molecular Sciences. 2025; 26(8):3586. https://doi.org/10.3390/ijms26083586
Chicago/Turabian StyleAynaci, Asena, Maxence Toussaint, Florentine Gilis, Martine Albert, Jean-François Gaussin, Michel Jadot, and Marielle Boonen. 2025. "Disruption of Man-6-P-Dependent Sorting to Lysosomes Confers IGF1R-Mediated Apoptosis Resistance" International Journal of Molecular Sciences 26, no. 8: 3586. https://doi.org/10.3390/ijms26083586
APA StyleAynaci, A., Toussaint, M., Gilis, F., Albert, M., Gaussin, J.-F., Jadot, M., & Boonen, M. (2025). Disruption of Man-6-P-Dependent Sorting to Lysosomes Confers IGF1R-Mediated Apoptosis Resistance. International Journal of Molecular Sciences, 26(8), 3586. https://doi.org/10.3390/ijms26083586