
Suitability of Real-Time PCR Methods for New Genomic Technique Detection in the Context of the European Regulations: A Case Study in Arabidopsis

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
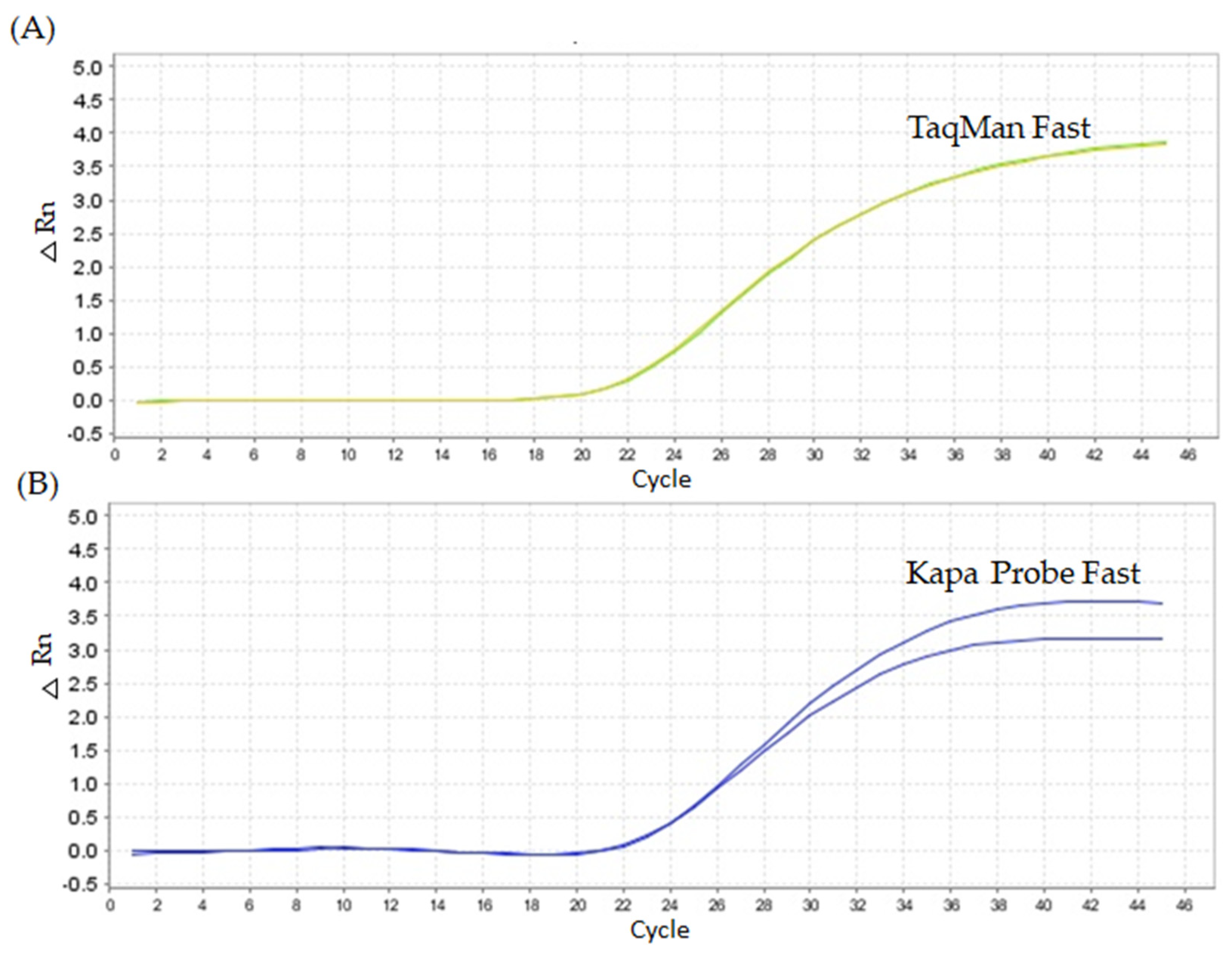
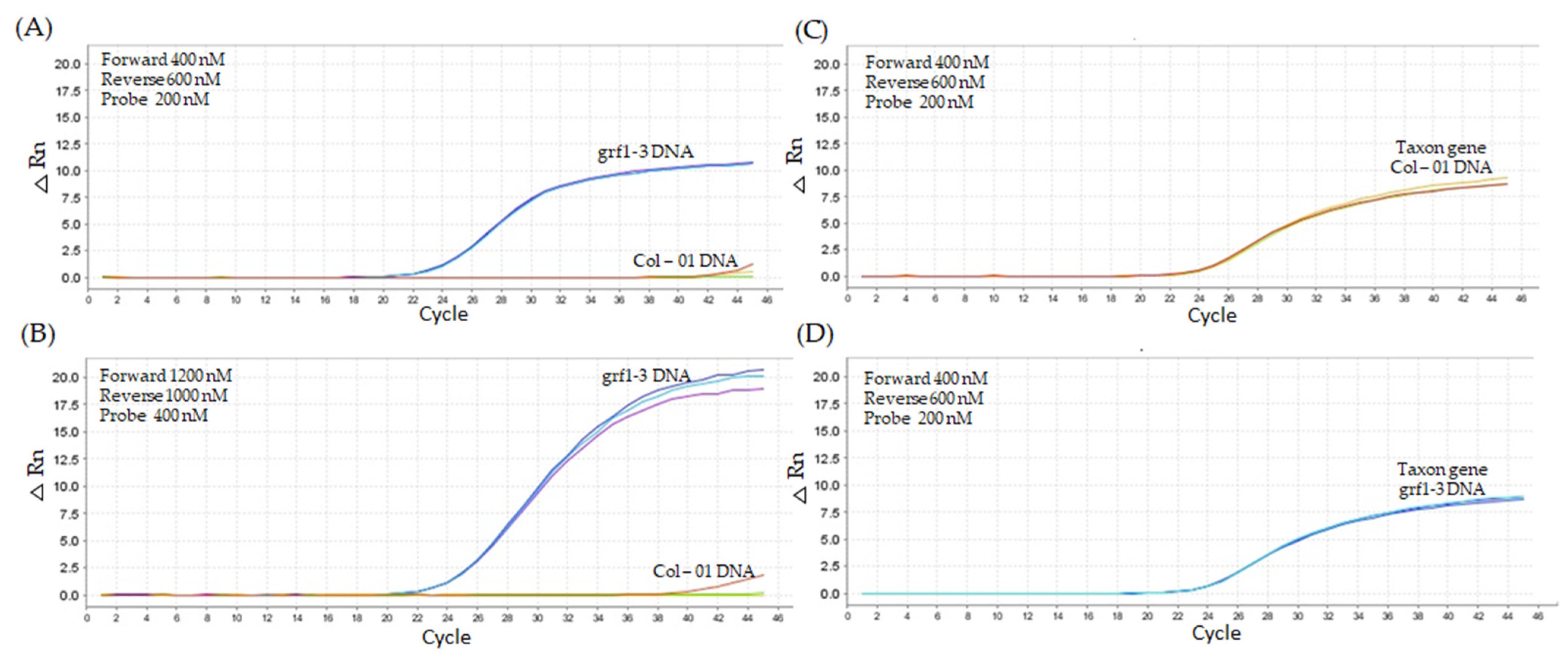
2.1. Quantitative PCR Method Using Unmodified Taqman™ Primers and Probe
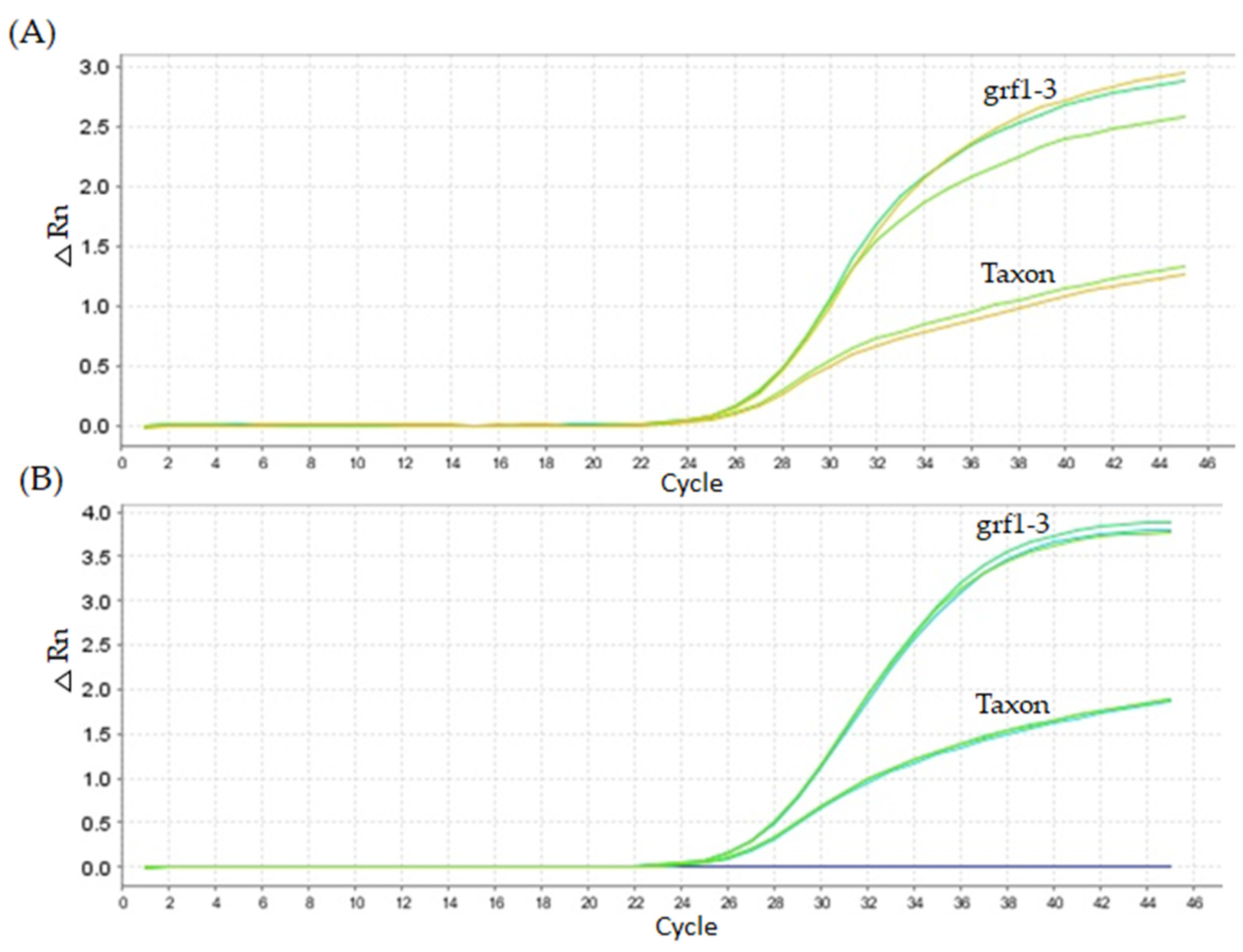
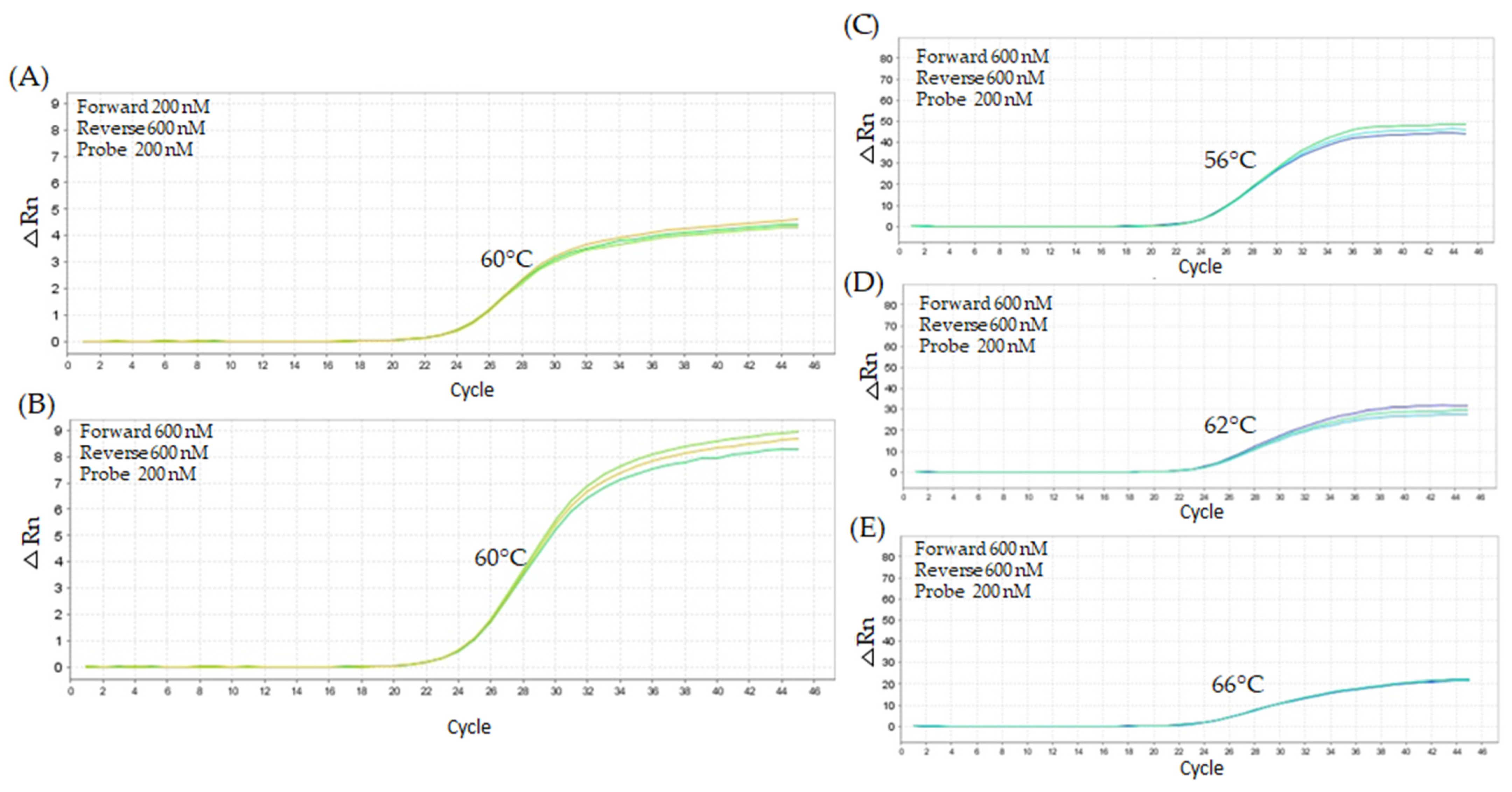
2.2. Quantitative PCR Method Using Modified LNA Primers
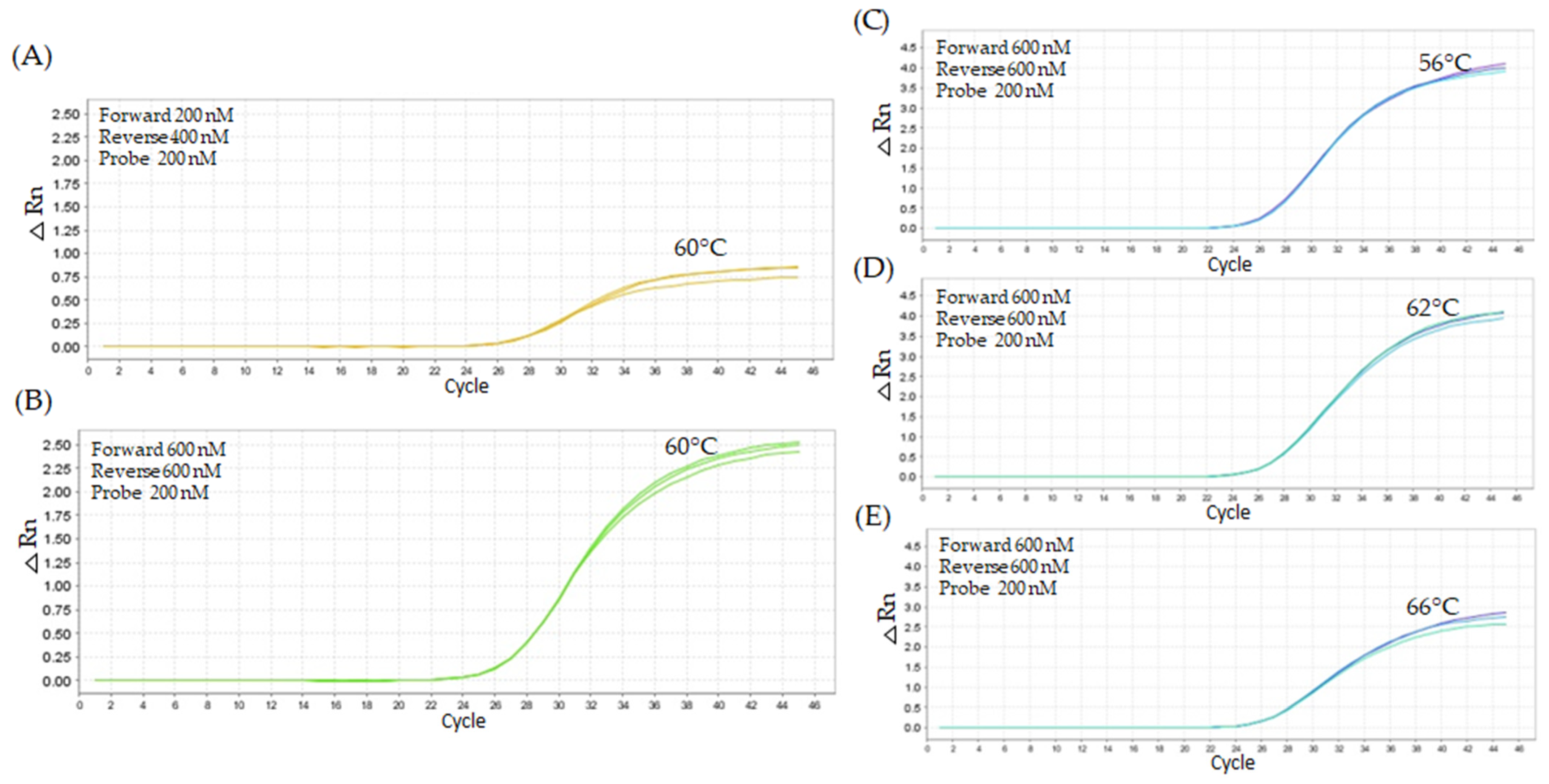
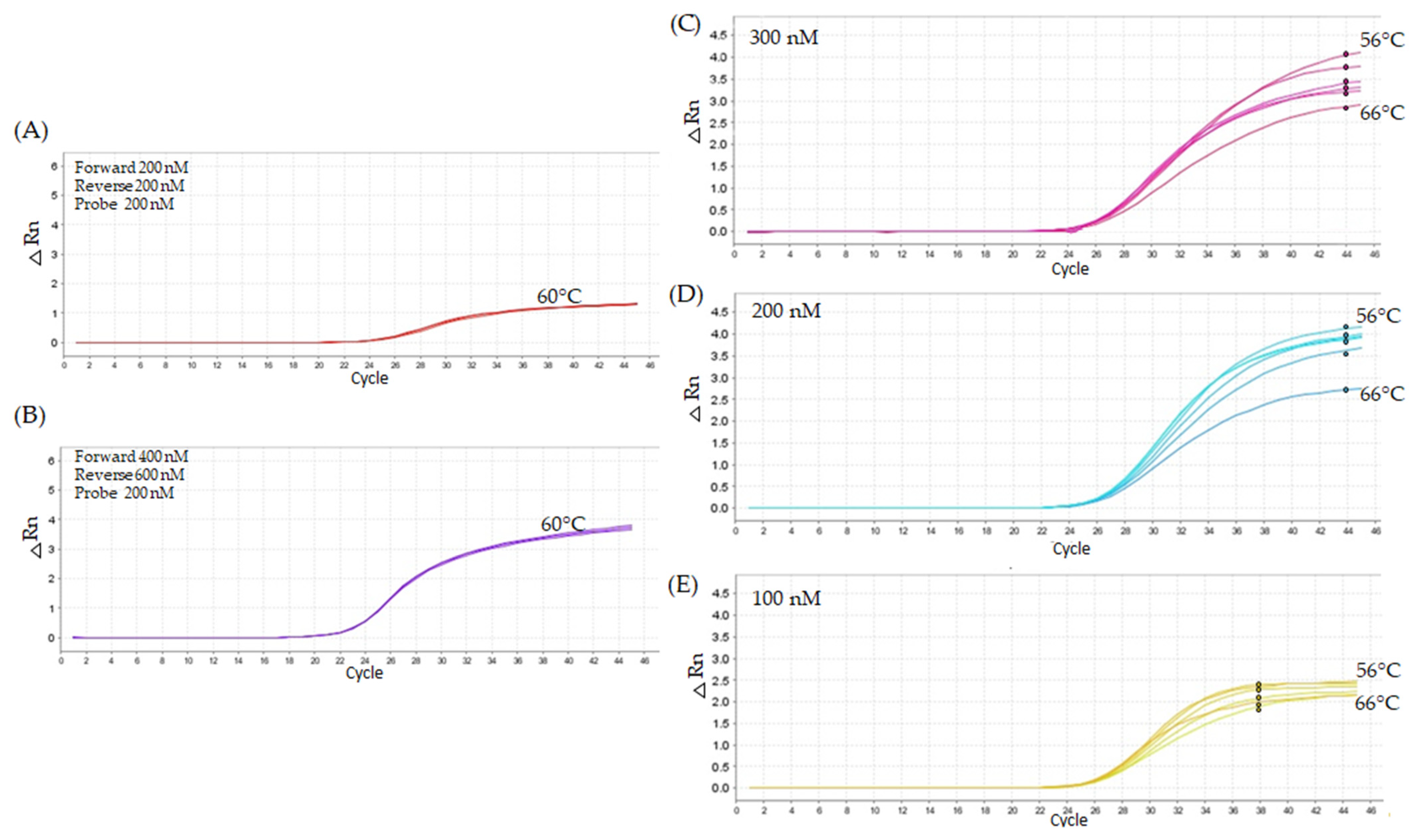
2.2.1. First Titration Assay—Lower Oligonucleotide Concentrations
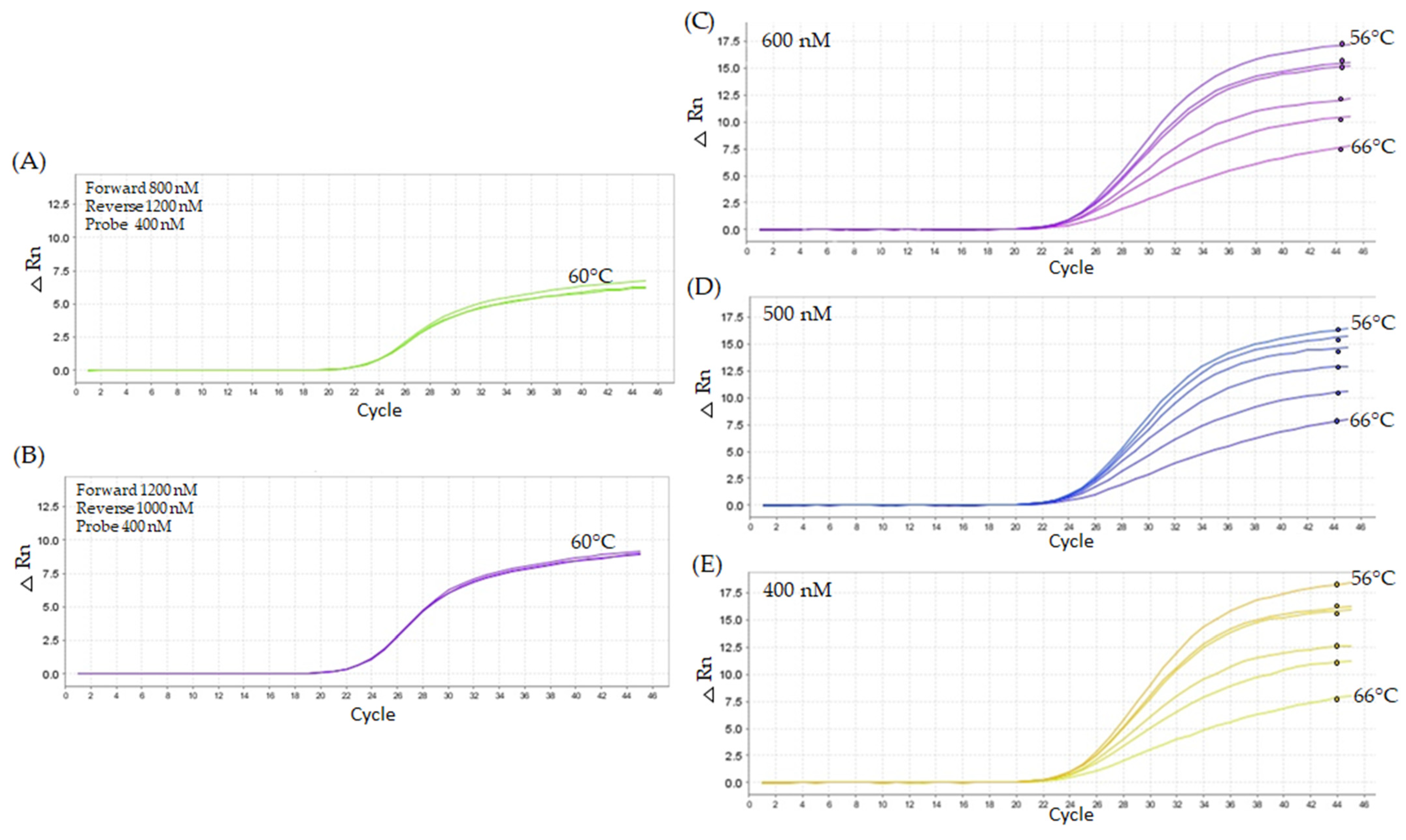
2.2.2. Second Titration Assay—Higher Oligonucleotide Concentrations
2.2.3. Third Assay—Replication of Best Titration Results
2.3. Interlaboratory Validation Results
2.3.1. Limit of Detection (LOD)
2.3.2. Robustness
2.3.3. Trueness and Precision
2.3.4. Limit of Quantification (LOQ)
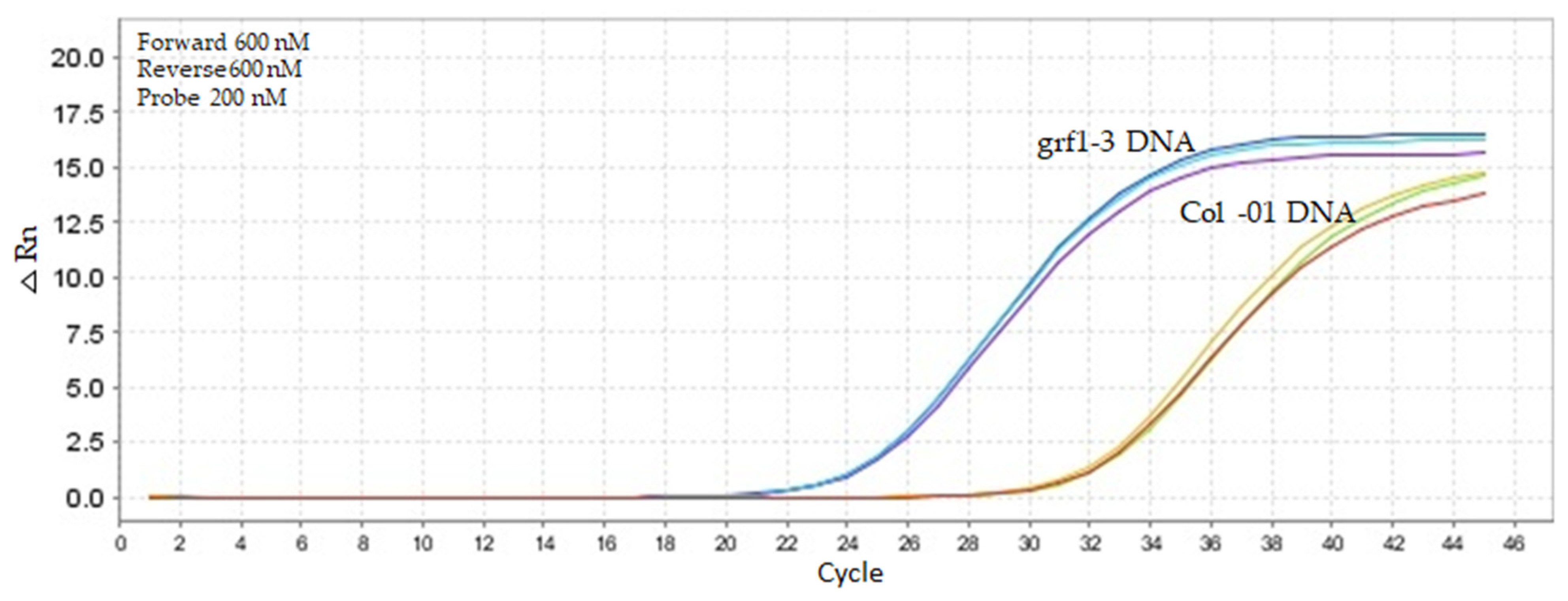
2.4. In Vitro Specificity Testing for the Newly Established qPCR Method
3. Discussion
3.1. Suitability of Enhanced Real-Time PCR Systems for NGT Detection
3.2. Advantages of Other Analytical Methods for NGT Detection
3.3. State of the Art for NGT Detection Methods in the Context of the European Regulations
4. Material and Methods
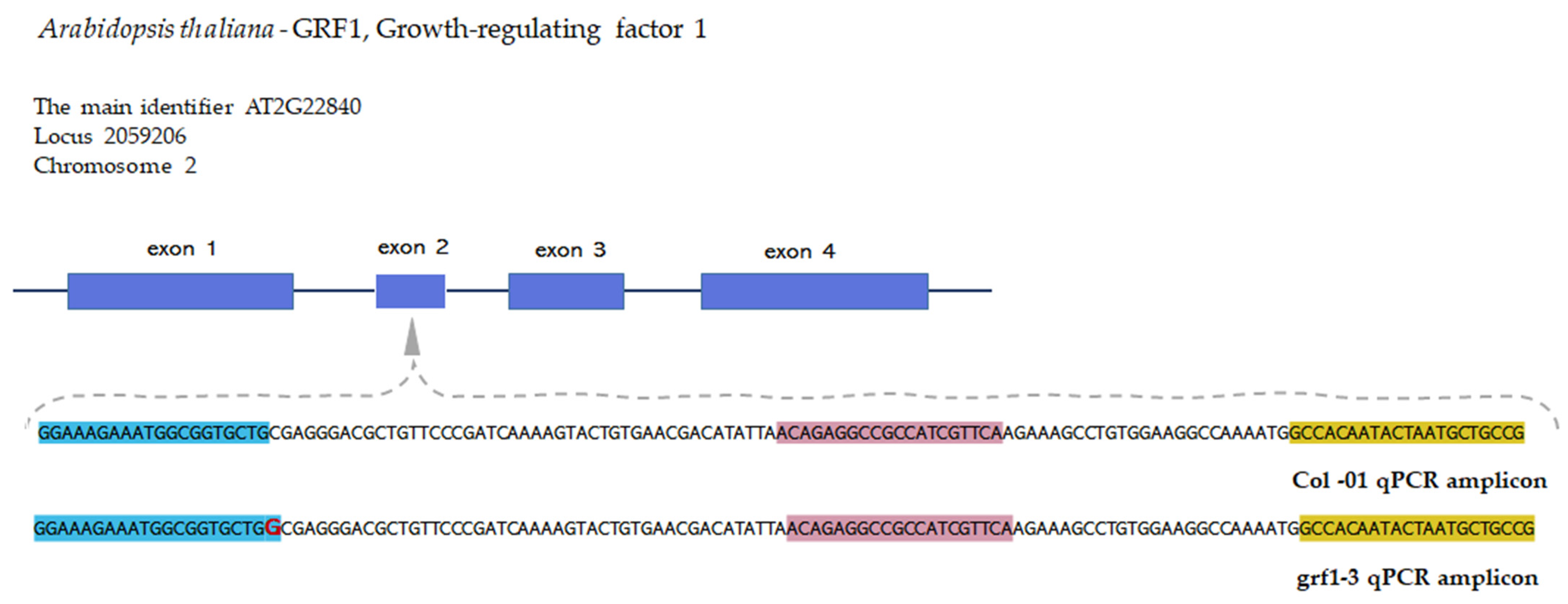
4.1. Arabidopsis Germplasm
4.2. Extraction of Genomic DNA
4.3. Oligonucleotides Primers and Probes
4.4. Real-Time Quantitative PCR Method Development
4.5. Inter-Laboratory Method Validation
4.5.1. Trueness and Precision Parameters
4.5.2. Robustness Parameters
4.5.3. LOD-LOQ Parameters
4.5.4. Specificity Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Parliament and Council of the European Union. Directive 2001/18/EC on the Deliberate Release into the Environment of Genetically Modified Organisms and Repealing Council Directive 90/220/EEC; EUR-Lex: EU law: 2001; European Union: Brussels, Belgium, 2001. [Google Scholar]
- Ribarits, A.; Eckerstorfer, M.; Simon, S.; Stepanek, W. Genome-edited plants: Opportunities and challenges for an anticipatory detection and identification framework. Foods 2021, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Eckerstorfer, M.; Zanon Agapito-Tenfen, S.; Kleter, G.A. Editorial: Genome-edited organisms for agriculture—Challenges and perspectives for development and regulation. Front. Genome Ed. 2023, 5, 1287973. [Google Scholar] [CrossRef] [PubMed]
- Dolezel, M.; Lang, A.; Greiter, A.; Miklau, M.; Eckerstorfer, M.; Heissenberger, A.; Willée, E.; Züghart, W. Challenges for the post-market environmental monitoring in the European Union imposed by novel applications of genetically modified and genome-edited organisms. BioTech 2024, 13, 14. [Google Scholar] [CrossRef]
- Teufel, J.; López Hernández, V.; Greiter, A.; Kampffmeyer, N.; Hilbert, I.; Eckerstorfer, M.; Narendja, F.; Heissenberger, A.; Simon, S. Strategies for traceability to prevent unauthorised GMOs (including NGTs) in the EU: State of the art and possible alternative approaches. Foods 2024, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, C.B.; Hoepers, A.M.; Nodari, R.O.; Agapito-Tenfen, S.Z. Specificity testing for NGT PCR-based detection methods in the context of the EU GMO regulations. Foods 2023, 12, 4298. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Guiderdoni, E.; Meunier, A.C.; Papazova, N.; Roosens, N. ddPCR strategy to detect a gene-edited plant carrying a single variation point: Technical feasibility and interpretation issues. Food Control. 2022, 137, 108904. [Google Scholar] [CrossRef]
- Peng, C.; Zheng, M.; Ding, L.; Chen, X.; Wang, X.; Feng, X.; Wang, J.; Xu, J. Accurate detection and evaluation of the gene-editing frequency in plants using droplet digital PCR. Front. Plant Sci. 2020, 11, 610790. [Google Scholar] [CrossRef]
- Fraiture, M.A.; D’aes, J.; Guiderdoni, E.; Meunier, A.C.; Delcourt, T.; Hoffman, S.; Vandermassen, E.; De Keersmaecker, S.C.J.; Vanneste, K.; Roosens, N.H.C. Targeted high-throughput sequencing enables the detection of single nucleotide variations in CRISPR/Cas9 gene-edited organisms. Foods 2023, 12, 455. [Google Scholar] [CrossRef]
- Zhang, Q.; Xing, H.L.; Wang, Z.P.; Zhang, H.Y.; Yang, F.; Wang, X.C.; Chen, Q.J. Potential high-frequency off-target mutagenesis induced by CRISPR/Cas9 in Arabidopsis and its prevention. Plant Mol. Biol. 2018, 96, 445–456. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, Y.; Yao, S.; Wei, Y. A PCR-based protocol for detecting indel mutations induced by TALENs and CRISPR/Cas9 in zebrafish. PLoS ONE 2014, 9, e98282. [Google Scholar] [CrossRef]
- Zhang, Y.; Iaffaldano, B.; Qi, Y. CRISPR ribonucleoprotein-mediated genetic engineering in plants. Plant Commun. 2021, 2, 100168. [Google Scholar] [CrossRef] [PubMed]
- Chhalliyil, P.; Ilves, H.; Kazakov, S.A.; Howard, S.J.; Johnston, B.H.; Fagan, J. A real-time quantitative PCR method specific for detection and quantification of the first commercialized genome-edited plant. Foods 2020, 9, 1245. [Google Scholar] [CrossRef]
- Weidner, C.; Edelmann, S.; Moor, D.; Lieske, K.; Savini, C.; Jacchia, S.; Sacco, M.G.; Mazzara, M.; Lämke, J.; Eckermann, K.N.; et al. Assessment of the real-time PCR method claiming to be specific for detection and quantification of the first commercialised genome-edited plant. Food Anal. Methods 2022, 15, 2107–2125. [Google Scholar] [CrossRef]
- Grohmann, L.; Broll, H.; Dagand, E.; Hildebrandt, S.; Hübert, P.; Kiesecker, H.; Lieske, K.; Mäde, D.; Mankertz, J.; Reiting, J.; et al. Guidelines for the Single-Laboratory Validation of Qualitative Real-Time PCR Methods; Bundesamt f ̧r Verbraucherschutz und Lebensmittelsicherheit (BVL): Braunschweig, Germany, 2016. [Google Scholar]
- Hougs, L.; Gatto, F.; Goerlich, O.; Grohmann, L.; Lieske, K.; Mazzara, M.; Narendja, F.; Ovesna, J.; Papazova, N.; Scholtens, I.; et al. Verification of Analytical Methods for GMO Testing When Implementing Interlaboratory Validated Methods: Version 2. EUR 29015 EN; JRC109940; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar] [CrossRef]
- Grohmann, L.; Broll, H.; Dagand, E.; Hildebrandt, S.; Hübert, P.; Kiesecker, H.; Lieske, K.; Mäde, D.; Mankertz, J.; Reiting, J.; et al. Guidelines for the Validation of Qualitative Real-Time PCR Methods by Means of a Collaborative Study; Bundesamt f ̧r Verbraucherschutz und Lebensmittelsicherheit (BVL): Braunschweig, Germany, 2016. [Google Scholar]
- Gatto, F.; Savini, C.; Baillie, C.K.; Broothaerts, W.; Burns, M.; Dagand, E.; Debode, F.; Dobnik, D.; Grantina-Levina, L.; Grohmann, L.; et al. Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing Part 2; Publications Office of the European Union: Luxembourg, 2023; 32p. [Google Scholar] [CrossRef]
- Weidner, C.; Köppel, R.; Freyer, R.; Richl, P.; Lieske, K.; Mankertz, J.; Waiblinger, H.U. Development and validation of a multiplex real-time PCR method for screening genetically modified plants. J. Consum. Prot. Food Saf. 2024, 19, 165–174. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Zhao, S.; Yan, X.; Si, N.; Gao, H.; Li, Y.; Zhai, S.; Xiao, F.; Wu, G.; et al. An editing-site-specific PCR method for detection and quantification of CAO1-edited rice. Foods 2021, 10, 1209. [Google Scholar] [CrossRef]
- Ishii, K.; Fukui, M. Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl. Environ. Microbiol. 2001, 67, 3753–3755. [Google Scholar] [CrossRef]
- Grohmann, L.; Keilwagen, J.; Duensing, N.; Dagand, E.; Hartung, F.; Wilhelm, R.; Bendiek, J.; Sprink, T. Detection and identification of genome editing in plants: Challenges and opportunities. Front. Plant Sci. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Bektaş, A.; Chapela, I. Loop-mediated isothermal amplification of single pollen grains. J. Integr. Plant Biol. 2014, 56, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Gibriel, A.A.; Adel, O. Advances in ligase chain reaction and ligation-based amplifications for genotyping assays: Detection and applications. Mutat. Res. Rev. Mutat. Res. 2017, 773, 66–90. [Google Scholar] [CrossRef]
- Dobosy, J.R.; Rose, S.D.; Beltz, K.R.; Rupp, S.M.; Powers, K.M.; Behlke, M.A.; Walder, J.A. RNase H-dependent PCR (rhPCR): Improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011, 11, 80. [Google Scholar] [CrossRef]
- Ayalew, H.; Tsang, P.W.; Chu, C.; Wang, J.; Liu, S.; Chen, C.; Ma, X.F. Comparison of TaqMan, KASP and rhAmp SNP genotyping platforms in hexaploid wheat. PLoS ONE 2019, 14, e0217222. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Mei, D.Y.; Liu, Q.N.; Qiao, X.H.; Ruan, W.M.; Huang, T.; Cao, G.S. Research of methods to detect genomic mutations induced by CRISPR/Cas systems. J. Biotechnol. 2015, 214, 128–132. [Google Scholar] [CrossRef]
- Eckerstorfer, M.F.; Engelhard, M.; Heissenberger, A.; Simon, S.; Teichmann, H. Plants developed by new genetic modification techniques—Comparison of existing regulatory frameworks in the EU and non-EU countries. Front. Bioeng. Biotechnol. 2019, 7, 26. [Google Scholar] [CrossRef]
- Gurtler, P.; Baillie, C.-K.; Goerlich, O.; Estendorfer-Rinner, S.; Baiker, A. Genome Editing; [Bayerisches Landesamt für Gesundheit und Lebensmittelsicherheit (LGL)]: Erlangen, Germany, 2019. [Google Scholar]
- Guertler, P.; Pallarz, S.; Belter, A.; Eckermann, K.N.; Grohmann, L. Detection of commercialized plant products derived from new genomic techniques (NGT)Practical examples and current perspectives. Food Control 2023, 152, 109869. [Google Scholar] [CrossRef]
- Zischewski, J.; Fischer, R.; Bortesi, L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017, 35, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Holst-Jensen, A.; Bertheau, Y.; de Loose, M.; Grohmann, L.; Hamels, S.; Hougs, L.; Morisset, D.; Pecoraro, S.; Pla, M.; Van den Bulcke, M.; et al. Detecting un-authorized genetically modified organisms (GMOs) and derived materials. Biotechnol. Adv. 2012, 30, 1318–1335. [Google Scholar] [CrossRef]
- Datukishvili, N.; Kutateladze, T.; Gabriadze, I.; Bitskinashvili, K.; Vishnepolsky, B. New multiplex PCR methods for rapid screening of genetically modified organisms in foods. Front. Microbiol. 2015, 6, 757. [Google Scholar] [CrossRef] [PubMed]
- Höijer, I.; Johansson, J.; Gudmundsson, S.; Chin, C.S.; Bunikis, I.; Häggqvist, S.; Emmanouilidou, A.; Wilbe, M.; den Hoed, M.; Bondeson, M.L.; et al. Amplification-free long-read sequencing reveals unforeseen CRISPR-Cas9 off-target activity. Genome Biol. 2020, 21, 290. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Y.; Yu, H.; Chen, X.; Xie, J. An integrated strategy for analyzing the complete complex integrated structure of maize MON810 and identification of an SNP in external insertion sequences. Plants 2024, 13, 2276. [Google Scholar] [CrossRef]
- Smelik, M.; Zhao, Y.; Li, X.; Loscalzo, J.; Sysoev, O.; Mahmud, F.; Mansour Aly, D.; Benson, M. An interactive atlas of genomic, proteomic, and metabolomic biomarkers promotes the potential of proteins to predict complex diseases. Sci. Rep. 2024, 14, 12710. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Herman, P.; Taverniers, I.; De Loose, M.; Deforce, D.; Roosens, N.H. Current and new approaches in GMO detection: Challenges and solutions. Biomed Res. Int. 2015, 2015, 392872. [Google Scholar] [CrossRef]
- Metje-Sprink, J.; Menz, J.; Modrzejewski, D.; Sprink, T. DNA-free genome editing: Past, present and future. Front. Plant Sci. 2019, 9, 1957. [Google Scholar] [CrossRef]
- Bonfini, L.; Van den Bulcke, M.H.; Mazzara, M.; Ben, E.; Patak, A. GMOMETHODS: The European Union database of reference methods for GMO analysis. J. AOAC Int. 2012, 95, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- European Network of GMO Laboratories. Detection of Food and Feed Plant Products Obtained by New Mutagenesis Techniques. 2020. Available online: https://gmo-crl.jrc.ec.europa.eu/doc/JRC116289-GE-report-ENGL.pdf (accessed on 28 October 2024).
- Wahler, D.; Schauser, L.; Bendiek, J.; Grohmann, L. Next-generation sequencing as a tool for detailed molecular characterisation of genomic insertions and flanking regions in genetically modified plants: A pilot study using a rice event unauthorised in the EU. Food Anal. Methods 2013, 6, 1718–1727. [Google Scholar] [CrossRef]
- Broothaerts, W.; Savini, C.; Mazzara, M.; Sowa, S. Detection of Food and Feed Plant Products Obtained by Targeted Mutagenesis and Cisgenesis; JRC133689; Publications Office of the European Union: Luxembourg, 2023. [Google Scholar] [CrossRef]
- European Network of GMO Laboratories (ENGL). Detection of Food and Feed Plant Products Obtained by New Mutagenesis Techniques; [European Union Reference Laboratory for GM Food and Feed]: Ispra, Italy, 2019. [Google Scholar]
- Angulo, J.; Astin, C.P.; Bauer, O.; Blash, K.J.; Bowen, N.M.; Chukwudinma, N.J.; DiNofrio, A.S.; Faletti, D.O.; Ghulam, A.M.; Gusinde-Duffy, C.M.; et al. CRISPR/Cas9 mutagenesis of the Arabidopsis GROWTH-REGULATING FACTOR (GRF) gene family. Front. Genome Ed. 2023, 5, 1251557. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, X.; Hasi, A.; Wang, Z. Structural and functional analysis of a bidirectional promoter from Gossypium hirsutum in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 3291. [Google Scholar] [CrossRef]
- Johnston, J.S.; Pepper, A.E.; Hall, A.E.; Chen, Z.J.; Hodnett, G.; Drabek, J.; Lopez, R.; Price, H.J. Evolution of genome size in Brassicaceae. Ann. Bot. 2005, 95, 229–235. [Google Scholar] [CrossRef]
- Joint Research Centre (European Commission); Köppel, R.; Scholtens, I.; Sovová, T.; Marmin, L.; Narendja, F.; Weidner, C.; Owen, H.; Eriksson, R.; Mazzara, M.; et al. Detection of Food and Feed Plant Products Obtained by Targeted Mutagenesis and Cisgenesis; Report No.: JRC133689; Publications Office of the European Union: Luxembourg, 2021. [Google Scholar] [CrossRef]
- Angers-Loustau, A.; Petrillo, M.; Bonfini, L.; Gatto, F.; Rosa, S.; Patak, A.; Kreysa, J. JRC GMO-Matrix: A web application to support Genetically Modified Organisms detection strategies. BMC Bioinform. 2014, 15, 417. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Forward Primer Concentration (nM) | Reverse Primer Concentration (nM) | Probe Concentration (nM) | Annealing Temperature (°C) | Cq Value | Fluorescence (ΔRn) |
|---|---|---|---|---|---|---|
| 1 | 200 | 200 | 200 | 60 | 26.02 | ~2 |
| 2 | 200 | 400 | 200 | 60 | 24.35 | ~2 |
| 3 | 200 | 600 | 200 | 60 | 24.49 | 2 |
| 4 | 400 | 200 | 200 | 60 | 23.14 | ~2 |
| 5 | 400 | 400 | 200 | 60 | 22.03 | ~3 |
| 6 | 400 | 600 | 200 | 60 | 22.19 | ~3 |
| 7 | 600 | 200 | 200 | 60 | 22.53 | 2 |
| 8 | 600 | 400 | 200 | 60 | 21.46 | 4 |
| 9 | 600 | 600 | 200 | 60 | 21.49 | 5 |
| Experiment | Forward Primer Concentration (nM) | Reverse Primer Concentration (nM) | Probe Concentration (nM) | Temperature (°C) | Cq Value | Fluorescence (ΔRn) |
|---|---|---|---|---|---|---|
| 1 | 800 | 800 | 400 | 60 | 23.60 | ~6 |
| 2 | 800 | 1000 | 400 | 60 | 23.04 | ~10 |
| 3 | 800 | 1200 | 400 | 60 | 23.93 | ~6 |
| 4 | 1000 | 800 | 400 | 60 | 23.55 | ~7.5 |
| 5 | 1000 | 1000 | 400 | 60 | 22.97 | ~11 |
| 6 | 1000 | 1200 | 400 | 60 | 23.36 | ~9 |
| 7 | 1200 | 800 | 400 | 60 | 23.37 | ~8.5 |
| 8 | 1200 | 1000 | 400 | 60 | 23.46 | ~9 |
| 9 | 1200 | 1200 | 400 | 60 | 23.04 | ~12 (replicates variation) |
| LOD | ||
|---|---|---|
| Nominal Copy Number of Target Sequence | N° of Replicates | N° of Positive |
| 40 | 12 | 12 |
| 20 | 12 | 12 |
| 10 | 12 | 12 |
| 5 | 12 | 12 |
| 1 | 12 | 9 |
| 0.1 | 12 | 2 |
| Factor-Level Combination | ||||||||
|---|---|---|---|---|---|---|---|---|
| Condition | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Real-time PCR equipment | ABI7500 | ABI7500 | ABI7500 | ABI7500 | CFX96 | CFX96 | CFX96 | CFX96 |
| PCR reagent kit | KAPA | KAPA | Qiagen | Qiagen | KAPA | KAPA | Qiagen | Qiagen |
| Annealing temperature (°C) | 63 | 61 | 63 | 61 | 63 | 61 | 63 | 61 |
| Master mix volume (µL) | 19 | 19 | 21 | 21 | 21 | 21 | 19 | 21 |
| Primer concentration (nM) | 1200 | 840 | 1200 | 840 | 700 | 1000 | 700 | 1000 |
| Probe concentration (nM) | 400 | 280 | 280 | 400 | 280 | 400 | 400 | 280 |
| Target % grf1-3 Level | Measured % grf1-3 Level | Precision (RSDr%) | Bias % of Target grf1-3 Level |
|---|---|---|---|
| 0.1 | 0.09 | 15.10 | −8.23 |
| 1 | 1.106 | 7.58 | 10.60 |
| 10 | 10.154 | 6.16 | 1.54 |
| LOQ | |||
|---|---|---|---|
| Nominal Copy Number of Target Sequence | Copy Number Measured | SD | RSDr |
| 40 | 48.4 | 9.9 | 20.6 |
| 20 | 24.5 | 9.3 | 37.9 |
| 10 | 14.1 | 5.2 | 36.5 |
| 5 | 6.9 | 3.6 | 52.5 |
| 1 | 2.2 | 1.3 | 58.3 |
| 0.1 | 1.2 | 0.2 | 16.5 |
| Species Hits | Gene ID | Forward Oligonucleotide Sequence | Reverse Oligonucleotide Sequence | Probe Sequence |
|---|---|---|---|---|
| Brassica napus | XM_0487528; XM_0138297; HG994367.1; HG994357.1. | GGAAAGAAATGGCGGTGCT- | GCCACAATACTAATGCTaCCG | ACAGAGGCCGCCATCGTTCA |
| Brassica rapa | LS974619.2 | GGAAAGAAATGGCGGTGCT- | GCCACAATACTAATGCTaCCG | ACAGAGGCCGCCATCGTTCA |
| Sample | DNA [] (ng) | Target Gene | Average Cq | Target Gene | Average Cq |
|---|---|---|---|---|---|
| grf3-9 | 15 | grf1-3 | 36.7 | RG | 21.8 |
| grf4-17 | 15 | grf1-3 | 34.7 | RG | 21.7 |
| grf8-61 | 15 | grf1-3 | 41.3 | RG | 26.79 |
| grf1-3 | 15 | grf1-3 | 18.9 | RG | 21.2 |
| Col-01 | 15 | grf1-3 | 35.8 | RG | 21.9 |
| GT73 | 100 | grf1-3 | 42.88 | cruA | 26.2 |
| MS8 | 100 | grf1-3 | 37.16 | cruA | 21.55 |
| RF3 | 100 | grf1-3 | 37 | cruA | 25.47 |
| 88302 | 100 | grf1-3 | 43.21 | cruA | 25.49 |
| 73496 | 100 | grf1-3 | 37.75 | cruA | 22.61 |
| non-GM oilseed rape | 100 | grf1-3 | 40.72 | cruA | 25.11 |
| Primer Name | Primer and Probe Sequence | Amplicon Length | Primer and Probe Concentration | PCR Condition | Reference |
|---|---|---|---|---|---|
| grf1-3 Taqman | F: 5′-GGAAAGAAATGGCGGTGCT-3′ R: 5′-CGGCAGCATTAGTATTGTGGC-3′ P: 5′-ACAGAGGCCGCCATCGTTCA-3′ | 130 | 600 nM forward 600 nM reverse 200 nM probe | 95 °C 10 min; 95 °C by 15 s [45×]; 62 °C by 1 min | This study |
| grf1-3 LNA | F: 5′-GGAAAGAAATGGCGGTGCT+G-3′ R: 5′-CGGCAGCATTAGTATTGTGGC-3′ P: 5′-ACAGAGGCCGCCATCGTTCA-3′ | 131 | 1000 nM forward 1200 nM reverse 400 nM probe | 95 °C 10 min; 95 °C by 15 s [45×]; 62 °C by 1 min | This study |
| Taxon specific primer (RG (AT1G03400.1) | F: 5′-GCGGAGCATAGGGTGATAGC-3′ R: 5′-TGTAACTTAGGAGCATCGAGCG-3′ P: 5′-ATGGGCCAATCAAAGATCTCCTGTCTGC-3′ | 209 | 400 nM forward 600 nM reverse 200 nM probe | 95 °C 10 min; 95 °C by 15 s [45×]; 62 °C by 1 min | [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanatta, C.B.; Narendja, F.; El Jawhary, H.; Abou-Sleymane, G.; Subburaj, S.; Nodari, R.O.; Agapito-Tenfen, S.Z. Suitability of Real-Time PCR Methods for New Genomic Technique Detection in the Context of the European Regulations: A Case Study in Arabidopsis. Int. J. Mol. Sci. 2025, 26, 3308. https://doi.org/10.3390/ijms26073308
Zanatta CB, Narendja F, El Jawhary H, Abou-Sleymane G, Subburaj S, Nodari RO, Agapito-Tenfen SZ. Suitability of Real-Time PCR Methods for New Genomic Technique Detection in the Context of the European Regulations: A Case Study in Arabidopsis. International Journal of Molecular Sciences. 2025; 26(7):3308. https://doi.org/10.3390/ijms26073308
Chicago/Turabian StyleZanatta, Caroline Bedin, Frank Narendja, Hilana El Jawhary, Gretta Abou-Sleymane, Saminathan Subburaj, Rubens Onofre Nodari, and Sarah Zanon Agapito-Tenfen. 2025. "Suitability of Real-Time PCR Methods for New Genomic Technique Detection in the Context of the European Regulations: A Case Study in Arabidopsis" International Journal of Molecular Sciences 26, no. 7: 3308. https://doi.org/10.3390/ijms26073308
APA StyleZanatta, C. B., Narendja, F., El Jawhary, H., Abou-Sleymane, G., Subburaj, S., Nodari, R. O., & Agapito-Tenfen, S. Z. (2025). Suitability of Real-Time PCR Methods for New Genomic Technique Detection in the Context of the European Regulations: A Case Study in Arabidopsis. International Journal of Molecular Sciences, 26(7), 3308. https://doi.org/10.3390/ijms26073308