_Kim.png)

The Burning Pain Transcriptome in the Mouse Primary Somatosensory Cortex




Abstract
1. Introduction
2. Results
2.1. Burn Injury Significantly Alters the Activity of Different Cortical Neuron Populations in the S1 Cortex, as Assessed by c-Fos Immunohistochemistry
2.2. Burn Injury Is Accompanied by Moderate Transcriptomic Changes in the Primary Somatosensory Cortex as Early as One-Hour Post-Burn Injury
2.3. Differential Cortical Mechanisms in Acute Pain Processing Within the Primary Somatosensory Cortex Following Burn Injury and Inflammatory Pain
2.4. Differential Pain-Related Signaling in the S1 Cortex: Insights from KEGG Pathway Analysis Across Sensory Modalities
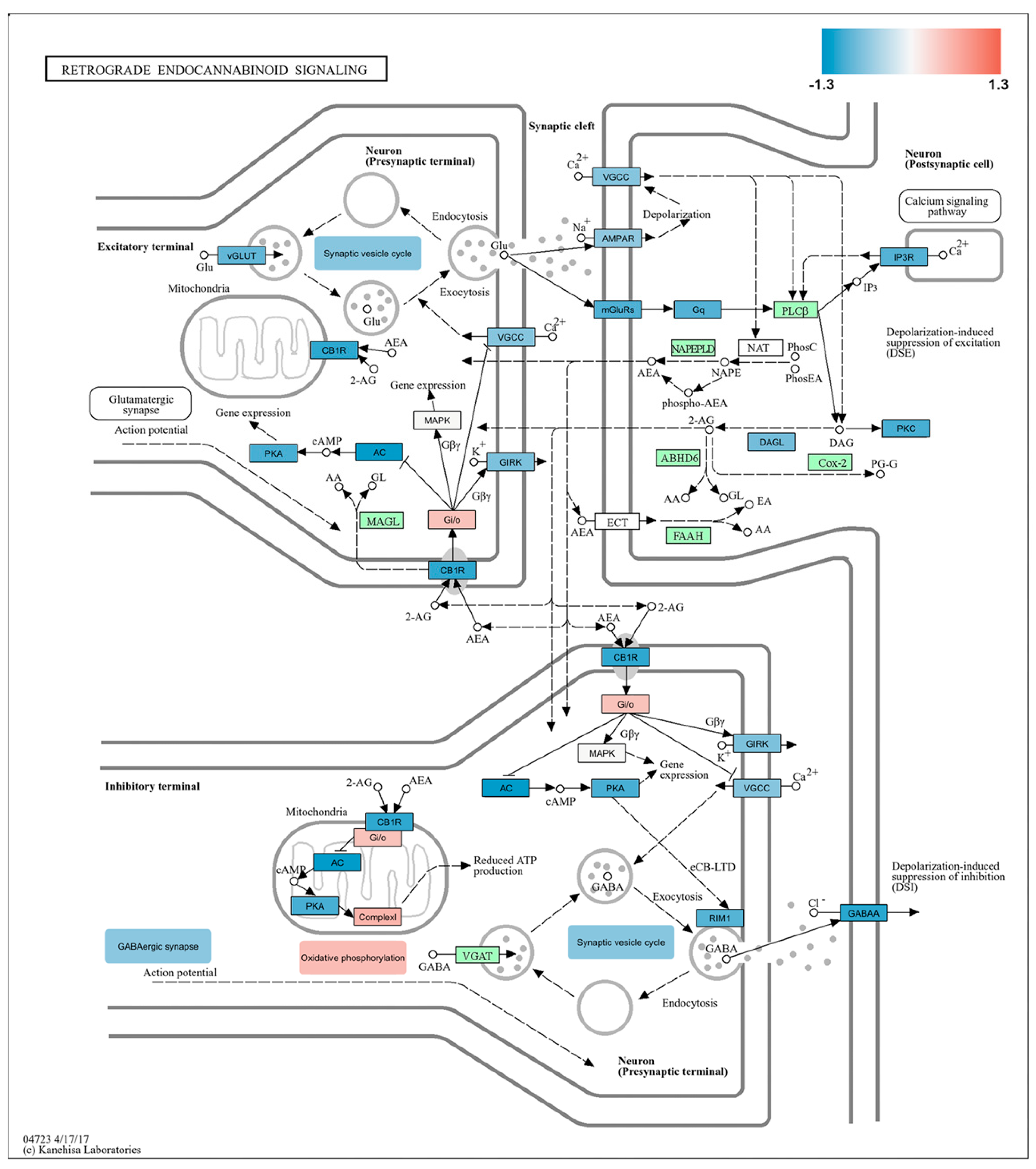
2.5. Impact of Burn Injury on the Components of the Retrograde Endocannabinoid Network in the S1 Cortex
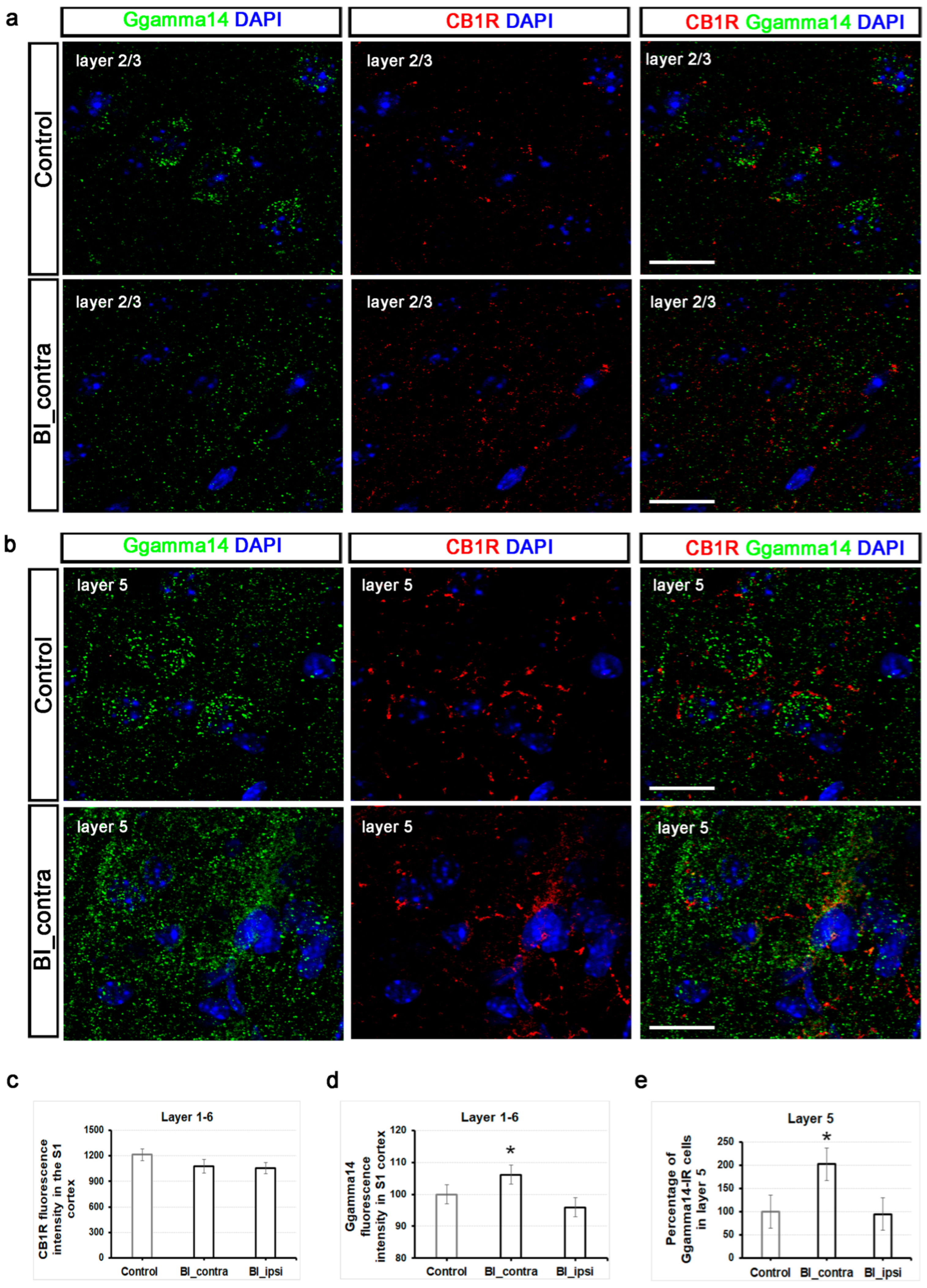
2.6. Unlike CB1 Receptors, Ggamma14 Fluorescence Intensity in the S1 Cortex and the Number of Ggamma14-IR Cells in Layer 5 of the S1 Cortex Show Significant Alterations One Hour After Burn Injury
3. Discussion
4. Materials and Methods
4.1. Animals, Ethical Considerations, and Study Design
4.2. Establishment of Burn Injury Model
4.3. Establishment of Formalin-Induced Inflammatory Pain Model
4.4. Immunofluorescent STAINING
4.5. Total RNA Isolation and RNA-Seq Analysis
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allahham, A.; Rowe, G.; Stevenson, A.; Fear, M.W.; Vallence, A.-M.; Wood, F.M. The Impact of Burn Injury on the Central Nervous System. Burns Trauma 2024, 12, tkad037. [Google Scholar] [CrossRef] [PubMed]
- Vetrichevvel, T.P.; Randall, S.M.; Fear, M.W.; Wood, F.M.; Boyd, J.H.; Duke, J.M. Burn Injury and Long-Term Nervous System Morbidity: A Population-Based Cohort Study. BMJ Open 2016, 6, e012668. [Google Scholar] [CrossRef]
- Xie, C.; Hu, J.; Cheng, Y.; Yao, Z. Researches on Cognitive Sequelae of Burn Injury: Current Status and Advances. Front. Neurosci. 2022, 16, 1026152. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, D.; Zhang, J.; Wang, Y. Pathological Changes in the Brain after Peripheral Burns. Burns Trauma 2023, 11, tkac061. [Google Scholar] [CrossRef]
- Seifert, F.; Maihöfner, C. Central Mechanisms of Experimental and Chronic Neuropathic Pain: Findings from Functional Imaging Studies. Cell. Mol. Life Sci. CMLS 2009, 66, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.S.; Park, H.; Kim, S.K. Neural Plasticity in the Brain during Neuropathic Pain. Biomedicines 2021, 9, 624. [Google Scholar] [CrossRef]
- Ong, W.-Y.; Stohler, C.S.; Herr, D.R. Role of the Prefrontal Cortex in Pain Processing. Mol. Neurobiol. 2019, 56, 1137–1166. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; McCartin, M.; Liu, W.; Zhang, Q.; Kenefati, G.; Chen, Z.S.; Wang, J. Temporal Pain Processing in the Primary Somatosensory Cortex and Anterior Cingulate Cortex. Mol. Brain 2023, 16, 3. [Google Scholar] [CrossRef]
- Quiton, R.L.; Masri, R.; Thompson, S.M.; Keller, A. Abnormal Activity of Primary Somatosensory Cortex in Central Pain Syndrome. J. Neurophysiol. 2010, 104, 1717–1725. [Google Scholar] [CrossRef]
- Timmermann, L.; Ploner, M.; Haucke, K.; Schmitz, F.; Baltissen, R.; Schnitzler, A. Differential Coding of Pain Intensity in the Human Primary and Secondary Somatosensory Cortex. J. Neurophysiol. 2001, 86, 1499–1503. [Google Scholar] [CrossRef]
- Tao, W.; Chen, C.; Wang, Y.; Zhou, W.; Jin, Y.; Mao, Y.; Wang, H.; Wang, L.; Xie, W.; Zhang, X.; et al. MeCP2 Mediates Transgenerational Transmission of Chronic Pain. Prog. Neurobiol. 2020, 189, 101790. [Google Scholar] [CrossRef] [PubMed]
- Orczyk, J.J.; Sethia, R.; Doster, D.; Garraghty, P.E. Transcriptome Response to Infraorbital Nerve Transection in the Gonadally Intact Male Rat Barrel Cortex: RNA-Seq. J. Comp. Neurol. 2016, 524, 152–159. [Google Scholar] [CrossRef]
- Ping, X.; Xie, J.; Yuan, C.; Jin, X. Electroacupuncture Induces Bilateral S1 and ACC Epigenetic Regulation of Genes in a Mouse Model of Neuropathic Pain. Biomedicines 2023, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Labus, J.S.; Osadchiy, V.; Hsiao, E.Y.; Tap, J.; Derrien, M.; Gupta, A.; Tillisch, K.; Le Nevé, B.; Grinsvall, C.; Ljungberg, M.; et al. Evidence for an Association of Gut Microbial Clostridia with Brain Functional Connectivity and Gastrointestinal Sensorimotor Function in Patients with Irritable Bowel Syndrome, Based on Tripartite Network Analysis. Microbiome 2019, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Blom, S.M.; Pfister, J.-P.; Santello, M.; Senn, W.; Nevian, T. Nerve Injury-Induced Neuropathic Pain Causes Disinhibition of the Anterior Cingulate Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 5754–5764. [Google Scholar] [CrossRef]
- Alvarado, S.; Tajerian, M.; Millecamps, M.; Suderman, M.; Stone, L.S.; Szyf, M. Peripheral Nerve Injury Is Accompanied by Chronic Transcriptome-Wide Changes in the Mouse Prefrontal Cortex. Mol. Pain 2013, 9, 21. [Google Scholar] [CrossRef]
- Zheng, Q.-M.; Zhou, Z.-R.; Hou, X.-Y.; Lv, N.; Zhang, Y.-Q.; Cao, H. Transcriptome Analysis of the Mouse Medial Prefrontal Cortex in a Chronic Constriction Injury Model. Neuromol. Med. 2023, 25, 375–387. [Google Scholar] [CrossRef]
- Li, H.; Li, X.; Wang, J.; Gao, F.; Wiech, K.; Hu, L.; Kong, Y. Pain-Related Reorganization in the Primary Somatosensory Cortex of Patients with Postherpetic Neuralgia. Hum. Brain Mapp. 2022, 43, 5167–5179. [Google Scholar] [CrossRef]
- Sneddon, L.U. Comparative Physiology of Nociception and Pain. Physiol. Bethesda Md 2018, 33, 63–73. [Google Scholar] [CrossRef]
- Mészár, Z.; Erdei, V.; Szücs, P.; Varga, A. Epigenetic Regulation and Molecular Mechanisms of Burn Injury-Induced Nociception in the Spinal Cord of Mice. Int. J. Mol. Sci. 2024, 25, 8510. [Google Scholar] [CrossRef]
- Lee, G.J.; Kim, Y.J.; Lee, K.; Oh, S.B. Patterns of Brain C-Fos Expression in Response to Feeding Behavior in Acute and Chronic Inflammatory Pain Condition. Neuroreport 2021, 32, 1269–1277. [Google Scholar] [CrossRef]
- Li, J.; Qin, Y.; Zhong, Z.; Meng, L.; Huang, L.; Li, B. Pain Experience Reduces Social Avoidance to Others in Pain: A c-Fos-Based Functional Connectivity Network Study in Mice. Cereb. Cortex 2024, 34, bhae207. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Qin, Y.; Yang, S.; Zhang, H.; Zheng, J.; Wen, S.; Li, W.; Liang, Z.; Zhang, X.; Li, B.; et al. Differences in the Neural Basis and Transcriptomic Patterns in Acute and Persistent Pain-Related Anxiety-like Behaviors. Front. Mol. Neurosci. 2023, 16, 1185243. [Google Scholar] [CrossRef] [PubMed]
- Hjornevik, T.; Leergaard, T.B.; Darine, D.; Moldestad, O.; Dale, A.M.; Willoch, F.; Bjaalie, J.G. Three-Dimensional Atlas System for Mouse and Rat Brain Imaging Data. Front. Neuroinform. 2007, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Donvito, G.; Nass, S.R.; Wilkerson, J.L.; Curry, Z.A.; Schurman, L.D.; Kinsey, S.G.; Lichtman, A.H. The Endogenous Cannabinoid System: A Budding Source of Targets for Treating Inflammatory and Neuropathic Pain. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 52–79. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid Signaling in the Brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef]
- Manzanares, J.; Julian, M.; Carrascosa, A. Role of the Cannabinoid System in Pain Control and Therapeutic Implications for the Management of Acute and Chronic Pain Episodes. Curr. Neuropharmacol. 2006, 4, 239–257. [Google Scholar] [CrossRef]
- Bodor, A.L.; Katona, I.; Nyíri, G.; Mackie, K.; Ledent, C.; Hájos, N.; Freund, T.F. Endocannabinoid Signaling in Rat Somatosensory Cortex: Laminar Differences and Involvement of Specific Interneuron Types. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 6845–6856. [Google Scholar] [CrossRef]
- Tennakoon, M.; Senarath, K.; Kankanamge, D.; Ratnayake, K.; Wijayaratna, D.; Olupothage, K.; Ubeysinghe, S.; Martins-Cannavino, K.; Hébert, T.E.; Karunarathne, A. Subtype-Dependent Regulation of Gβγ Signalling. Cell. Signal. 2021, 82, 109947. [Google Scholar] [CrossRef]
- Tennakoon, M.; Senarath, K.; Kankanamge, D.; Chadee, D.N.; Karunarathne, A. A Short C-Terminal Peptide in Gγ Regulates Gβγ Signaling Efficacy. Mol. Biol. Cell 2021, 32, 1446–1458. [Google Scholar] [CrossRef]
- Chen, K.-H.; Lin, C.-R.; Cheng, J.-T.; Cheng, J.-K.; Liao, W.-T.; Yang, C.-H. Altered Mitochondrial ATP Synthase Expression in the Rat Dorsal Root Ganglion after Sciatic Nerve Injury and Analgesic Effects of Intrathecal ATP. Cell. Mol. Neurobiol. 2014, 34, 51–59. [Google Scholar] [CrossRef]
- Hozumi, T.; Sawai, S.; Jitsuishi, T.; Kitajo, K.; Inage, K.; Eguchi, Y.; Shiga, Y.; Narita, M.; Orita, S.; Ohtori, S.; et al. Gene Expression Profiling of the Spinal Cord at the Chronic Pain Phase Identified CDKL5 as a Candidate Gene for Neural Remodeling. Neurosci. Lett. 2021, 749, 135772. [Google Scholar] [CrossRef] [PubMed]
- Schneiders, F.I.; Maertens, B.; Böse, K.; Li, Y.; Brunken, W.J.; Paulsson, M.; Smyth, N.; Koch, M. Binding of Netrin-4 to Laminin Short Arms Regulates Basement Membrane Assembly. J. Biol. Chem. 2007, 282, 23750–23758. [Google Scholar] [CrossRef]
- Kuner, R.; Flor, H. Structural Plasticity and Reorganisation in Chronic Pain. Nat. Rev. Neurosci. 2016, 18, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Dresbach, T.; Hempelmann, A.; Spilker, C.; tom Dieck, S.; Altrock, W.D.; Zuschratter, W.; Garner, C.C.; Gundelfinger, E.D. Functional Regions of the Presynaptic Cytomatrix Protein Bassoon: Significance for Synaptic Targeting and Cytomatrix Anchoring. Mol. Cell. Neurosci. 2003, 23, 279–291. [Google Scholar] [CrossRef]
- Terry-Lorenzo, R.T.; Torres, V.I.; Wagh, D.; Galaz, J.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Waites, C.L.; Gundelfinger, E.D.; Reimer, R.J.; et al. Trio, a Rho Family GEF, Interacts with the Presynaptic Active Zone Proteins Piccolo and Bassoon. PLoS ONE 2016, 11, e0167535. [Google Scholar] [CrossRef] [PubMed]
- Vaskovsky, A.; Lupowitz, Z.; Erlich, S.; Pinkas-Kramarski, R. ErbB-4 Activation Promotes Neurite Outgrowth in PC12 Cells. J. Neurochem. 2000, 74, 979–987. [Google Scholar] [CrossRef]
- Wang, H.; Chen, W.; Dong, Z.; Xing, G.; Cui, W.; Yao, L.; Zou, W.-J.; Robinson, H.L.; Bian, Y.; Liu, Z.; et al. A Novel Spinal Neuron Connection for Heat Sensation. Neuron 2022, 110, 2315–2333.e6. [Google Scholar] [CrossRef]
- Yu, H.; Cranfill, S.L.; Luo, W. ErbB4+ Spinal Cord Dorsal Horn Neurons Process Heat Pain. Neuron 2022, 110, 2206–2208. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Goscik, J.; Swiergiel, A.H.; Majewska, A.; Wieczorek, M.; Juszczak, G.R.; Lisowski, P. Social Stress Increases Expression of Hemoglobin Genes in Mouse Prefrontal Cortex. BMC Neurosci. 2014, 15, 130. [Google Scholar] [CrossRef]
- Sugeno, A.; Piao, W.; Yamazaki, M.; Takahashi, K.; Arikawa, K.; Matsunaga, H.; Hosokawa, M.; Tominaga, D.; Goshima, Y.; Takeyama, H.; et al. Cortical Transcriptome Analysis after Spinal Cord Injury Reveals the Regenerative Mechanism of Central Nervous System in CRMP2 Knock-in Mice. Neural Regen. Res. 2021, 16, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.K.Y.; Rone, M.B.; Lee, S.; Antel, J.P.; Zhang, J. Mitochondrial and Bioenergetic Dysfunction in Trauma-Induced Painful Peripheral Neuropathy. Mol. Pain 2015, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; He, J.; Cui, Z.; Wang, R.; Bao, K.; Huang, Y.; Wang, R.; Liu, T. Central 5-HTR2C in the Control of Metabolic Homeostasis. Front. Endocrinol. 2021, 12, 694204. [Google Scholar] [CrossRef]
- Nakae, A.; Nakai, K.; Tanaka, T.; Hosokawa, K.; Mashimo, T. Serotonin 2C Receptor Alternative Splicing in a Spinal Cord Injury Model. Neurosci. Lett. 2013, 532, 49–54. [Google Scholar] [CrossRef]
- Quintão, N.L.M.; Passos, G.F.; Medeiros, R.; Paszcuk, A.F.; Motta, F.L.; Pesquero, J.B.; Campos, M.M.; Calixto, J.B. Neuropathic Pain-like Behavior after Brachial Plexus Avulsion in Mice: The Relevance of Kinin B1 and B2 Receptors. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2856–2863. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Banerjee, R.; Marinelli, F.; Silberberg, S.; Faraldo-Gómez, J.D.; Hattori, M.; Swartz, K.J. Molecular Mechanisms of Human P2X3 Receptor Channel Activation and Modulation by Divalent Cation Bound ATP. eLife 2019, 8, e47060. [Google Scholar] [CrossRef]
- Prüss, H.; Derst, C.; Lommel, R.; Veh, R.W. Differential Distribution of Individual Subunits of Strongly Inwardly Rectifying Potassium Channels (Kir2 Family) in Rat Brain. Mol. Brain Res. 2005, 139, 63–79. [Google Scholar] [CrossRef]
- Bishnoi, M.; Jain, A.; Hurkat, P.; Jain, S.K. Chondroitin Sulphate: A Focus on Osteoarthritis. Glycoconj. J. 2016, 33, 693–705. [Google Scholar] [CrossRef]
- Stenina-Adognravi, O.; Plow, E.F. Thrombospondin-4 in Tissue Remodeling. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 75–76, 300–313. [Google Scholar] [CrossRef]
- Crosby, N.D.; Zaucke, F.; Kras, J.V.; Dong, L.; Luo, Z.D.; Winkelstein, B.A. Thrombospondin-4 and Excitatory Synaptogenesis Promote Spinal Sensitization after Painful Mechanical Joint Injury. Exp. Neurol. 2015, 264, 111–120. [Google Scholar] [CrossRef]
- Düzenli, N.; Ülker, S.; Şengül, G.; Kayhan, B.; Önal, A. Effects of Cyanocobalamin and Its Combination with Morphine on Neuropathic Rats and the Relationship between These Effects and Thrombospondin-4 Expression. Korean J. Pain 2022, 35, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, M.; Xu, X.; Gao, Y.; Li, X.; Li, Y.; Su, S.; Xie, X.; Yang, Z.; Ke, C. Thrombospondin 4, a Mediator and Candidate Indicator of Pain. Eur. J. Cell Biol. 2024, 103, 151395. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Noriega-Prieto, J.A.; Kofuji, P.; Araque, A. Endocannabinoid Signaling in Synaptic Function. Glia 2023, 71, 36–43. [Google Scholar] [CrossRef]
- Wolf, J.; Urits, I.; Orhurhu, V.; Peck, J.; Orhurhu, M.S.; Giacomazzi, S.; Smoots, D.; Piermarini, C.; Manchikanti, L.; Kaye, A.D.; et al. The Role of the Cannabinoid System in Pain Control: Basic and Clinical Implications. Curr. Pain Headache Rep. 2020, 24, 35. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Pacher, P.; Tegeder, I.; Amaya, F.; Constantin, C.E.; Brenner, G.J.; Rubino, T.; Michalski, C.W.; Marsicano, G.; Monory, K.; et al. Cannabinoids Mediate Analgesia Largely via Peripheral Type 1 Cannabinoid Receptors in Nociceptors. Nat. Neurosci. 2007, 10, 870–879. [Google Scholar] [CrossRef]
- Bishay, P.; Schmidt, H.; Marian, C.; Häussler, A.; Wijnvoord, N.; Ziebell, S.; Metzner, J.; Koch, M.; Myrczek, T.; Bechmann, I.; et al. R-Flurbiprofen Reduces Neuropathic Pain in Rodents by Restoring Endogenous Cannabinoids. PLoS ONE 2010, 5, e10628. [Google Scholar] [CrossRef]
- Wu, J.; Hua, L.; Liu, W.; Yang, X.; Tang, X.; Yuan, S.; Zhou, S.; Ye, Q.; Cui, S.; Wu, Z.; et al. Electroacupuncture Exerts Analgesic Effects by Restoring Hyperactivity via Cannabinoid Type 1 Receptors in the Anterior Cingulate Cortex in Chronic Inflammatory Pain. Mol. Neurobiol. 2024, 61, 2949–2963. [Google Scholar] [CrossRef]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef]
- Howlett, A.C.; Mukhopadhyay, S. Cellular Signal Transduction by Anandamide and 2-Arachidonoylglycerol. Chem. Phys. Lipids 2000, 108, 53–70. [Google Scholar] [CrossRef]
- Xu, Z.; Lv, X.-A.; Dai, Q.; Ge, Y.-Q.; Xu, J. Acute Upregulation of Neuronal Mitochondrial Type-1 Cannabinoid Receptor and It’s Role in Metabolic Defects and Neuronal Apoptosis after TBI. Mol. Brain 2016, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, L.; Urbinati, C.; Lanzillotta, C.; De Rasmo, D.; Valenti, D.; Pellas, M.; Quattrini, M.C.; Piscitelli, F.; Kostrzewa, M.; Di Domenico, F.; et al. Pharmacological Inhibition of the CB1 Cannabinoid Receptor Restores Abnormal Brain Mitochondrial CB1 Receptor Expression and Rescues Bioenergetic and Cognitive Defects in a Female Mouse Model of Rett Syndrome. Mol. Autism 2024, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Colmers, P.L.W.; Bains, J.S. Presynaptic mGluRs Control the Duration of Endocannabinoid-Mediated DSI. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 10444–10453. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB₁ Receptors Regulate Neuronal Energy Metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Harkany, T.; Horvath, T.L. (S)Pot on Mitochondria: Cannabinoids Disrupt Cellular Respiration to Limit Neuronal Activity. Cell Metab. 2017, 25, 8–10. [Google Scholar] [CrossRef]
- Guo, J.; Ikeda, S.R. Endocannabinoids Modulate N-Type Calcium Channels and G-Protein-Coupled Inwardly Rectifying Potassium Channels via CB1 Cannabinoid Receptors Heterologously Expressed in Mammalian Neurons. Mol. Pharmacol. 2004, 65, 665–674. [Google Scholar] [CrossRef]
- Takemura, Y.; Sudo, Y.; Saeki, T.; Kurata, S.; Suzuki, T.; Mori, T.; Uezono, Y. Involvement of Spinal G-Protein Inwardly Rectifying Potassium (GIRK) Channels in the Enhanced Antinociceptive Effects of the Activation of Both μ-Opioid and Cannabinoid CB1 Receptors. J. Pharmacol. Sci. 2022, 149, 85–92. [Google Scholar] [CrossRef]
- Imai, S.; Ikegami, D.; Yamashita, A.; Shimizu, T.; Narita, M.; Niikura, K.; Furuya, M.; Kobayashi, Y.; Miyashita, K.; Okutsu, D.; et al. Epigenetic Transcriptional Activation of Monocyte Chemotactic Protein 3 Contributes to Long-Lasting Neuropathic Pain. Brain J. Neurol. 2013, 136, 828–843. [Google Scholar] [CrossRef]
- Liang, L.; Tao, Y.-X. Expression of Acetyl-Histone H3 and Acetyl-Histone H4 in Dorsal Root Ganglion and Spinal Dorsal Horn in Rat Chronic Pain Models. Life Sci. 2018, 211, 182–188. [Google Scholar] [CrossRef]
- Wang, G.-Q.; Cen, C.; Li, C.; Cao, S.; Wang, N.; Zhou, Z.; Liu, X.-M.; Xu, Y.; Tian, N.-X.; Zhang, Y.; et al. Deactivation of Excitatory Neurons in the Prelimbic Cortex via Cdk5 Promotes Pain Sensation and Anxiety. Nat. Commun. 2015, 6, 7660. [Google Scholar] [CrossRef]
- Varga, A.; Mészár, Z.; Sivadó, M.; Bácskai, T.; Végh, B.; Kókai, É.; Nagy, I.; Szücs, P. Spinal Excitatory Dynorphinergic Interneurons Contribute to Burn Injury-Induced Nociception Mediated by Phosphorylated Histone 3 at Serine 10 in Rodents. Int. J. Mol. Sci. 2021, 22, 2297. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.M.; Ko, C.W.; Fidalgo, A.R.; Cibelli, M.; Paule, C.C.; Anderson, P.J.; Cruz, C.; Gomba, S.; Matesz, K.; Veress, G.; et al. Severe Burn Injury Induces a Characteristic Activation of Extracellular Signal-Regulated Kinase 1/2 in Spinal Dorsal Horn Neurons. Eur. J. Pain Lond. Engl. 2011, 15, 683–690. [Google Scholar] [CrossRef]
- Bruen, K.J.; Campbell, C.A.; Schooler, W.G.; deSerres, S.; Cairns, B.A.; Hultman, C.S.; Meyer, A.A.; Randell, S.H. Real-Time Monitoring of Keratin 5 Expression during Burn Re-Epithelialization. J. Surg. Res. 2004, 120, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Tjølsen, A.; Berge, O.-G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The Formalin Test: An Evaluation of the Method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Mészár, Z.; Kókai, É.; Varga, R.; Ducza, L.; Papp, T.; Béresová, M.; Nagy, M.; Szücs, P.; Varga, A. CRISPR/Cas9-Based Mutagenesis of Histone H3.1 in Spinal Dynorphinergic Neurons Attenuates Thermal Sensitivity in Mice. Int. J. Mol. Sci. 2022, 23, 3178. [Google Scholar] [CrossRef]
- Echeazarra, L.; Barrondo, S.; García Del Caño, G.; Bonilla-Del Río, I.; Egaña-Huguet, J.; Puente, N.; Aretxabala, X.; Montaña, M.; López de Jesús, M.; González-Burguera, I.; et al. Up-Regulation of CB1 Cannabinoid Receptors Located at Glutamatergic Terminals in the Medial Prefrontal Cortex of the Obese Zucker Rat. Front. Neuroanat. 2022, 16, 1004702. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinform. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A Free Online Platform for Data Visualization and Graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
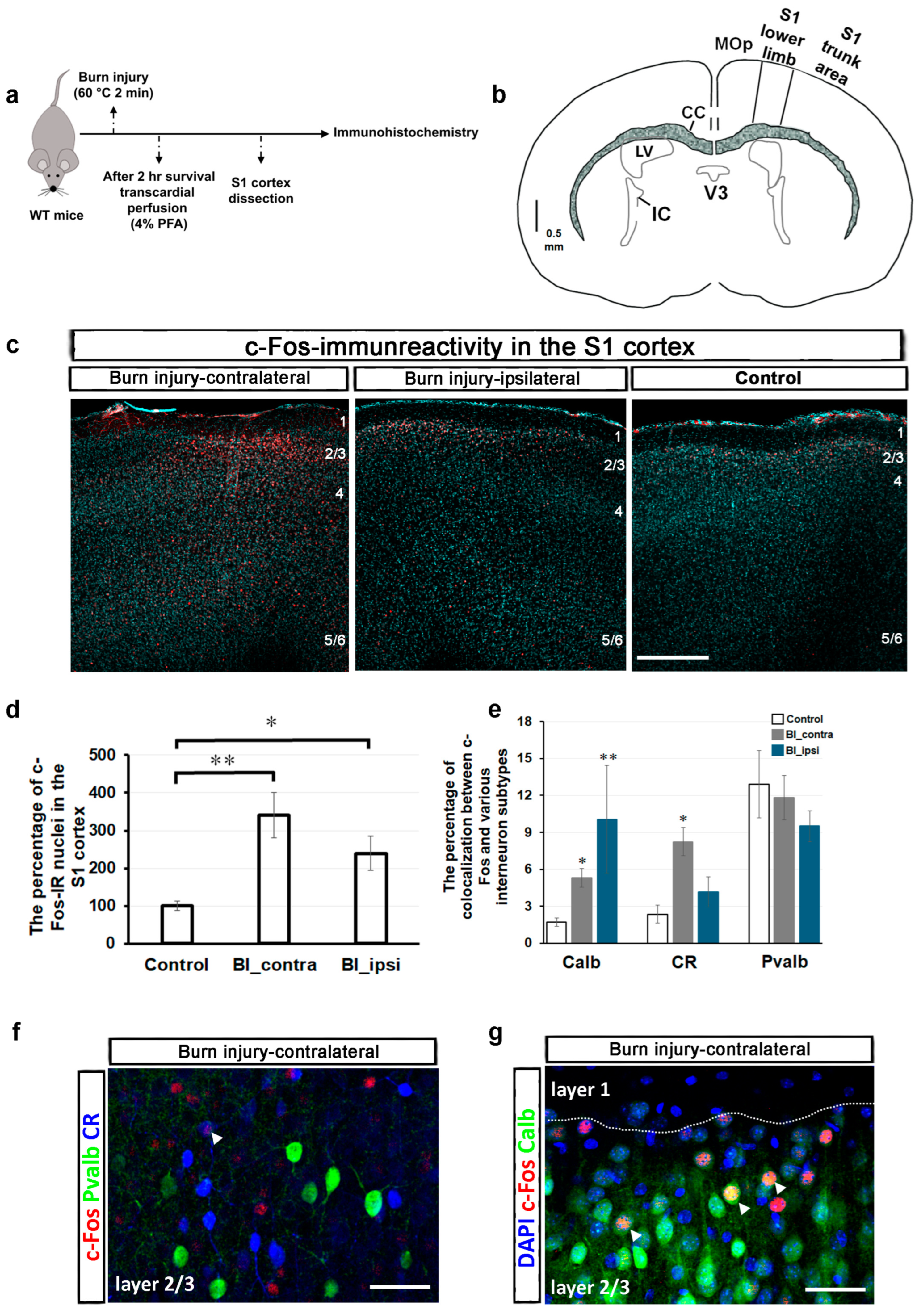
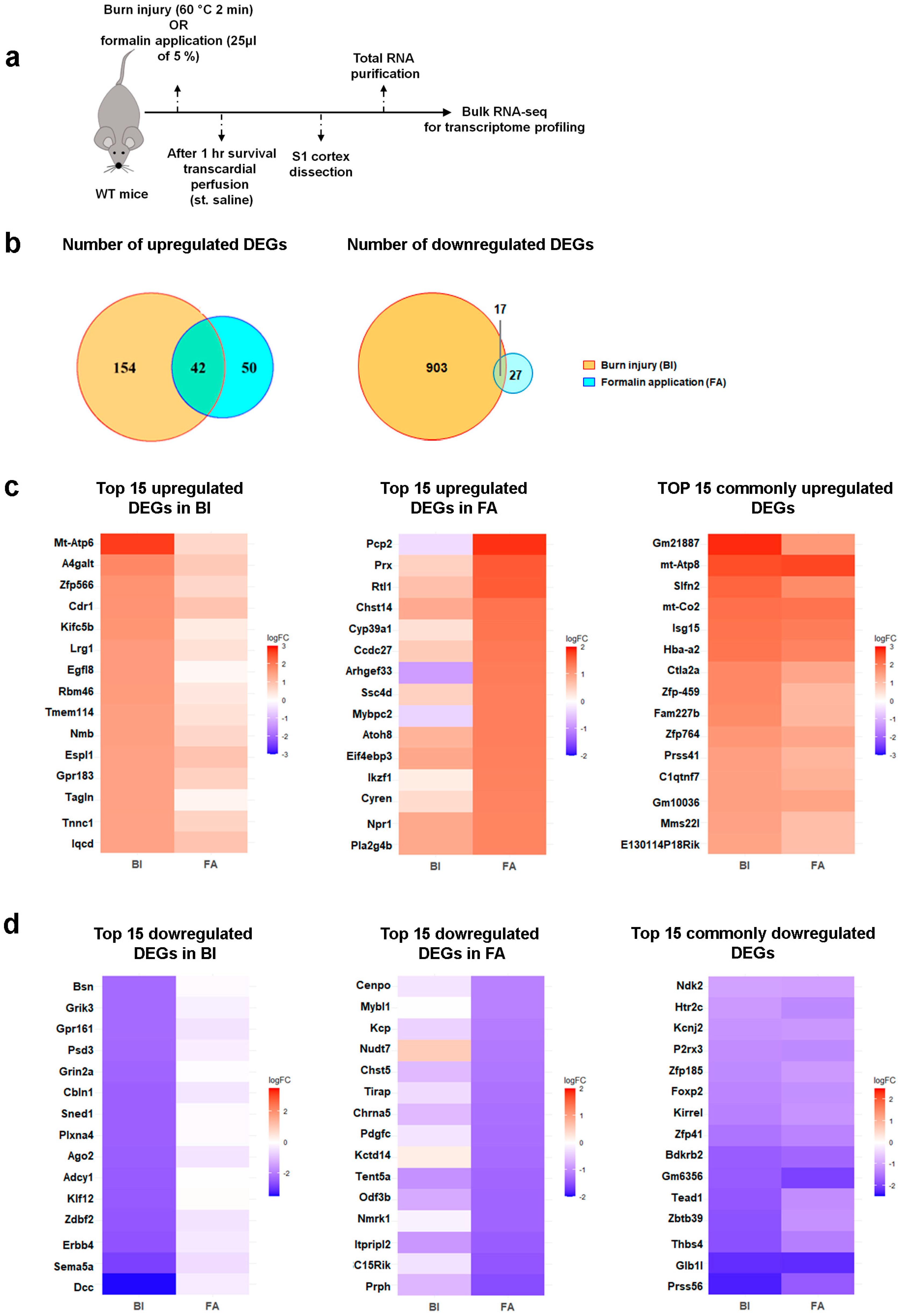
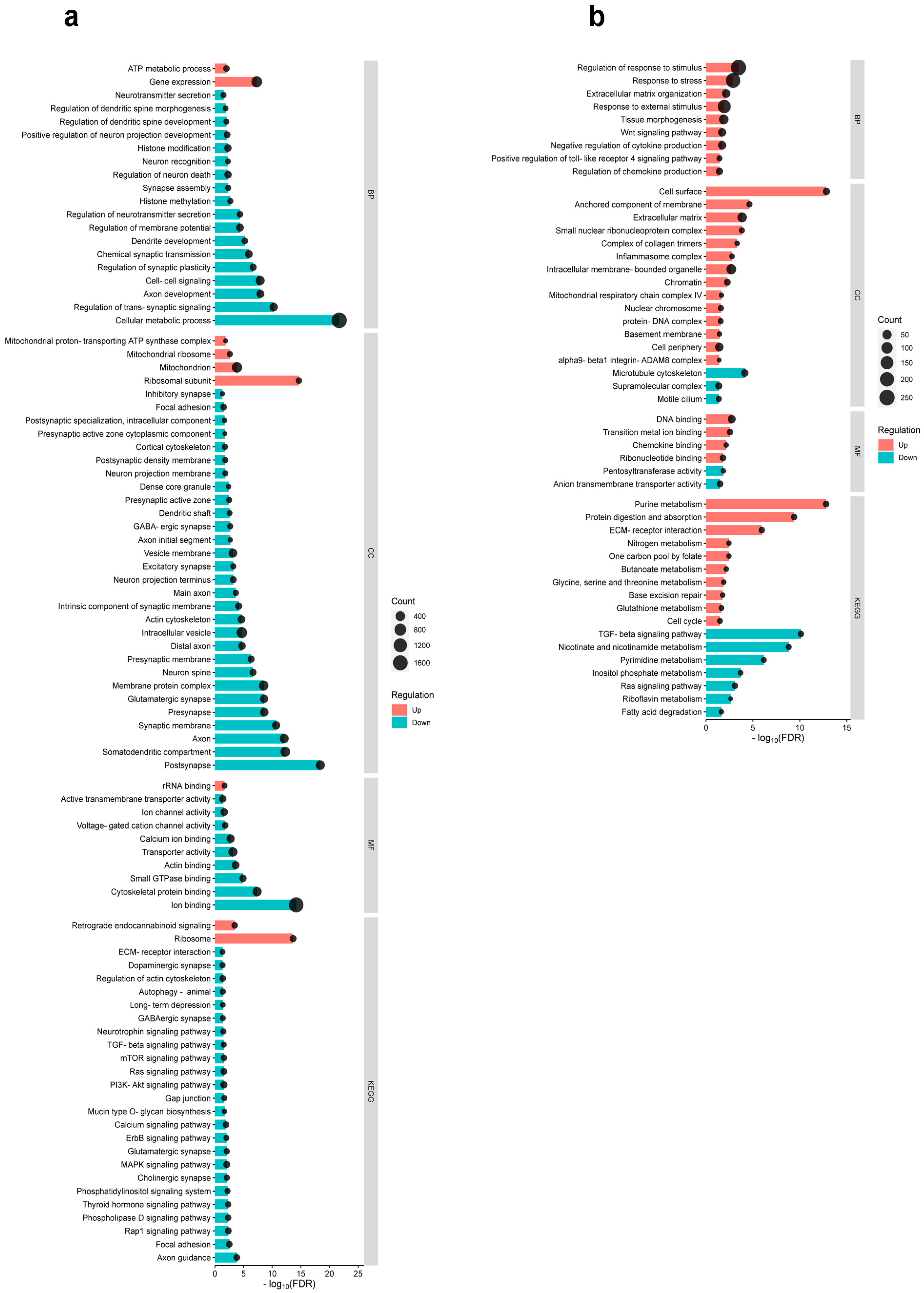
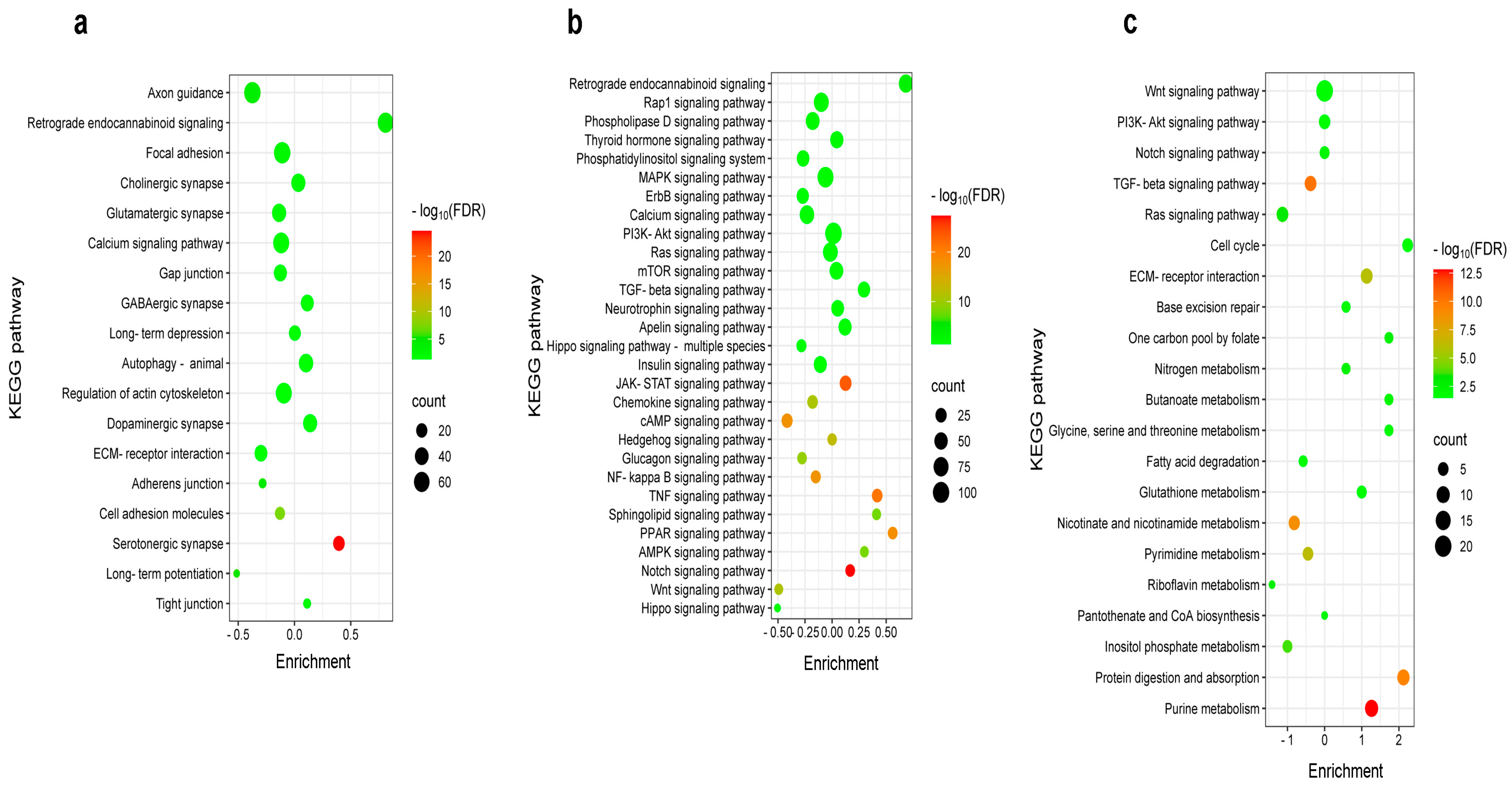

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Species | Dilution | Supplier | Cat. No. |
|---|---|---|---|---|
| * c-Fos (K-25) | rabbit polyclonal IgG | 1:1000 | Santa Cruz Biotechnology, Inc., Dallas, TX, USA | sc-253 |
| € CB1 receptor | goat polyclonal IgG | 1:1000 | Frontiers Institute Co., Ltd., Ishikari, Japan | CB1-Go-Af450 |
| Ggamma14 | rabbit polyclonal IgG | 1:250 | Novus Biologicals, Centennial, CO, USA | NBP2-58646 |
| calbindin D28k | guinea pig polyclonal IgG | 1:250 | Synaptic Systems GmbH.; Göttingen, Germany | 214004 |
| calretinin | guinea pig polyclonal IgG | 1:500 | Swant; Burgdorf, Switzerland | CRGp7 |
| parvalbumin | mouse monoclonal IgG1 | 1:1000 | Swant | PV-235 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erdei, V.; Mészár, Z.; Varga, A. The Burning Pain Transcriptome in the Mouse Primary Somatosensory Cortex. Int. J. Mol. Sci. 2025, 26, 3538. https://doi.org/10.3390/ijms26083538
Erdei V, Mészár Z, Varga A. The Burning Pain Transcriptome in the Mouse Primary Somatosensory Cortex. International Journal of Molecular Sciences. 2025; 26(8):3538. https://doi.org/10.3390/ijms26083538
Chicago/Turabian StyleErdei, Virág, Zoltán Mészár, and Angelika Varga. 2025. "The Burning Pain Transcriptome in the Mouse Primary Somatosensory Cortex" International Journal of Molecular Sciences 26, no. 8: 3538. https://doi.org/10.3390/ijms26083538
APA StyleErdei, V., Mészár, Z., & Varga, A. (2025). The Burning Pain Transcriptome in the Mouse Primary Somatosensory Cortex. International Journal of Molecular Sciences, 26(8), 3538. https://doi.org/10.3390/ijms26083538