
Unraveling the IDH3A: Expanding the Genotypic Spectrum of Macular Pseudocoloboma
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Case Description
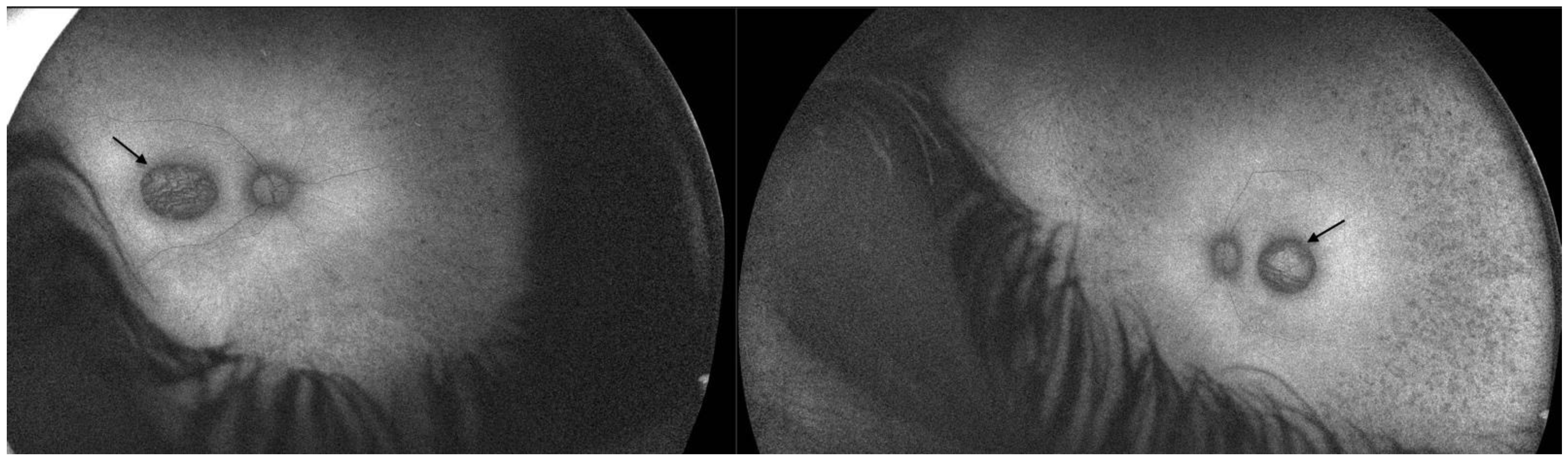
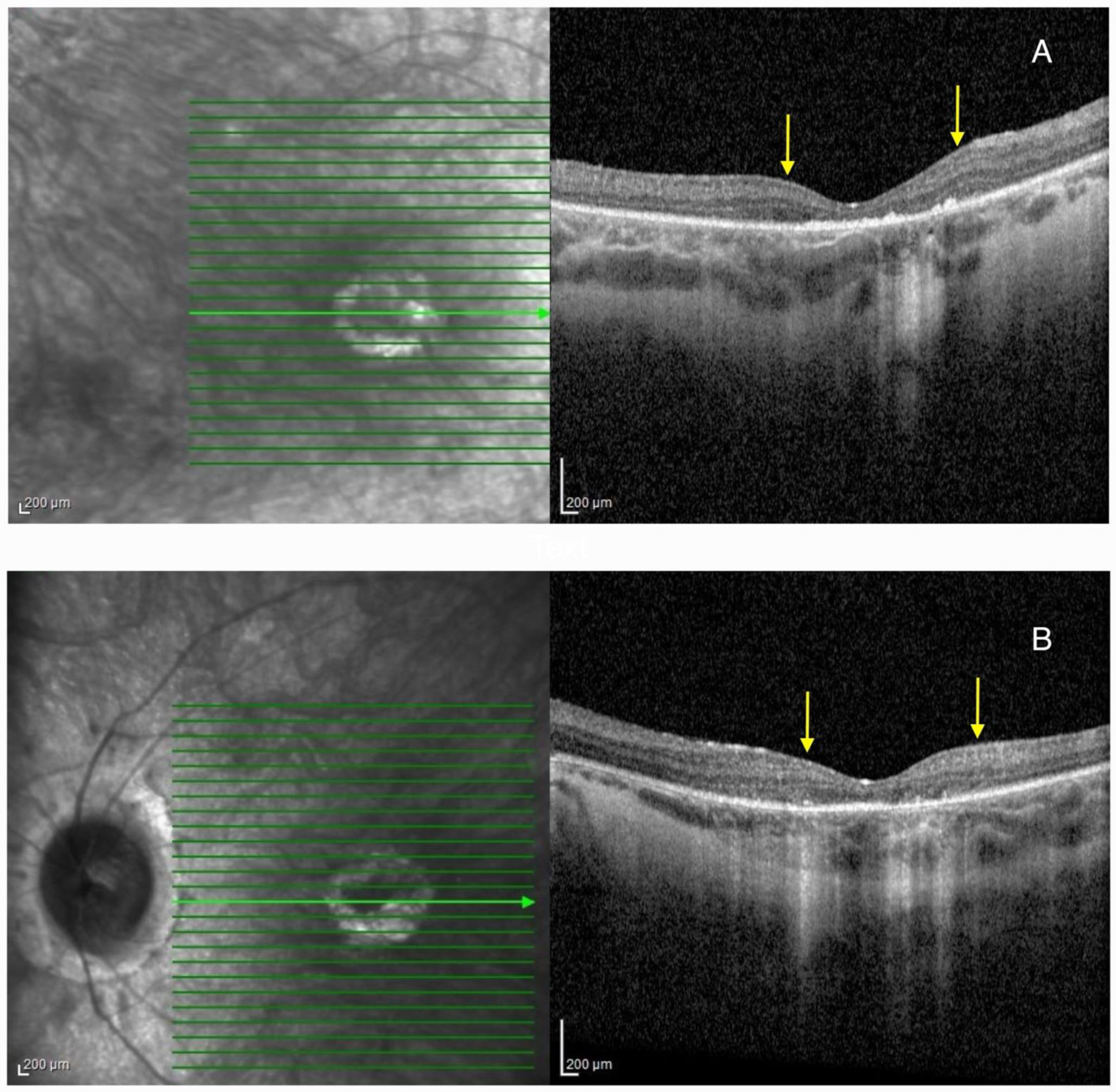
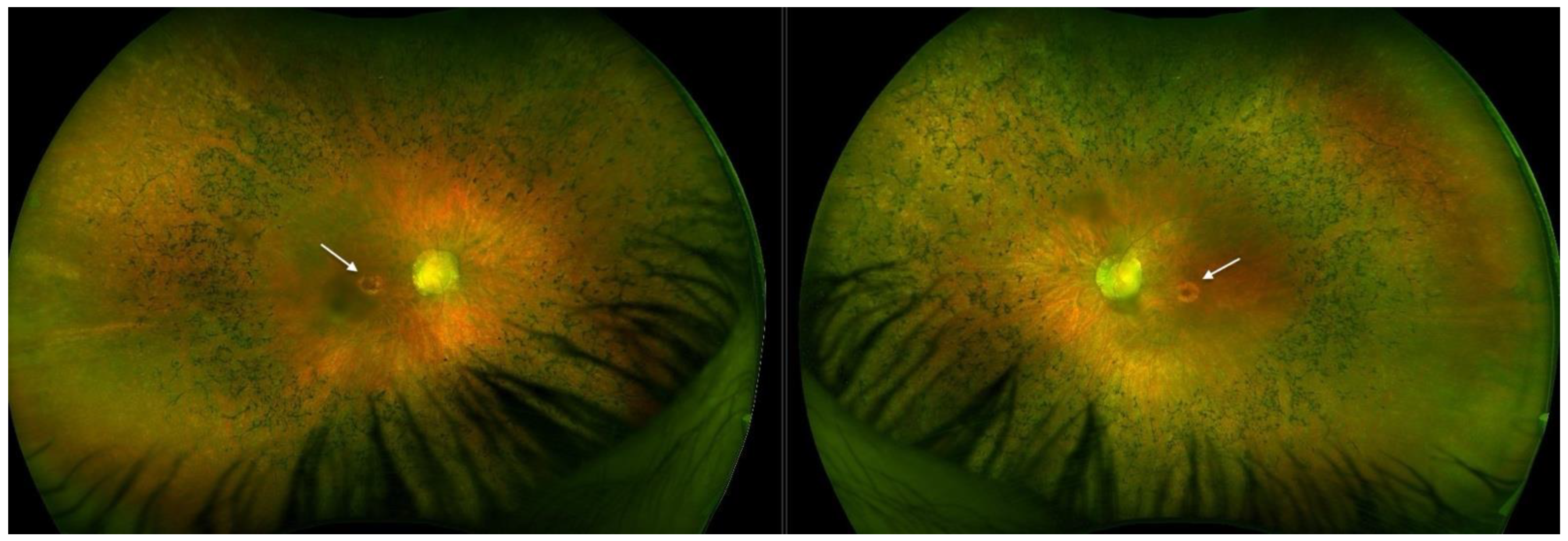
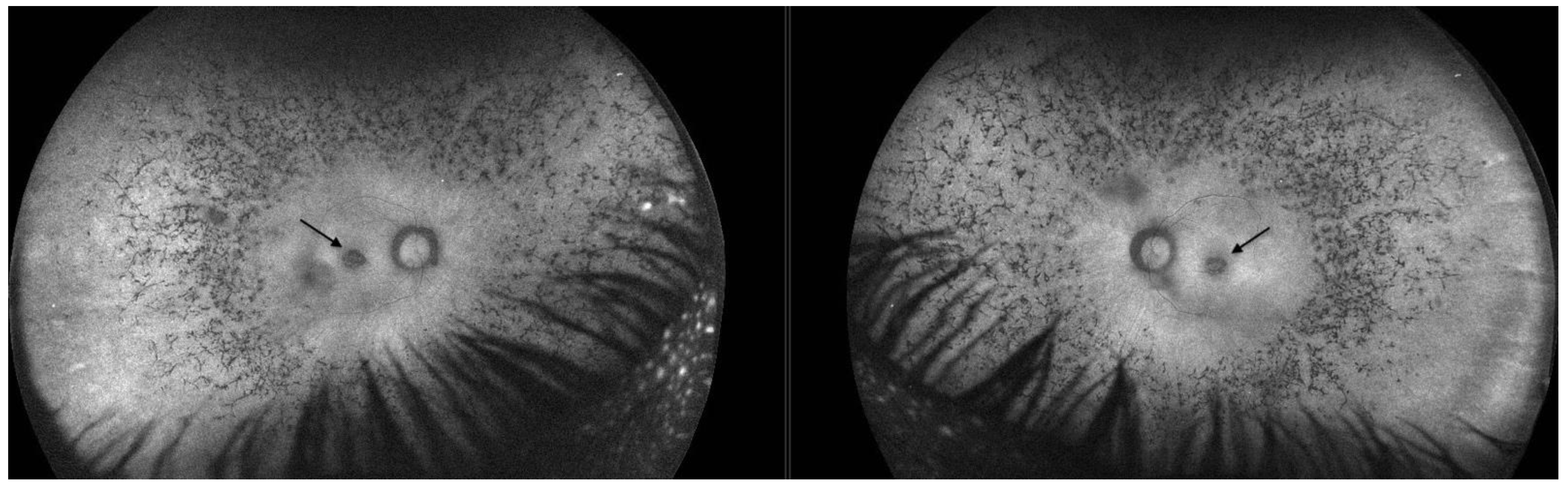
2.1. Patient 1
2.2. Patient 2
3. Discussion
3.1. IDH3A
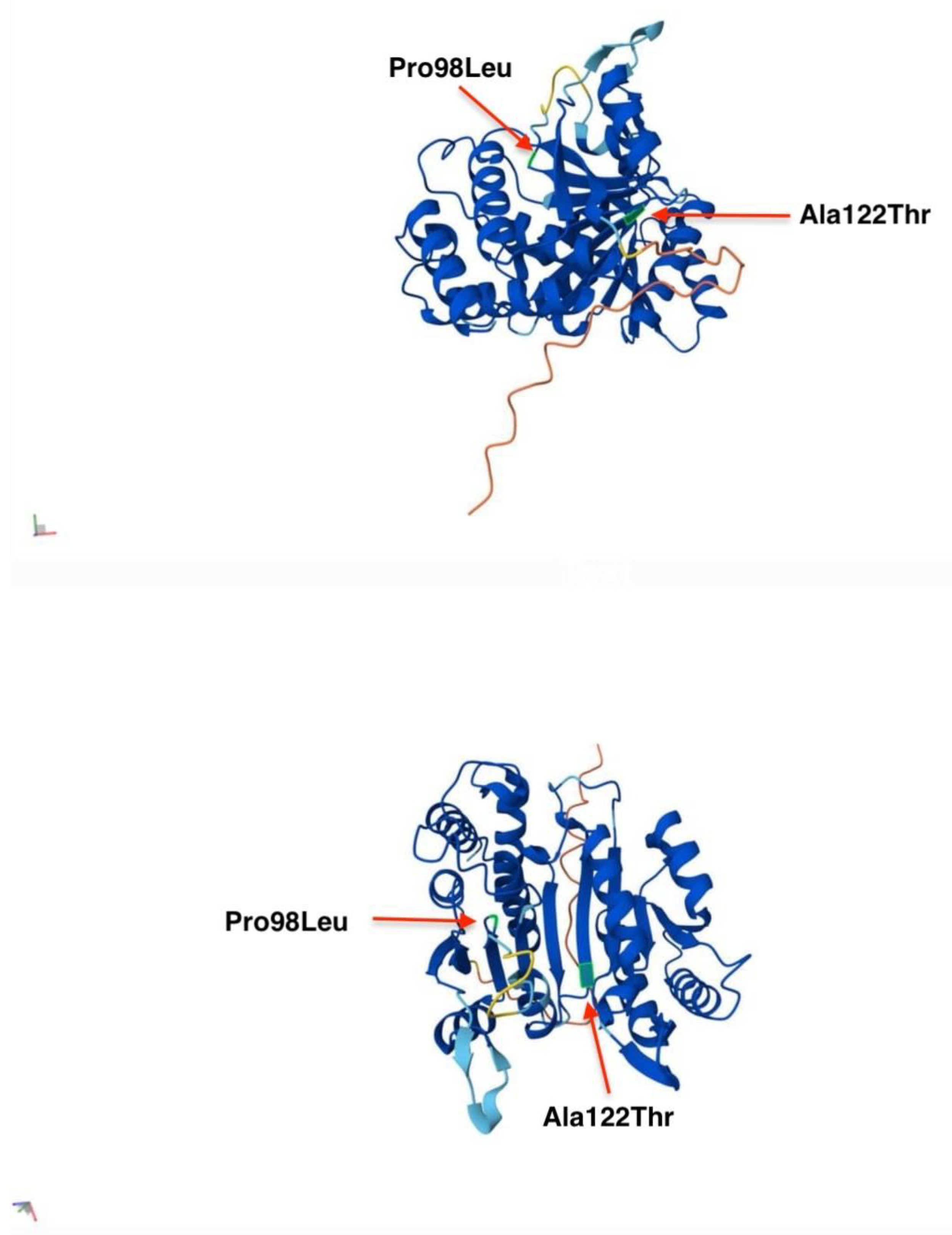
3.2. IDH3A c.364G>A, p.(Ala122Thr)
3.3. IDH3A c.293C>T, p.(Pro98Leu)
3.4. Macular Pseudocoloboma
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fattal-Valevski, A.; Eliyahu, H.; Fraenkel, N.D.; Elmaliach, G.; Hausman-Kedem, M.; Shaag, A.; Mandel, D.; Pines, O.; Elpeleg, O. Homozygous mutation, p.Pro304His, in IDH3A, encoding isocitrate dehydrogenase subunit is associated with severe encephalopathy in infancy. Neurogenetics 2017, 18, 57–61. [Google Scholar] [PubMed]
- OMIM. ISOCITRATE DEHYDROGENASE, NAD(+), 3, CATALYTIC SUBUNIT ALPHA; IDH3A. Available online: https://www.omim.org/entry/601149?search=idh3a&highlight=idh3a (accessed on 1 March 2025).
- Hartong, D.T.; Dange, M.; McGee, T.L.; Berson, E.L.; Dryja, T.P.; Colman, R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008, 40, 1230–1234. [Google Scholar] [PubMed]
- The Human Gene Mutation Database. Available online: https://www.hgmd.cf.ac.uk/ac/index.php (accessed on 1 March 2025).
- Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Mizota, A.; Tormene, A.P.; International Society for Clinical Electrophysiology of Vision. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc. Ophthalmol. 2016, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar]
- Peter, V.G.; Nikopoulos, K.; Quinodoz, M.; Granse, L.; Farinelli, P.; Superti-Furga, A.; Andréasson, S.; Rivolta, C. A novel missense variant in IDH3A causes autosomal recessive retinitis pigmentosa. Ophthalmic. Genet. 2019, 40, 177–181. [Google Scholar] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [PubMed]
- Pierrache, L.H.M.; Kimchi, A.; Ratnapriya, R.; Roberts, L.; Astuti, G.D.N.; Obolensky, A.; Beryozkin, A.; Tjon-Fo-Sang, M.J.H.; Schuil, J.; Klaver, C.C.W.; et al. Whole-exome sequencing identifies biallelic IDH3A variants as a cause of retinitis pigmentosa accompanied by pseudocoloboma. Ophthalmology 2017, 124, 992–1003. [Google Scholar] [PubMed]
- Sun, W.; Zhang, Q. A novel variant in IDH3A identified in a case with Leber congenital amaurosis accompanied by macular pseudocoloboma. Ophthalmic. Genet. 2018, 39, 662–663. [Google Scholar]
- GnomAD. SNV: 15-78161655-G-A(GRCh38). Available online: https://gnomad.broadinstitute.org/variant/15-78161655-G-A?dataset=gnomad_r4 (accessed on 2 March 2025).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/977474/?oq=977474&m=NM_005530.3 (accessed on 2 March 2025).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Clausen, W. Typisches, beiderseitiges hereditares Makula Kolobom. Klin. Monatsbl. Augenheilkd. 1921, 67, 116. [Google Scholar]
- Foxman, S.G.; Heckenlively, J.R.; Bateman, J.B.; Wirtschafter, J.D. Classification of congenital and early onset retinitis pigmentosa. Arch. Ophthalmol. 1985, 103, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Budde, B.S.; Nürnberg, P.; Kawalia, A.; Lenzner, S.; Bolz, H.J. Genome-wide linkage and sequence analysis challenge CCDC66 as a human retinal dystrophy candidate gene and support a distinct NMNAT1-related fundus phenotype. Clin. Genet. 2018, 93, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Guo, Y.; Liu, J.; Li, S.; Fan, W.; Lin, M.; Rokohl, A.C.; Heindl, L.M. A Systematic Review of the Clinical Manifestations and Diagnostic Methods for Macular Coloboma. Curr. Eye. Res. 2021, 46, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Patel, N.; Ghazi, N.G.; Alzahrani, S.S.; Arold, S.T.; Alkuraya, F.S. Familial non-syndromic macular pseudocoloboma secondary to homozygous CLDN19 mutation. Ophthalmic Genet. 2018, 39, 577–583. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjeloš, M.; Ćurić, A.; Rak, B.; Kuzmanović Elabjer, B.; Bušić, M.; Rončević, K. Unraveling the IDH3A: Expanding the Genotypic Spectrum of Macular Pseudocoloboma. Int. J. Mol. Sci. 2025, 26, 3364. https://doi.org/10.3390/ijms26073364
Bjeloš M, Ćurić A, Rak B, Kuzmanović Elabjer B, Bušić M, Rončević K. Unraveling the IDH3A: Expanding the Genotypic Spectrum of Macular Pseudocoloboma. International Journal of Molecular Sciences. 2025; 26(7):3364. https://doi.org/10.3390/ijms26073364
Chicago/Turabian StyleBjeloš, Mirjana, Ana Ćurić, Benedict Rak, Biljana Kuzmanović Elabjer, Mladen Bušić, and Katja Rončević. 2025. "Unraveling the IDH3A: Expanding the Genotypic Spectrum of Macular Pseudocoloboma" International Journal of Molecular Sciences 26, no. 7: 3364. https://doi.org/10.3390/ijms26073364
APA StyleBjeloš, M., Ćurić, A., Rak, B., Kuzmanović Elabjer, B., Bušić, M., & Rončević, K. (2025). Unraveling the IDH3A: Expanding the Genotypic Spectrum of Macular Pseudocoloboma. International Journal of Molecular Sciences, 26(7), 3364. https://doi.org/10.3390/ijms26073364