
SRSF9-Mediated Exon Recognition Promotes Exon 2 Inclusion in Mecp2 Pre-mRNA Alternative Splicing



Abstract

1. Introduction
2. Results
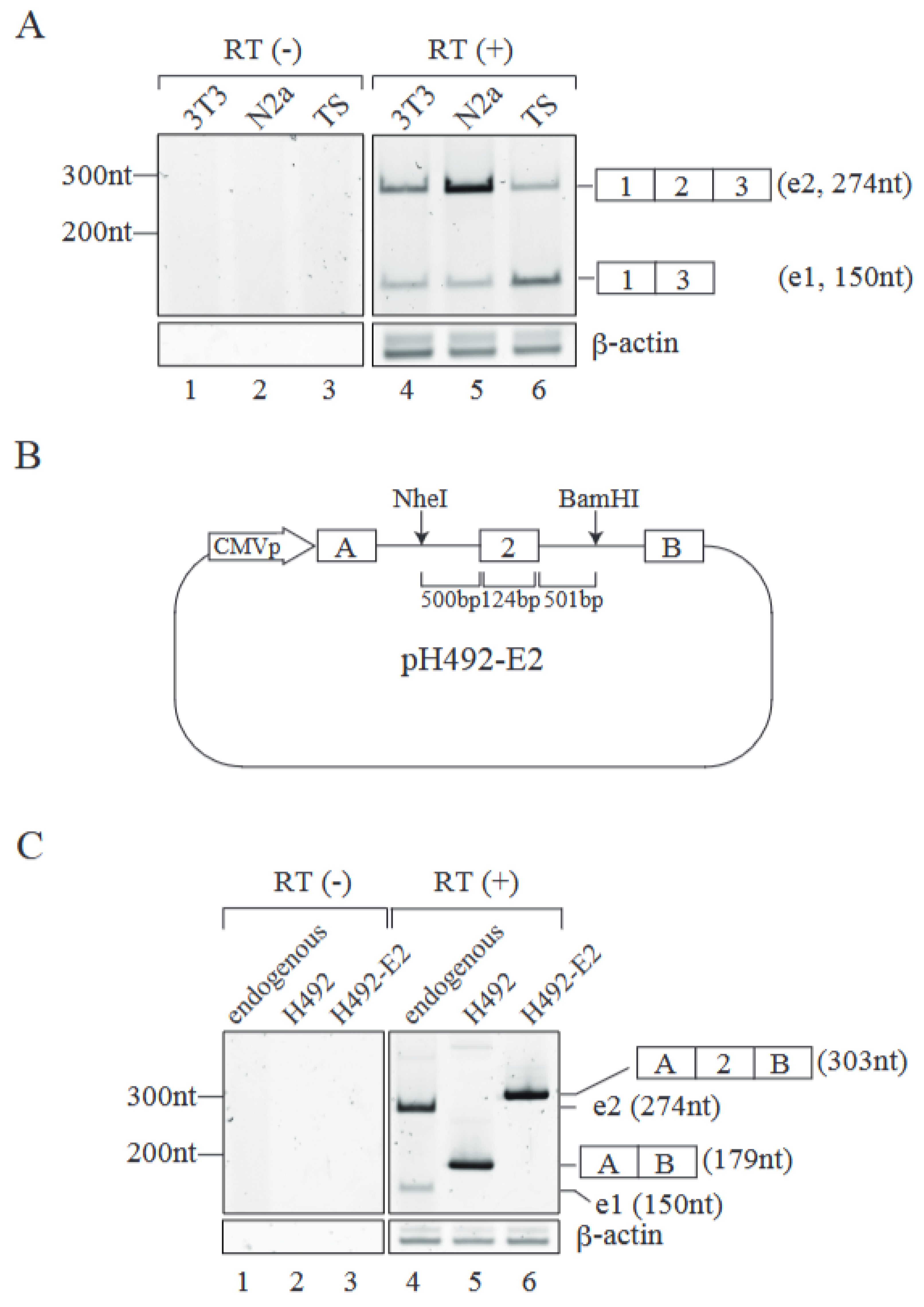
2.1. Establishment of an Alternative Splicing Reporter for Mecp2 Pre-mRNA
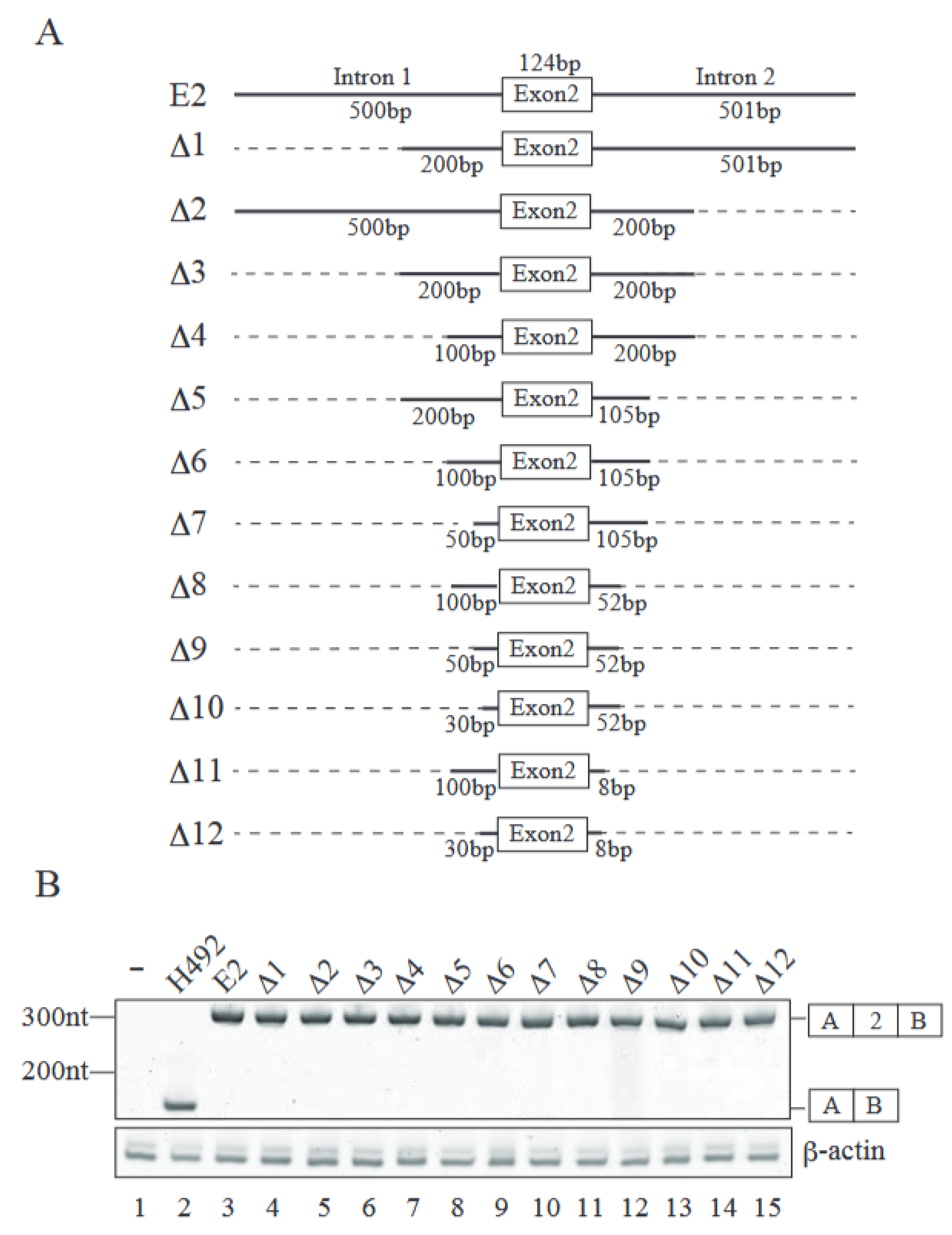
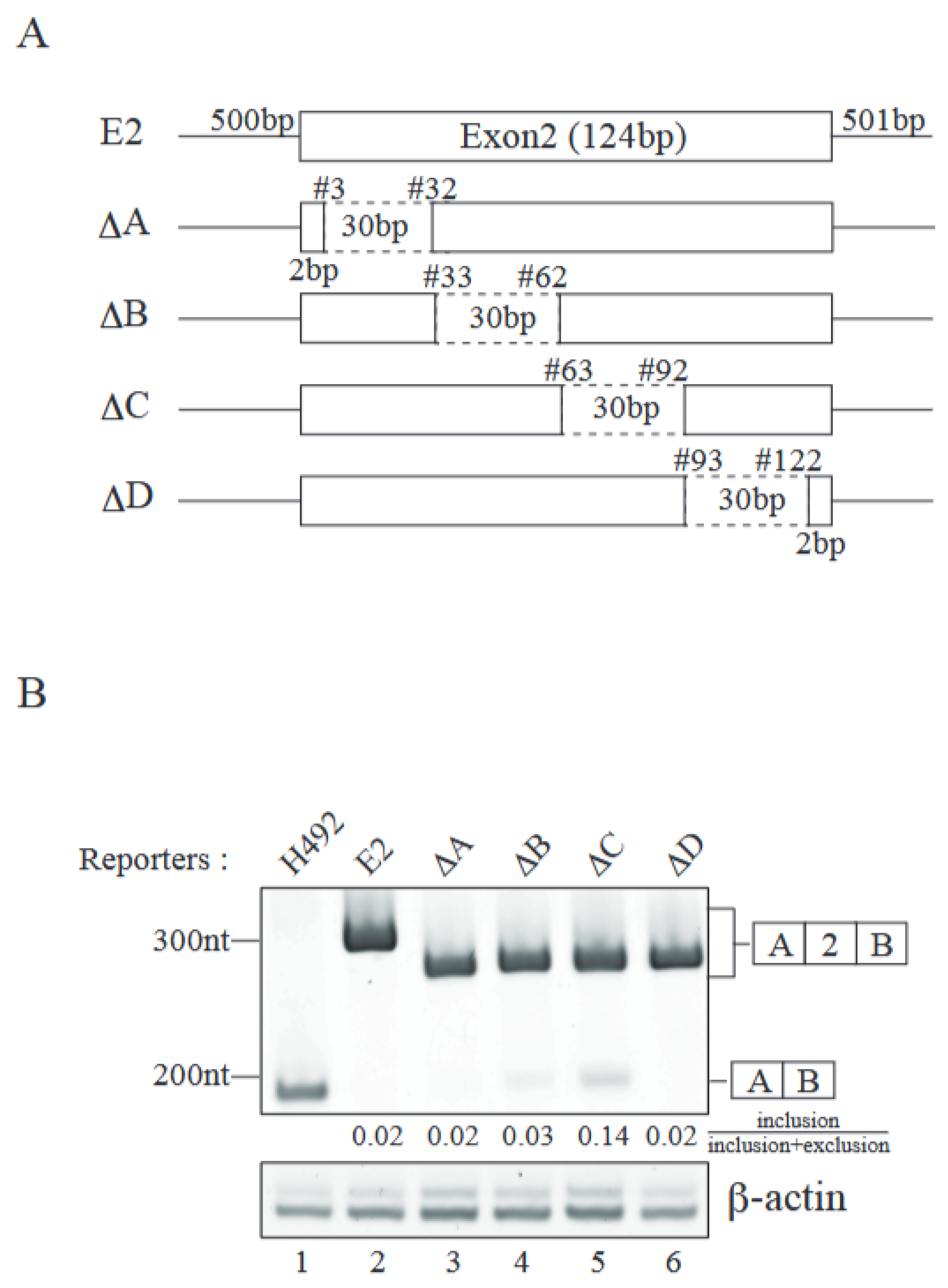
2.2. Identification of Cis-Regulatory Element(s) Required for Exon 2 Inclusion
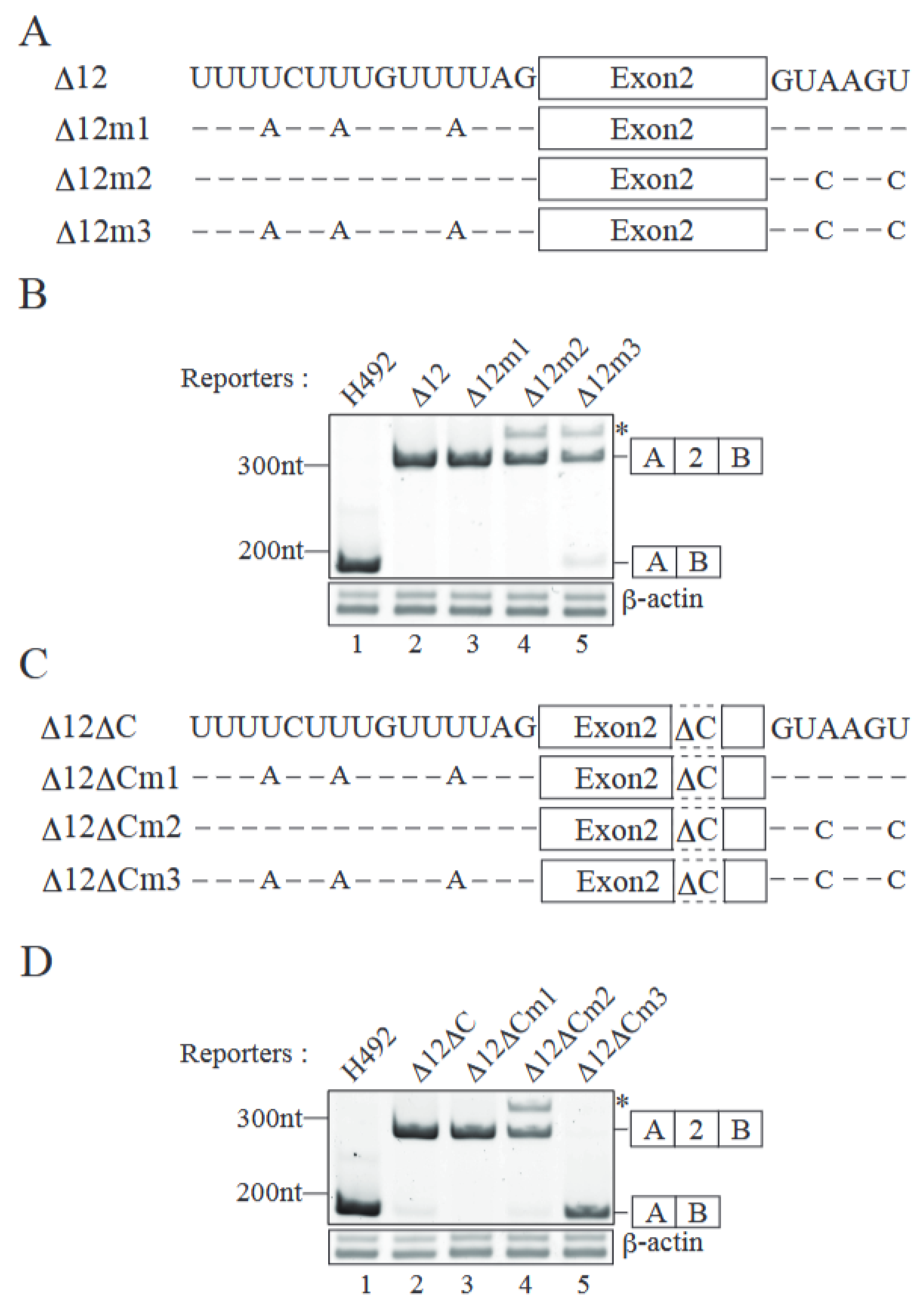
2.3. Both Splice Sites Adjacent to Exon 2 Contribute to Exon 2 Inclusion
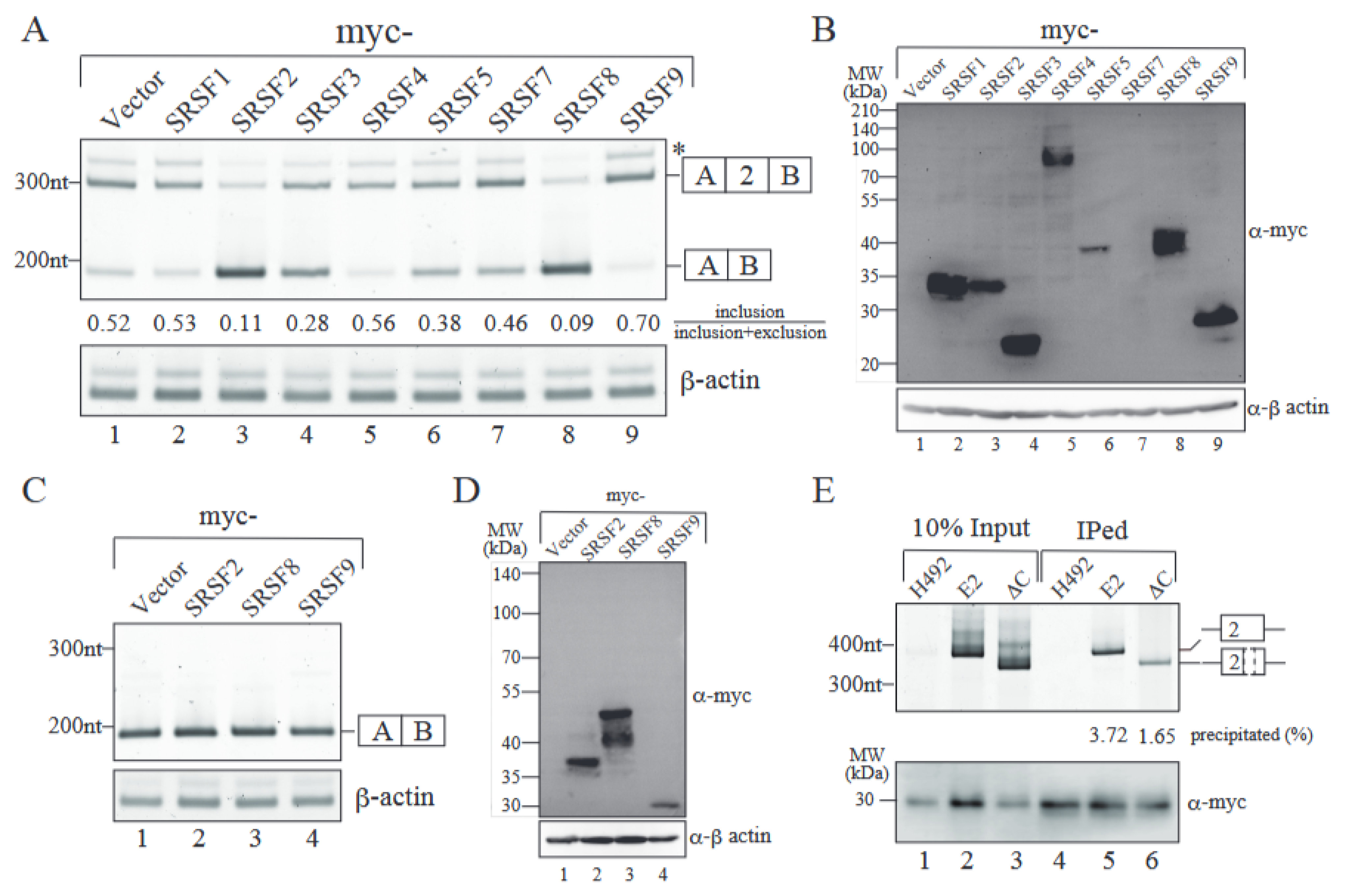
2.4. SRSF9 Promotes Exon 2 Inclusion Through Binding to Exon 2 Sequence
3. Discussion
4. Materials and Methods
4.1. Enzymes and Reagents
4.2. Plasmid Construction
4.3. Cell Culture and Transfection
4.4. RNA Preparation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.5. Antibodies and Immunoblotting Analyses
4.6. Calculation of the Splice Site Scores by MaxEntScan Program
4.7. RNA Immunoprecipitation (RIP) Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Berget, S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995, 270, 2411–2414. [Google Scholar] [CrossRef]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef]
- Manley, J.L.; Krainer, A.R. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes. Dev. 2010, 24, 1073–1074. [Google Scholar] [CrossRef]
- Collins, B.E.; Neul, J.L. Rett Syndrome and MECP2 Duplication Syndrome: Disorders of MeCP2 Dosage. Neuropsychiatr. Dis. Treat. 2022, 18, 2813–2835. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Bird, A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [PubMed]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Olson, C.O.; Ezeonwuka, C.; Rastegar, M. Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and astrocytes. PLoS ONE 2012, 7, e49763. [Google Scholar] [CrossRef]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.; Soto, C.J.; Saez, M.; Abrams, A.; Walz, K.; Young, J.I. Transgenic complementation of MeCP2 deficiency: Phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. Eur. J. Hum. Genet. 2012, 20, 69–76. [Google Scholar] [CrossRef]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Carolyn Schanen, N.; Olson, C.O.; et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef]
- Itoh, M.; Tahimic, C.G.; Ide, S.; Otsuki, A.; Sasaoka, T.; Noguchi, S.; Oshimura, M.; Goto, Y.; Kurimasa, A. Methyl CpG-binding protein isoform MeCP2_e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. J. Biol. Chem. 2012, 287, 13859–13867. [Google Scholar] [CrossRef]
- Martinez de Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 63. [Google Scholar] [CrossRef]
- Hayakawa, K.; Himeno, E.; Tanaka, S.; Kunath, T. Isolation and manipulation of mouse trophoblast stem cells. Curr. Protoc. Stem Cell Biol. 2015, 32, 1E.4.1–1E.4.32. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [PubMed]
- Nakura, T.; Ozoe, A.; Narita, Y.; Matsuo, M.; Hakuno, F.; Kataoka, N.; Takahashi, S.I. Rbfox2 mediates exon 11 inclusion in insulin receptor pre-mRNA splicing in hepatoma cells. Biochimie 2021, 187, 25–32. [Google Scholar] [CrossRef]
- Ohe, K.; Yoshida, M.; Nakano-Kobayashi, A.; Hosokawa, M.; Sako, Y.; Sakuma, M.; Okuno, Y.; Usui, T.; Ninomiya, K.; Nojima, T.; et al. RBM24 promotes U1 snRNP recognition of the mutated 5′ splice site in the IKBKAP gene of familial dysautonomia. RNA 2017, 23, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Shiota, K.; Kogo, Y.; Ohgane, J.; Imamura, T.; Urano, A.; Nishino, K.; Tanaka, S.; Hattori, N. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes. Cells 2002, 7, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Oshizuki, S.; Matsumoto, E.; Tanaka, S.; Kataoka, N. Mutations equivalent to Drosophila mago nashi mutants imply reduction of Magoh protein incorporation into exon junction complex. Genes. Cells 2022, 27, 505–511. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Construction | ||
| Plasmid | Primer Name | Primer (5′-3′) |
| E2 | F | AAAGCTAGCGAAGCAGAAGAGAGACTTTTGCCT |
| R | AAAGGATCCCTCCATTGTGCCGTGACACTTTCA | |
| ∆1 | F | AAAGCTAGCTTCTGACAGCCATAACTCTTGTCT |
| R | AAAGGATCCCTCCATTGTGCCGTGACACTTTCA | |
| ∆2 | F | AAAGCTAGCGAAGCAGAAGAGAGACTTTTGCCT |
| R | AAAGGATCCAAACTGAACTGAACTTTCAGGGCT | |
| ∆3 | F | AAAGCTAGCTTCTGACAGCCATAACTCTTGTCT |
| R | AAAGGATCCAAACTGAACTGAACTTTCAGGGCT | |
| ∆4 | F | AAAGCTAGCACATAGTATGTTGTCTGTTTATC |
| R | AAAGGATCCAAACTGAACTGAACTTTCAGGGCT | |
| ∆5 | F | AAAGCTAGCTTCTGACAGCCATAACTCTTGTCT |
| R | AAAGGATCCCATACTGTCCAAGAATAGTATATC | |
| ∆6 | F | AAAGCTAGCACATAGTATGTTGTCTGTTTATC |
| R | AAAGGATCCCATACTGTCCAAGAATAGTATATC | |
| ∆7 | F | AAAGCTAGCAAAGGTGCAGCTCAATGGGG |
| R | AAAGGATCCCATACTGTCCAAGAATAGTATATC | |
| ∆8 | F | AAAGCTAGCACATAGTATGTTGTCTGTTTATC |
| R | AAAGGATCCAGATGGCCAAACCAGGACATATAC | |
| ∆9 | F | AAAGCTAGCAAAGGTGCAGCTCAATGGGG |
| R | AAAGGATCCAGATGGCCAAACCAGGACATATAC | |
| ∆10 | F | AAAGCTAGCACTTTCAACTTACGATTTTCTTTG |
| R | AAAGGATCCAGATGGCCAAACCAGGACATATAC | |
| ∆11 | F | AAGCTAGCAAAGGTGCAGCTCAATGGGG |
| R | AAAGGATCCTTACTTACCTGAGCCCTAACATC | |
| ∆12 | F | AAAGCTAGCACTTTCAACTTACGATTTTCTTTG |
| R | AAAGGATCCTTACTTACCTGAGCCCTAACATC | |
| ∆A | F | GCCTAAAACAAAGAAAATCGTAAGTTG |
| R | CTTTGATGTGACATGTGACTCCC | |
| ∆B | F | CAGGAACTGGTGAGTCTGTATTTTTATG |
| R | CACCTTGCTTCTGTAGACCAGCTC | |
| ∆C | F | TATTCTGGGGAGTCACATGTCACATC |
| R | GATTCCATGGTAGCTGGGATGTTAG | |
| ∆D | F | CTGTTGGAGCTGGTCTACAGAAG |
| R | AGGTAAGTAACCTTCCTTTTTTTTTT | |
| ∆12m1 ∆12∆Cm1 | F | AAAGCTAGCACTTTCAACTTACGATTTACTATGTTATAG |
| R | AAAGGATCCTTACTTACCTGAGCCCTAACATC | |
| ∆12m2 ∆12∆Cm2 | F | AAAGCTAGCACTTTCAACTTACGATTTTCTTTG |
| R | AAAGGATCCTTGCTGACCTGAGCCCTAAC | |
| ∆12m3 ∆12∆Cm3 | F | AAAGCTAGCACTTTCAACTTACGATTTACTATGTTATAG |
| R | AAAGGATCCTTGCTGACCTGAGCCCTAAC | |
| RT-PCR | ||
| Target | Primer Name | Primer Sequence (5′-3′) |
| Mecp2 | F | GGTAAAACCCGTCCGGAAAATG |
| R | TTCAGTGGCTTGTCTCTGAG | |
| Actb | F | TTCTACAATGAGCTGCGTGTGG |
| R | ATGGCTGGGGTGTTGAAGGT | |
| Reporters | F | ATTACTCGCTCAGAAGCTGTGTTGC |
| R | AAGTCTCTCACTTAGCAACTGGCAG | |
| SP6 promoter | SP6 promoter primer | GATTTAGGTGACACTATAG |
| MeCP2 pre-mRNA | F | AAAGCTAGCACATAGTATGTTGTCTGTTTATC |
| MeCP2 pre-mRNA | R | AAAGGATCCCATACTGTCCAAGAATAGTATATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oshizuki, S.; Masaki, S.; Tanaka, S.; Kataoka, N. SRSF9-Mediated Exon Recognition Promotes Exon 2 Inclusion in Mecp2 Pre-mRNA Alternative Splicing. Int. J. Mol. Sci. 2025, 26, 3319. https://doi.org/10.3390/ijms26073319
Oshizuki S, Masaki S, Tanaka S, Kataoka N. SRSF9-Mediated Exon Recognition Promotes Exon 2 Inclusion in Mecp2 Pre-mRNA Alternative Splicing. International Journal of Molecular Sciences. 2025; 26(7):3319. https://doi.org/10.3390/ijms26073319
Chicago/Turabian StyleOshizuki, Saya, So Masaki, Satoshi Tanaka, and Naoyuki Kataoka. 2025. "SRSF9-Mediated Exon Recognition Promotes Exon 2 Inclusion in Mecp2 Pre-mRNA Alternative Splicing" International Journal of Molecular Sciences 26, no. 7: 3319. https://doi.org/10.3390/ijms26073319
APA StyleOshizuki, S., Masaki, S., Tanaka, S., & Kataoka, N. (2025). SRSF9-Mediated Exon Recognition Promotes Exon 2 Inclusion in Mecp2 Pre-mRNA Alternative Splicing. International Journal of Molecular Sciences, 26(7), 3319. https://doi.org/10.3390/ijms26073319