

Extracellular Cold-Inducible RNA-Binding Protein: Progress from Discovery to Present


Abstract
1. Introduction
2. Discovery
3. Secretion
4. Receptors
5. Signal-Transduction
6. Pathophysiology
7. Therapeutics
8. Perspectives
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DAMP | damage associated molecular pattern |
| eCIRP | extracellular cold-inducible RNA protein |
| TLR4 | toll-like receptor 4 |
| I/R | ischemia–reperfusion |
| NETs | neutrophil extracellular traps |
| TREM-1 | triggering receptor expressed on myeloid cells-1 |
| APANs | antigen-presenting aged neutrophils |
| Siglec-G | sialic acid binding Ig-like lectin |
| ZBP1 | Z-DNA-binding protein 1 |
| cGAS | cyclic GMP-AMP synthase |
| STING | stimulator of interferon genes |
| ALI | acute lung injury |
| AD | Alzheimer’s disease |
| ER | endoplasmic reticulum |
References
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A. An endogenous factor mediates shock-induced injury. Nat. Med. 2013, 19, 1368–1369. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H.; Itoh, K.; Kaneko, Y.; Kishishita, M.; Yoshida, O.; Fujita, J. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J. Cell. Biol. 1997, 137, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Yang, W.L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef]
- Yang, W.L.; Sharma, A.; Wang, Z.; Li, Z.; Fan, J.; Wang, P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 2016, 6, 26571. [Google Scholar] [CrossRef]
- Khan, M.M.; Yang, W.L.; Brenner, M.; Bolognese, A.C.; Wang, P. Cold-inducible RNA-binding protein (CIRP) causes sepsis-associated acute lung injury via induction of endoplasmic reticulum stress. Sci. Rep. 2017, 7, 41363. [Google Scholar] [CrossRef]
- Chen, K.; Murao, A.; Arif, A.; Takizawa, S.; Jin, H.; Jiang, J.; Aziz, M.; Wang, P. Inhibition of Efferocytosis by Extracellular CIRP-Induced Neutrophil Extracellular Traps. J. Immunol. 2021, 206, 797–806. [Google Scholar] [CrossRef]
- Zhou, M.; Aziz, M.; Yen, H.T.; Ma, G.; Murao, A.; Wang, P. Extracellular CIRP dysregulates macrophage bacterial phagocytosis in sepsis. Cell Mol. Immunol. 2023, 20, 80–93. [Google Scholar] [CrossRef]
- Takizawa, S.; Lee, Y.; Jacob, A.; Aziz, M.; Wang, P. Neutrophil trogocytosis during their trans-endothelial migration: Role of extracellular CIRP. Mol. Med. 2022, 28, 91. [Google Scholar] [CrossRef]
- Lapin, D.; Sharma, A.; Wang, P. Extracellular cold-inducible RNA-binding protein in CNS injury: Molecular insights and therapeutic approaches. J. Neuroinflammation 2025, 22, 12. [Google Scholar] [CrossRef]
- Han, J.; Zhang, Y.; Ge, P.; Dakal, T.C.; Wen, H.; Tang, S.; Luo, Y.; Yang, Q.; Hua, B.; Zhang, G.; et al. Exosome-derived CIRP: An amplifier of inflammatory diseases. Front. Immunol. 2023, 14, 1066721. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H.; Higashitsuji, H.; Yokoi, H.; Itoh, K.; Danno, S.; Matsuda, T.; Fujita, J. Cloning and characterization of human CIRP (cold-inducible RNA-binding protein) cDNA and chromosomal assignment of the gene. Gene 1997, 204, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Itoh, K.; Higashitsuji, H.; Higashitsuji, H.; Nakazawa, N.; Sakurai, T.; Liu, Y.; Tokuchi, H.; Fujita, T.; Zhao, Y.; et al. Cold-inducible RNA-binding protein (Cirp) interacts with Dyrk1b/Mirk and promotes proliferation of immature male germ cells in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10885–10890. [Google Scholar] [CrossRef]
- Wellmann, S.; Bührer, C.; Moderegger, E.; Zelmer, A.; Kirschner, R.; Koehne, P.; Fujita, J.; Seeger, K. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J. Cell Sci. 2004, 117, 1785–1794. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, H.; Zhong, Y.; Huang, J.; Lv, J.; Li, J. The Cold-Inducible RNA-Binding Protein (CIRP) Level in Peripheral Blood Predicts Sepsis Outcome. PLoS ONE 2015, 10, e0137721. [Google Scholar] [CrossRef]
- Gurien, S.D.; Aziz, M.; Jin, H.; Wang, H.; He, M.; Al-Abed, Y.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Extracellular microRNA 130b-3p inhibits eCIRP-induced inflammation. EMBO Rep. 2020, 21, e48075. [Google Scholar] [CrossRef]
- Li, M.; Yao, M.; Shao, K.; Shen, X.; Ge, Z.; Li, Y. Serum cold-inducible RNA-binding protein (CIRP) levels as a prognostic indicator in patients with acute ischemic stroke. Front. Neurol. 2023, 14, 1211108. [Google Scholar]
- Yu, L.; Li, Q.H.; Deng, F.; Yu, Z.W.; Luo, X.Z.; Sun, J.L. Synovial fluid concentrations of cold-inducible RNA-binding protein are associated with severity in knee osteoarthritis. Clin. Chim. Acta 2017, 464, 44–49. [Google Scholar] [CrossRef]
- Yamaga, S.; Murao, A.; Ma, G.; Brenner, M.; Aziz, M.; Wang, P. Radiation upregulates macrophage TREM-1 expression to exacerbate injury in mice. Front. Immunol. 2023, 14, 1151250. [Google Scholar] [CrossRef]
- Murao, A.; Tan, C.; Jha, A.; Wang, P.; Aziz, M. Exosome-Mediated eCIRP Release From Macrophages to Induce Inflammation in Sepsis. Front. Pharmacol. 2021, 12, 791648. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Reilly, B.; Jha, A.; Murao, A.; Lee, Y.; Brenner, M.; Aziz, M.; Wang, P. Active Release of eCIRP via Gasdermin D Channels to Induce Inflammation in Sepsis. J. Immunol. 2022, 208, 2184–2195. [Google Scholar] [CrossRef]
- Reilly, B.; Tan, C.; Murao, A.; Nofi, C.; Jha, A.; Aziz, M.; Wang, P. Necroptosis-Mediated eCIRP Release in Sepsis. J. Inflamm. Res. 2022, 15, 4047–4059. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Aziz, M.; Wang, P. The vitals of NETs. J. Leukoc. Biol. 2021, 110, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Murao, A.; Gurien, S.D.; Ochani, M.; Prince, J.M.; Wang, P. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight 2020, 5, e134172. [Google Scholar] [CrossRef]
- Murao, A.; Arif, A.; Brenner, M.; Denning, N.L.; Jin, H.; Takizawa, S.; Nicastro, B.; Wang, P.; Aziz, M. Extracellular CIRP and TREM-1 axis promotes ICAM-1-Rho-mediated NETosis in sepsis. FASEB J. 2020, 34, 9771–9786. [Google Scholar] [CrossRef]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef]
- Zhou, M.; Aziz, M.; Denning, N.L.; Yen, H.T.; Ma, G.; Wang, P. Extracellular CIRP induces macrophage endotoxin tolerance through IL-6R-mediated STAT3 activation. JCI Insight 2020, 5, e133715. [Google Scholar] [CrossRef]
- Bolognese, A.C.; Sharma, A.; Yang, W.L.; Nicastro, J.; Coppa, G.F.; Wang, P. Cold-inducible RNA-binding protein activates splenic T cells during sepsis in a TLR4-dependent manner. Cell Mol. Immunol. 2018, 15, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Ode, Y.; Aziz, M.; Wang, P. CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J. Leukoc. Biol. 2018, 103, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Aziz, M.; Murao, A.; Kobritz, M.; Shih, A.J.; Adelson, R.P.; Brenner, M.; Wang, P. Antigen-presenting aged neutrophils induce CD4+ T cells to exacerbate inflammation in sepsis. J. Clin. Investig. 2023, 133, e164585. [Google Scholar] [CrossRef] [PubMed]
- Royster, W.; Jin, H.; Wang, P.; Aziz, M. Extracellular CIRP decreases Siglec-G expression on B-1a cells skewing them towards a pro-inflammatory phenotype in sepsis. Mol. Med. 2021, 27, 55. [Google Scholar] [CrossRef]
- Villanueva, L.; Silva, L.; Llopiz, D.; Ruiz, M.; Iglesias, T.; Lozano, T.; Casares, N.; Hervas-Stubbs, S.; Rodríguez, M.J.; Carrascosa, J.L.; et al. The Toll like receptor 4 ligand cold-inducible RNA-binding protein as vaccination platform against cancer. Oncoimmunology 2018, 7, e1409321. [Google Scholar] [CrossRef]
- Shimizu, J.; Murao, A.; Nofi, C.; Wang, P.; Aziz, M. Extracellular CIRP Promotes GPX4-Mediated Ferroptosis in Sepsis. Front. Immunol. 2022, 13, 903859. [Google Scholar] [CrossRef]
- Lee, Y.; Reilly, B.; Tan, C.; Wang, P.; Aziz, M. Extracellular CIRP Induces Macrophage Extracellular Trap Formation via Gasdermin D Activation. Front. Immunol. 2021, 12, 780210. [Google Scholar] [CrossRef]
- Li, Z.; Fan, E.K.; Liu, J.; Scott, M.J.; Li, Y.; Li, S.; Xie, W.; Billiar, T.R.; Wilson, M.A.; Jiang, Y.; et al. Cold-inducible RNA-binding protein through TLR4 signaling induces mitochondrial DNA fragmentation and regulates macrophage cell death after trauma. Cell Death Dis. 2017, 8, e2775. [Google Scholar] [CrossRef]
- Chen, K.; Cagliani, J.; Aziz, M.; Tan, C.; Brenner, M.; Wang, P. Extracellular CIRP activates STING to exacerbate hemorrhagic shock. JCI Insight 2021, 6, e143715. [Google Scholar] [CrossRef]
- Gong, T.; Wang, Q.D.; Loughran, P.A.; Li, Y.H.; Scott, M.J.; Billiar, T.R.; Liu, Y.-T.; Fan, J. Mechanism of lactic acidemia-promoted pulmonary endothelial cells death in sepsis: Role for CIRP-ZBP1-PANoptosis pathway. Mil. Med. Res. 2024, 11, 71. [Google Scholar] [CrossRef]
- Pandian, N.; Kanneganti, T.D. PANoptosis: A Unique Innate Immune Inflammatory Cell Death Modality. J. Immunol. 2022, 209, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; VanPortfliet, J.J.; Chen, Y.F.; Bryant, J.D.; Li, Y.; Fails, D.; Torres-Odio, S.; Ragan, K.B.; Deng, J.; Mohan, A.; et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell 2023, 186, 3013–3032.e22. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Gurien, S.D.; Royster, W.; Aziz, M.; Wang, P. Extracellular CIRP Induces Inflammation in Alveolar Type II Cells via TREM-1. Front. Cell Dev. Biol. 2020, 8, 579157. [Google Scholar] [CrossRef]
- Zhong, P.; Zhou, M.; Zhang, J.; Peng, J.; Zeng, G.; Huang, H. The role of Cold-Inducible RNA-binding protein in respiratory diseases. J. Cell Mol. Med. 2022, 26, 957–965. [Google Scholar] [CrossRef]
- Siskind, S.; Zhang, F.; Brenner, M.; Wang, P. Extracellular CIRP induces acute kidney injury via endothelial TREM-1. Front. Physiol. 2022, 13, 954815. [Google Scholar] [CrossRef]
- Cen, C.; McGinn, J.; Aziz, M.; Yang, W.L.; Cagliani, J.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Deficiency in cold-inducible RNA-binding protein attenuates acute respiratory distress syndrome induced by intestinal ischemia-reperfusion. Surgery 2017, 162, 917–927. [Google Scholar] [CrossRef]
- Godwin, A.; Yang, W.L.; Sharma, A.; Khader, A.; Wang, Z.; Zhang, F.; Nicastro, J.; Coppa, G.F.; Wang, P. Blocking cold-inducible RNA-binding protein protects liver from ischemia-reperfusion injury. Shock 2015, 43, 24–30. [Google Scholar] [CrossRef]
- Cen, C.; Yang, W.L.; Yen, H.T.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Deficiency of cold-inducible ribonucleic acid-binding protein reduces renal injury after ischemia-reperfusion. Surgery 2016, 160, 473–483. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, W.L.; Ji, Y.; Qiang, X.; Wang, P. Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim. Biophys. Acta 2014, 1840, 2253–2261. [Google Scholar] [CrossRef]
- Yamaga, S.; Murao, A.; Zhou, M.; Aziz, M.; Brenner, M.; Wang, P. Radiation-induced eCIRP impairs macrophage bacterial phagocytosis. J. Leukoc. Biol. 2024, 116, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Brenner, M.; Jacob, A.; Marambaud, P.; Wang, P. Extracellular CIRP Activates the IL-6Rα/STAT3/Cdk5 Pathway in Neurons. Mol. Neurobiol. 2021, 58, 3628–3640. [Google Scholar] [CrossRef]
- Sharma, A.; Brenner, M.; Wang, P. Potential Role of Extracellular CIRP in Alcohol-Induced Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 5000–5010. [Google Scholar] [CrossRef]
- Jacob, A.; Wang, P. Alcohol Intoxication and Cognition: Implications on Mechanisms and Therapeutic Strategies. Front. Neurosci. 2020, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, J.P.; Yao, F.H.; Cao, Y.; Li, S.C.; Liu, Y.Y.; Wen, S.X.; Liu, Y.X.; Liu, A.J. Cold Inducible RNA-Binding Protein Promotes the Development of Alzheimer’s Disease Partly by Inhibition of uPA in Astrocytes. Degener. Neurol. Neuromuscul. Dis. 2024, 14, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Aylar, D.; Lee, Y.; Brenner, M.; Marambaud, P.; Wang, P. Extracellular CIRP induces neurotoxic A1 astrocytes via TREM-1 in Alzheimer’s disease. Alzheimers. Dement. 2025, 20 (Suppl. S8), e094835. [Google Scholar] [CrossRef]
- Sharma, A.; Sari, E.; Lee, Y.; Patel, S.; Brenner, M.; Marambaud, P.; Wang, P. Extracellular CIRP Induces Calpain Activation in Neurons via PLC-IP(3)-Dependent Calcium Pathway. Mol. Neurobiol. 2023, 60, 3311–3328. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Bolourani, S.; Sari, E.; Brenner, M.; Wang, P. The role of eCIRP in bleomycin-induced pulmonary fibrosis in mice. PLoS ONE 2022, 17, e0266163. [Google Scholar] [CrossRef]
- Bolourani, S.; Sari, E.; Brenner, M.; Wang, P. Extracellular CIRP Induces an Inflammatory Phenotype in Pulmonary Fibroblasts via TLR4. Front. Immunol. 2021, 12, 721970. [Google Scholar] [CrossRef]
- Sakurai, T.; Kashida, H.; Watanabe, T.; Hagiwara, S.; Mizushima, T.; Iijima, H.; Nishida, N.; Higashitsuji, H.; Fujita, J.; Kudo, M.; et al. Stress response protein cirp links inflammation and tumorigenesis in colitis-associated cancer. Cancer Res. 2014, 74, 6119–6128. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Huang, H. Recent progress in the research of cold-inducible RNA-binding protein. Future Sci. OA 2017, 3, FSO246. [Google Scholar] [CrossRef]
- De Meo, M.L.; Spicer, J.D. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin. Immunol. 2021, 57, 101595. [Google Scholar] [CrossRef]
- Borjas, T.; Jacob, A.; Kobritz, M.; Ma, G.; Tan, C.; Patel, V.; Coppa, G.F.; Aziz, M.; Wang, P. An engineered miRNA PS-OMe miR130 inhibits acute lung injury by targeting eCIRP in sepsis. Mol. Med. 2023, 29, 21. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Jha, A.; Ma, G.; Chaung, W.; Aziz, M.; Wang, P. A Synthetic Poly(A) Tail Targeting Extracellular CIRP Inhibits Sepsis. J. Immunol. 2023, 211, 1144–1153. [Google Scholar] [CrossRef]
- Liu, W.; Bi, J.; Ren, Y.; Chen, H.; Zhang, J.; Wang, T.; Wang, M.; Zhang, L.; Zhao, J.; Wu, Z.; et al. Targeting extracellular CIRP with an X-aptamer shows therapeutic potential in acute pancreatitis. iScience 2023, 26, 107043. [Google Scholar] [CrossRef]
- Nofi, C.P.; Tan, C.; Ma, G.; Kobritz, M.; Prince, J.M.; Wang, H.; Aziz, M.; Wang, P. A novel opsonic eCIRP inhibitor for lethal sepsis. J. Leukoc. Biol. 2024, 115, 385–400. [Google Scholar] [CrossRef]
- Hollis, R.; Li, J.; Lee, Y.; Jin, H.; Zhou, M.; Nofi, C.P.; Sfakianos, M.; Coppa, G.; Aziz, M.; Wang, P. A Novel Opsonic Extracellular Cirp Inhibitor Mop3 Alleviates Gut Ischemia/Reperfusion Injury. Shock 2025, 63, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Nofi, C.P.; Prince, J.M.; Aziz, M.; Wang, P. The Novel MFG-E8-derived Oligopeptide, MOP3, Improves Outcomes in a Preclinical Murine Model of Neonatal Sepsis. J. Pediatr. Surg. 2024, 59, 1282–1290. [Google Scholar] [CrossRef]
- Nofi, C.P.; Prince, J.M.; Brewer, M.R.; Aziz, M.; Wang, P. An anti-eCIRP strategy for necrotizing enterocolitis. Mol. Med. 2024, 30, 156. [Google Scholar] [CrossRef]
- Gurien, S.D.; Aziz, M.; Cagliani, J.; Denning, N.L.; Last, J.; Royster, W.; Coppa, G.F.; Wang, P. An extracellular cold-inducible RNA-binding protein-derived small peptide targeting triggering receptor expressed on myeloid cells-1 attenuates hemorrhagic shock. J. Trauma Acute Care Surg. 2020, 88, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Ochani, M.; Prince, J.M.; Wang, P. Inhibition of a triggering receptor expressed on myeloid cells-1 (TREM-1) with an extracellular cold-inducible RNA-binding protein (eCIRP)-derived peptide protects mice from intestinal ischemia-reperfusion injury. Surgery 2020, 168, 478–485. [Google Scholar] [CrossRef] [PubMed]
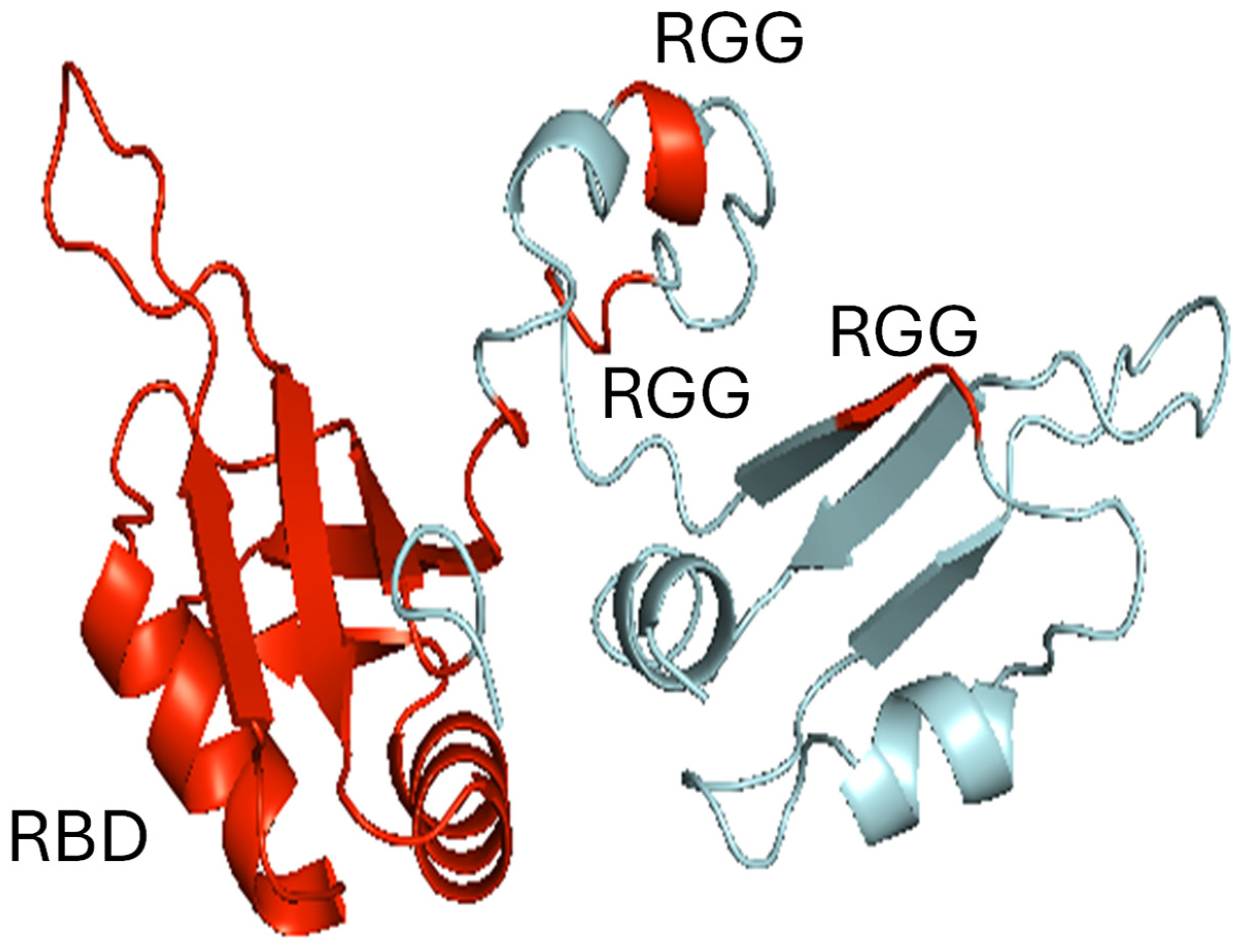
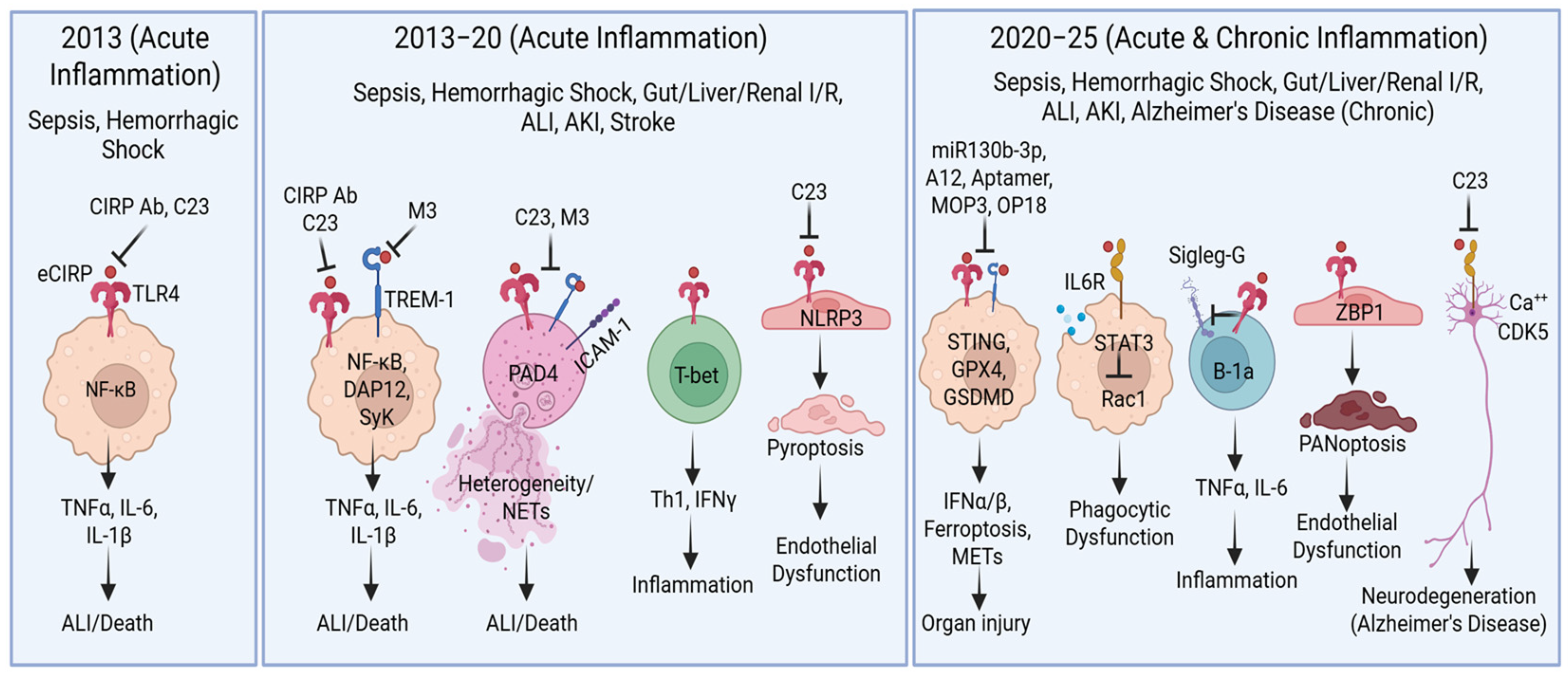

{kind=link}
{kind=link}
| Disease Conditions/ Stimulants | Release Pathways | Underlying Mechanisms | Cell Types | References |
|---|---|---|---|---|
| Sepsis/hemorrhagic shock, LPS, hypoxia, ER stress | Stress granule, lysosomal exocytosis | GSK3β, CKII | Macrophages | [4,13] |
| Sepsis, LPS, ATP, nigericin | Pyroptosis | Inflammasome, GSDMD | Macrophages | [23] |
| Sepsis, LPS, TNFα | Necroptosis | RIPK, MLKL | Macrophages | [24] |
| Sepsis, LPS | Exosomal | CD63 (Tetraspanin) | Macrophages | [11,22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, M.; Chaudry, I.H.; Wang, P. Extracellular Cold-Inducible RNA-Binding Protein: Progress from Discovery to Present. Int. J. Mol. Sci. 2025, 26, 3524. https://doi.org/10.3390/ijms26083524
Aziz M, Chaudry IH, Wang P. Extracellular Cold-Inducible RNA-Binding Protein: Progress from Discovery to Present. International Journal of Molecular Sciences. 2025; 26(8):3524. https://doi.org/10.3390/ijms26083524
Chicago/Turabian StyleAziz, Monowar, Irshad H. Chaudry, and Ping Wang. 2025. "Extracellular Cold-Inducible RNA-Binding Protein: Progress from Discovery to Present" International Journal of Molecular Sciences 26, no. 8: 3524. https://doi.org/10.3390/ijms26083524
APA StyleAziz, M., Chaudry, I. H., & Wang, P. (2025). Extracellular Cold-Inducible RNA-Binding Protein: Progress from Discovery to Present. International Journal of Molecular Sciences, 26(8), 3524. https://doi.org/10.3390/ijms26083524