Clinical and Genetic Characteristics of 18 Patients from Southeast China with ABCA4-Associated Stargardt Disease


Abstract
1. Introduction
2. Results
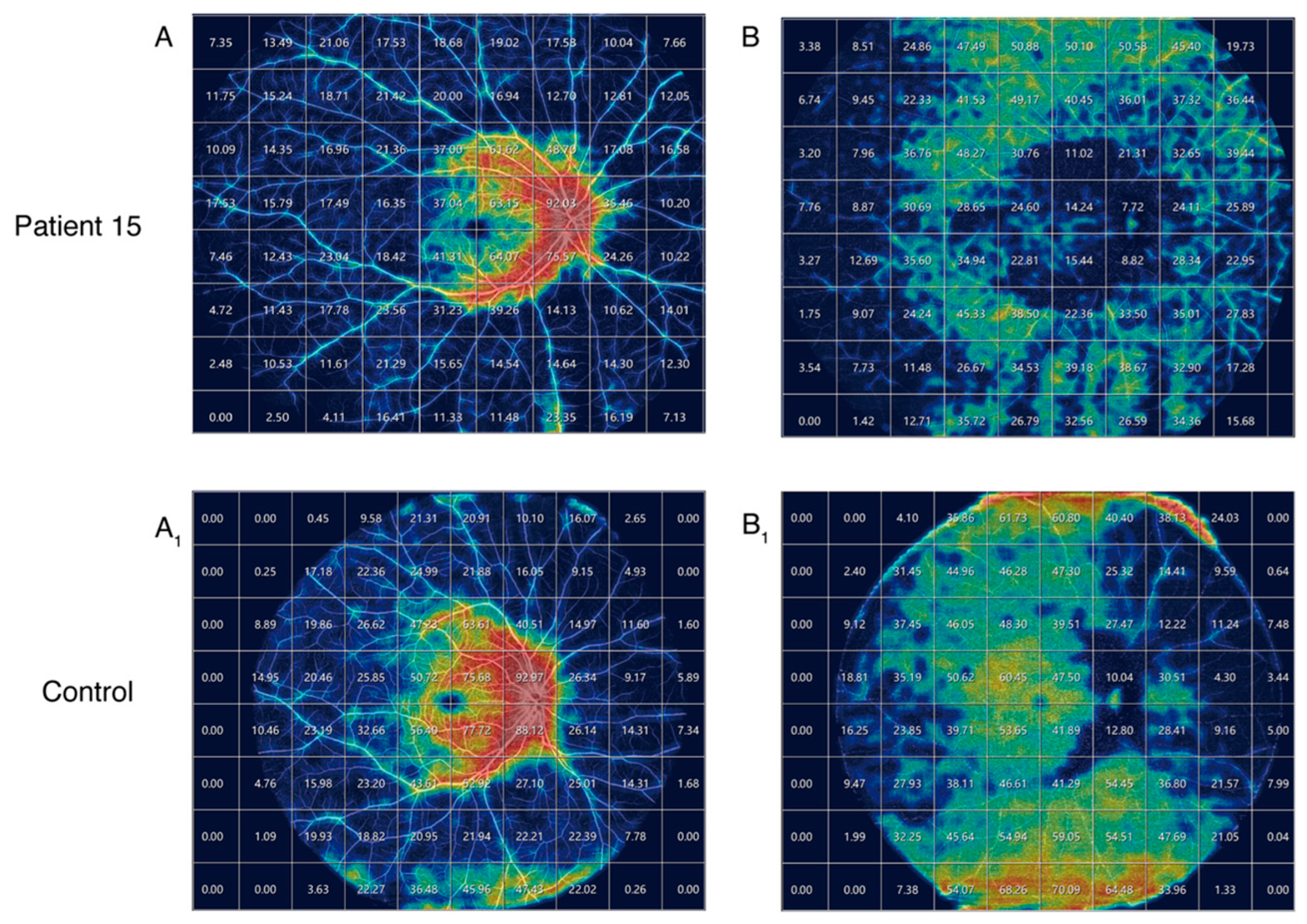
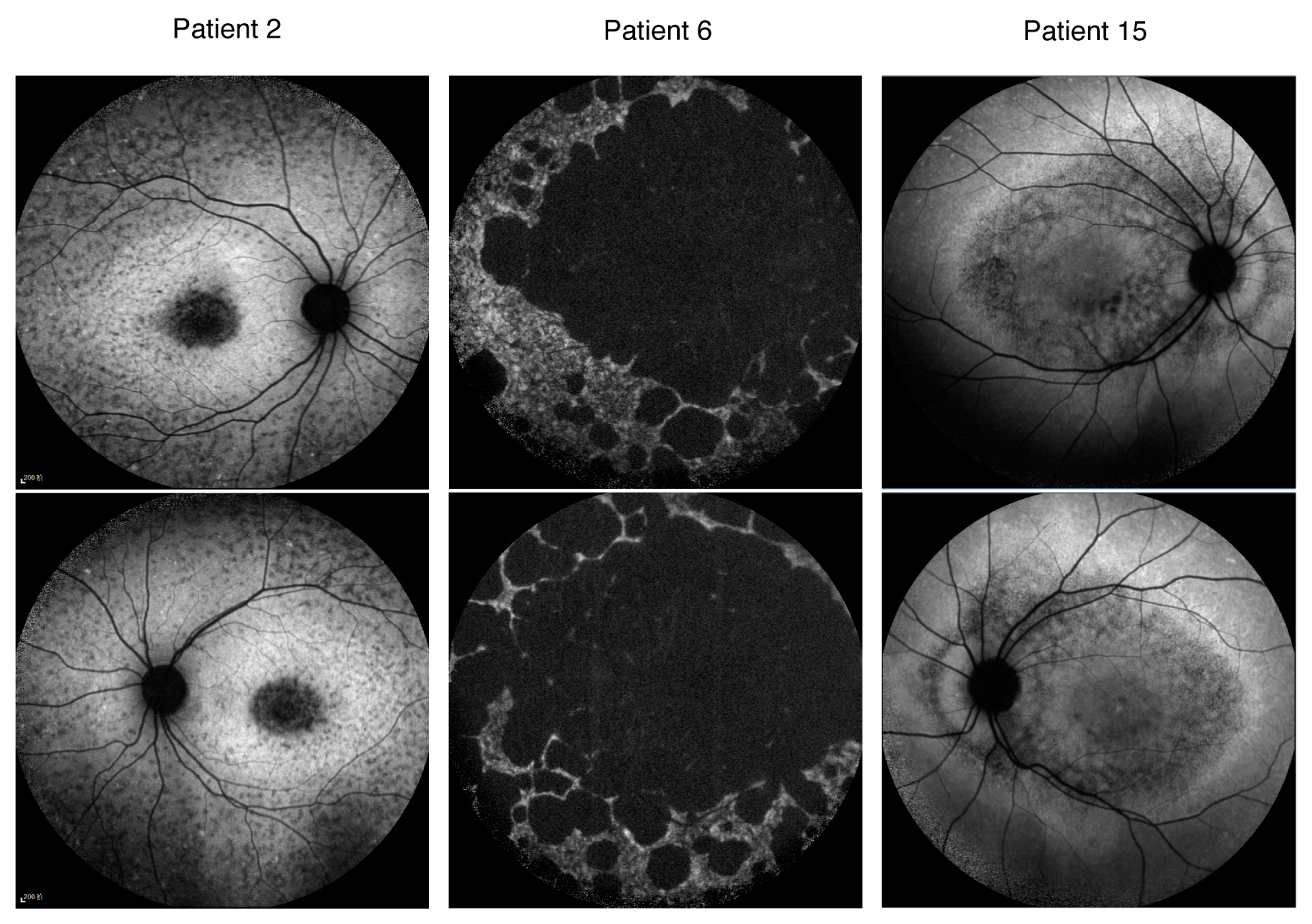
2.1. Clinical Manifestations of the Patients
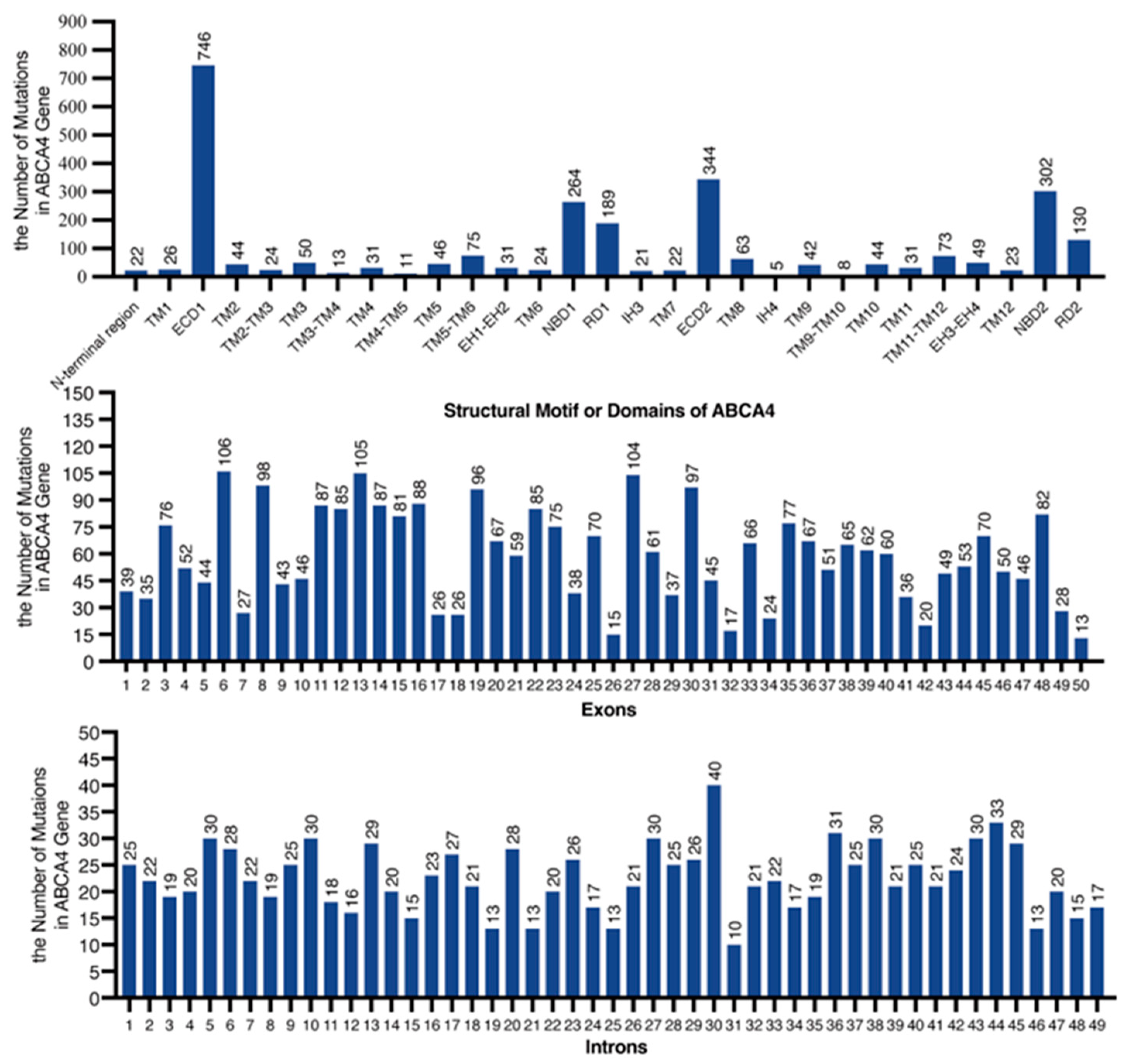
2.2. Genetic Mutation Screening and Bioinformatic Analysis
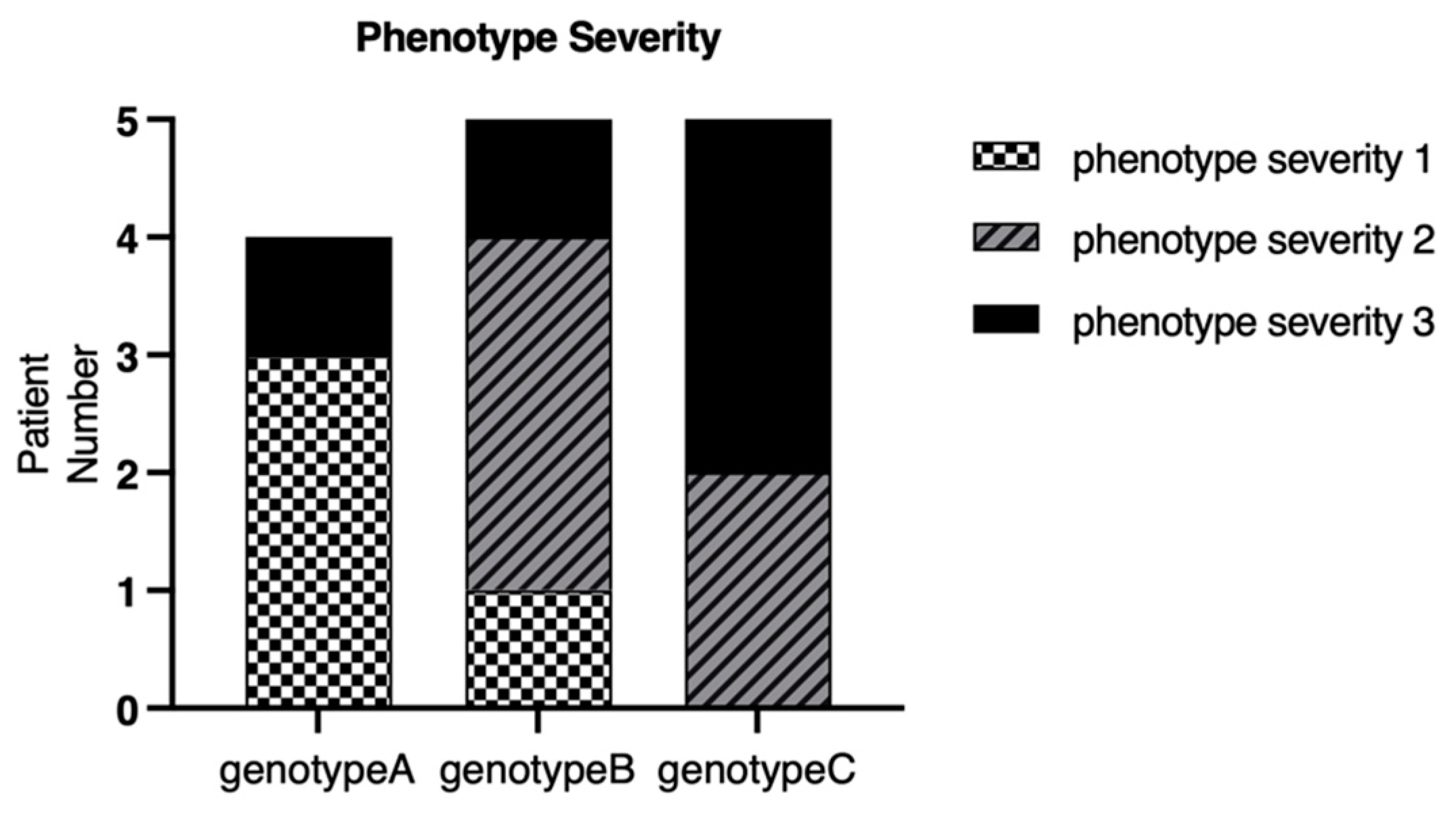
2.3. Genotype–Phenotype Correlation
3. Discussion
4. Materials and Methods
4.1. Participants and Clinical Investigations
4.2. Fundus Autofluorescence Type Classifications
4.3. Mutation Classifications
4.4. Classification of Severity for the Phenotype of Stargardt Disease
4.5. Whole-Exome Sequencing and Sanger Sequencing of the ABCA4 Gene
4.6. In Silico Molecular Genetic Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The genetics of inherited macular dystrophies. J. Med. Genet. 2003, 40, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Lois, N.; Mukherjee, R.; McBain, V.A.; Tsunoda, K.; Tsubota, K.; Stone, E.M.; Fitzke, F.W.; Bunce, C.; Moore, A.T.; et al. A longitudinal study of Stargardt disease: Quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8181–8190. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef]
- Martinez-Mir, A.; Paloma, E.; Allikmets, R.; Ayuso, C.; Del, R.T.; Dean, M.; Vilageliu, L.; Gonzalez-Duarte, R.; Balcells, S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet. 1998, 18, 11–12. [Google Scholar] [CrossRef]
- Bertelsen, M.; Zernant, J.; Larsen, M.; Duno, M.; Allikmets, R.; Rosenberg, T. Generalized choriocapillaris dystrophy, a distinct phenotype in the spectrum of ABCA4-associated retinopathies. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2766–2776. [Google Scholar] [CrossRef]
- Tanaka, K.; Lee, W.; Zernant, J.; Schuerch, K.; Ciccone, L.; Tsang, S.H.; Sparrow, J.R.; Allikmets, R. The Rapid-Onset Chorioretinopathy Phenotype of ABCA4 Disease. Ophthalmology 2018, 125, 89–99. [Google Scholar] [CrossRef]
- Molday, R.S.; Garces, F.A.; Scortecci, J.F.; Molday, L.L. Structure and function of ABCA4 and its role in the visual cycle and Stargardt macular degeneration. Prog. Retin. Eye Res. 2022, 89, 101036. [Google Scholar] [CrossRef]
- Xu, T.; Molday, L.L.; Molday, R.S. Retinal-phospholipid Schiff-base conjugates and their interaction with ABCA4, the ABC transporter associated with Stargardt disease. J. Biol. Chem. 2023, 299, 104614. [Google Scholar] [CrossRef]
- Al-Khuzaei, S.; Broadgate, S.; Foster, C.R.; Shah, M.; Yu, J.; Downes, S.M.; Halford, S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes 2021, 12, 1241. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Okano, K.; Maeda, T.; Chauhan, V.; Golczak, M.; Maeda, A.; Palczewski, K. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J. Biol. Chem. 2012, 287, 5059–5069. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar] [CrossRef] [PubMed]
- Westeneng-van, H.S.; Boon, C.J.; Cremers, F.P.; Hoefsloot, L.H.; den Hollander, A.I.; Hoyng, C.B. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012, 119, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; van Driel, M.; den Hollander, A.I.; van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef]
- Jiang, F.; Pan, Z.; Xu, K.; Tian, L.; Xie, Y.; Zhang, X.; Chen, J.; Dong, B.; Li, Y. Screening of ABCA4 Gene in a Chinese Cohort with Stargardt Disease or Cone-Rod Dystrophy With a Report on 85 Novel Mutations. Investig. Ophthalmol. Vis. Sci. 2016, 57, 145–152. [Google Scholar] [CrossRef]
- Fujinami, K.; Strauss, R.W.; Chiang, J.P.; Audo, I.S.; Bernstein, P.S.; Birch, D.G.; Bomotti, S.M.; Cideciyan, A.V.; Ervin, A.M.; Marino, M.J.; et al. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol. 2019, 103, 390–397. [Google Scholar] [CrossRef]
- Rosenberg, T.; Klie, F.; Garred, P.; Schwartz, M. N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol. Vis. 2007, 13, 1962–1969. [Google Scholar]
- Lange, C.; Feltgen, N.; Junker, B.; Schulze-Bonsel, K.; Bach, M. Resolving the clinical acuity categories “hand motion” and “counting fingers” using the Freiburg Visual Acuity Test (FrACT). Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 137–142. [Google Scholar] [CrossRef]
- Cremers, F.; Lee, W.; Collin, R.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- Tian, L.; Chen, C.; Song, Y.; Zhang, X.; Xu, K.; Xie, Y.; Jin, Z.B.; Li, Y. Phenotype-Based Genetic Analysis Reveals Missing Heritability of ABCA4-Related Retinopathy: Deep Intronic Variants and Copy Number Variations. Investig. Ophthalmol. Vis. Sci. 2022, 63, 5. [Google Scholar] [CrossRef]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Schubert, C.; Im, K.M.; Burke, T.; Brown, C.M.; Fishman, G.A.; Tsang, S.H.; Gouras, P.; Dean, M.; Allikmets, R. Analysis of the ABCA4 gene by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8479–8487. [Google Scholar] [CrossRef]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Mackay, D.S.; Tsunoda, K.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Moore, A.T.; Michaelides, M.; et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology 2013, 120, 2324–2331. [Google Scholar] [CrossRef]
- de Sainte, A.J.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef]
- Eng, L.; Coutinho, G.; Nahas, S.; Yeo, G.; Tanouye, R.; Babaei, M.; Dork, T.; Burge, C.; Gatti, R.A. Nonclassical splicing mutations in the coding and noncoding regions of the ATM Gene: Maximum entropy estimates of splice junction strengths. Hum. Mutat. 2004, 23, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.; Hunt, S.E.; Waddell, N.; Spurdle, A.B. A plugin for the Ensembl Variant Effect Predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt. No | Sex | Age at Onset | Age | BCVA (logMAR) OD OS | Mutation Status | Mutation Group | Phenotype Severity | AF Pattern | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 7 | 8 | 1.40 | 1.30 | c.5554del(p.Gln1852ArgfsTer3) * | C | 3 | 1 |
| c.260dup(p.Gly88ArgfsTer11) | |||||||||
| 2 | F | 12 | 16 | 1.30 | 1.30 | c.6006-3C>A | C | 2 | 2 |
| 3 | F | 7 | 8 | 1.12 | 1.12 | c.4853G>A:(p.Trp1618Ter) | C | 3 | 1 |
| chr1:94480267-94482198del * | |||||||||
| 4 | M | 55 | 57 | 0.40 | 0.15 | c.1378del(p.Val460Ter) * | B | 1 | 3 |
| c.71G>A:(p.Arg24His) | |||||||||
| 5 | F | 7 | 13 | 1 | 1 | c.4253+5G>A | C | 3 | N/A |
| c.3172G>T(p.Glu1058Ter) * | |||||||||
| c.1531C>T(p.Arg511Cys) | |||||||||
| 6 | M | 52 | 52 | 1.98 | 1.98 | c.4217A>G(p.His1406Arg) | A | 3 | 3 |
| 7 | F | 8 | 9 | 0.92 | 1 | c.4217A>G(p.His1406Arg) | B | 2 | 2 |
| c.1561del(p.Val521SerfsTer47) | |||||||||
| 8 | M | 11 | 13 | 0.70 | 0.82 | c.5761G>A(p.Val1921Met) | A | 1 | 1 |
| c.1804C>T(p.Arg602Trp) | |||||||||
| 9 | M | 11 | 31 | 1 | 1 | c.2894A>G(p.Asn965Ser) | B | 2 | 2 |
| c.2424C>G(p.Tyr808Ter) | |||||||||
| 10 | M | 6 | 6 | 1.15 | 1 | c.5646G>A(p.Met1882Ile) | B | 3 | 1 |
| c.1761-2A>G | |||||||||
| 11 | M | 25 | 25 | 1.10 | 1 | c.1006del(p.Ser336ProfsTer38) | C | N/A | N/A |
| c.303-2A>G | |||||||||
| 12 | M | 17 | 17 | 1 | 0.92 | c.2894A>G(p.Asn965Ser) | B | 2 | 2 |
| c.1222C>T(p.Arg408Ter) | |||||||||
| 13 | F | 14 | 19 | 1 | 1 | c.3385C>T(p.Arg1129Cys) | B | N/A | N/A |
| c.858+2T>A | |||||||||
| 14 | F | 41 | 41 | 0 | 0.10 | c.2894A>G(p.Asn965Ser) | A | 1 | N/A |
| c.1531C>T(p.Arg511Cys) | |||||||||
| 15 | M | 7 | 13 | 0.49 | 0.60 | c.2919-1G>A * | C | 2 | 2 |
| 16 | F | 46 | 47 | 0.40 | 0.60 | c.42C>A(p.Asn14Lys) | A | 1 | 2 |
| c.1937+392G>A * | |||||||||
| 17 | F | 47 | 50 | 0.60 | 0.49 | c.6119G>A(p.Arg1040Gln) | A | 1 | 3 |
| c.2160+782T>C * | |||||||||
| 18 | M | 8 | 15 | 1.70 | 1.15 | c.6689del(p.Leu2230ProfsTer17) * | C | 3 | 3 |
| c.2633C>A(p.Ser878Ter) | |||||||||
| Variant | Type | SpliceAI Delta Score | Position | ΔMaxEntScan |
|---|---|---|---|---|
| c.6006-3C>A ⇒1:94471141 G>T | Acceptor Loss | 0.42 * | −3 bp | −3.702 |
| c.4253+5G>A ⇒1:94496547 C>T | Donor Loss | 0.44 * | +5 bp | −4.809 |
| c.1937+392G>A ⇒1:94527741 C>T | N/A | 0.01 | −4 bp | N/A |
| c.2160+782T>C ⇒1:94525311 A>G | N/A | 0.00 | N/A | N/A |
| Age of Onset (yrs) | BCVA in the Better Eye (logMAR) | Fundus Appearance | AF Type | ERG Type | |
|---|---|---|---|---|---|
| Mild (Group 1) | Later onset (>15) | <0.78 | 1 | 1 | 1 |
| Moderate (Group 2) | Patients who did not meet at least 2 criteria of either mild or severe phenotype | ||||
| Severe (Group 3) | Early onset (<10) | >1.0 | 3 | 3 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Liu, Z.; Cui, J.; Tan, C.; Sun, W.; Lin, Y. Clinical and Genetic Characteristics of 18 Patients from Southeast China with ABCA4-Associated Stargardt Disease. Int. J. Mol. Sci. 2025, 26, 3354. https://doi.org/10.3390/ijms26073354
Liu X, Liu Z, Cui J, Tan C, Sun W, Lin Y. Clinical and Genetic Characteristics of 18 Patients from Southeast China with ABCA4-Associated Stargardt Disease. International Journal of Molecular Sciences. 2025; 26(7):3354. https://doi.org/10.3390/ijms26073354
Chicago/Turabian StyleLiu, Xinyu, Zehao Liu, Jinli Cui, Chen Tan, Wenmin Sun, and Ying Lin. 2025. "Clinical and Genetic Characteristics of 18 Patients from Southeast China with ABCA4-Associated Stargardt Disease" International Journal of Molecular Sciences 26, no. 7: 3354. https://doi.org/10.3390/ijms26073354
APA StyleLiu, X., Liu, Z., Cui, J., Tan, C., Sun, W., & Lin, Y. (2025). Clinical and Genetic Characteristics of 18 Patients from Southeast China with ABCA4-Associated Stargardt Disease. International Journal of Molecular Sciences, 26(7), 3354. https://doi.org/10.3390/ijms26073354