
Growth Hormone-Releasing Hormone Antagonists Increase Radiosensitivity in Non-Small Cell Lung Cancer Cells

 , , , , ,
, , , , ,  , , ,
, , ,  , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
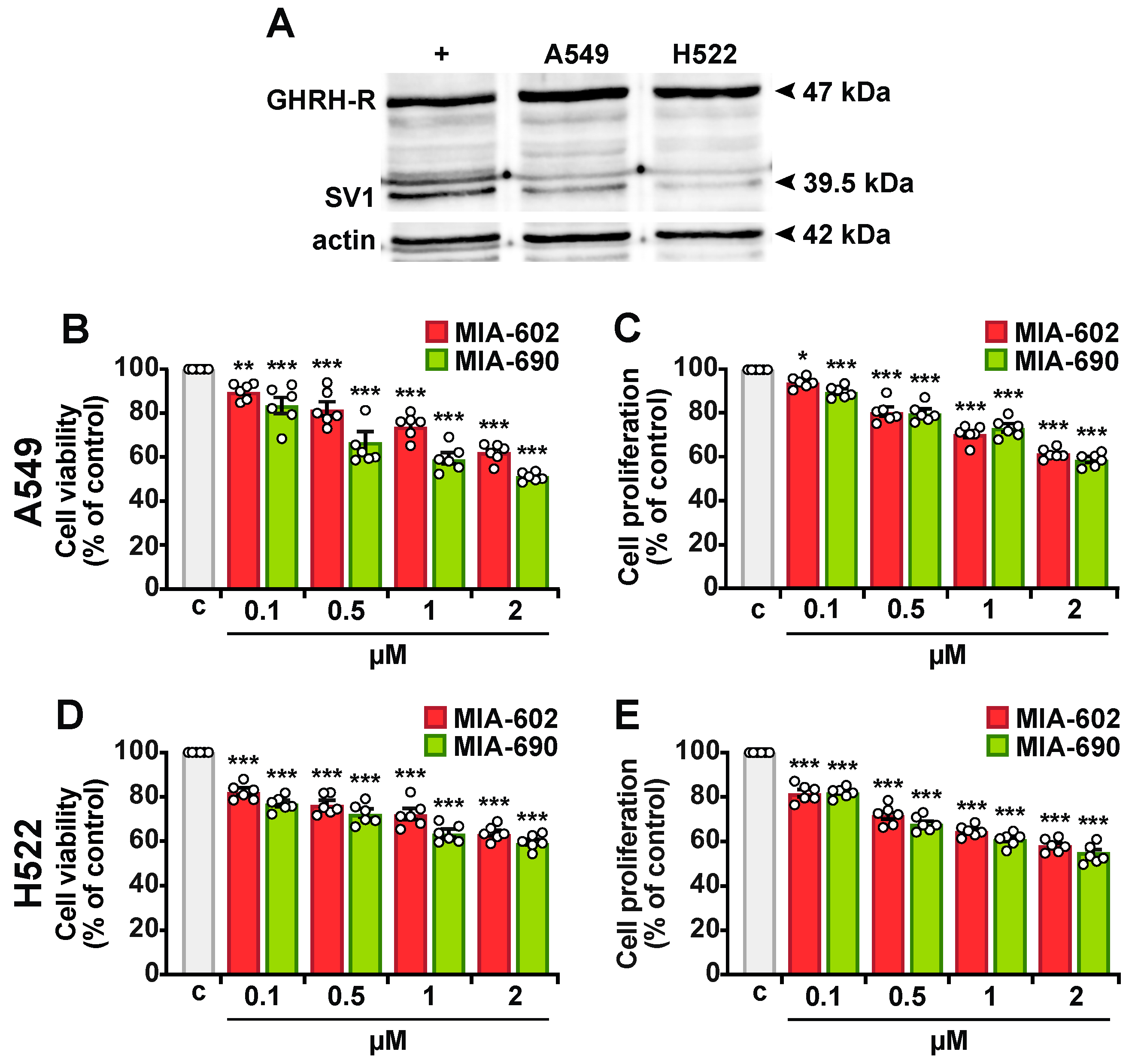
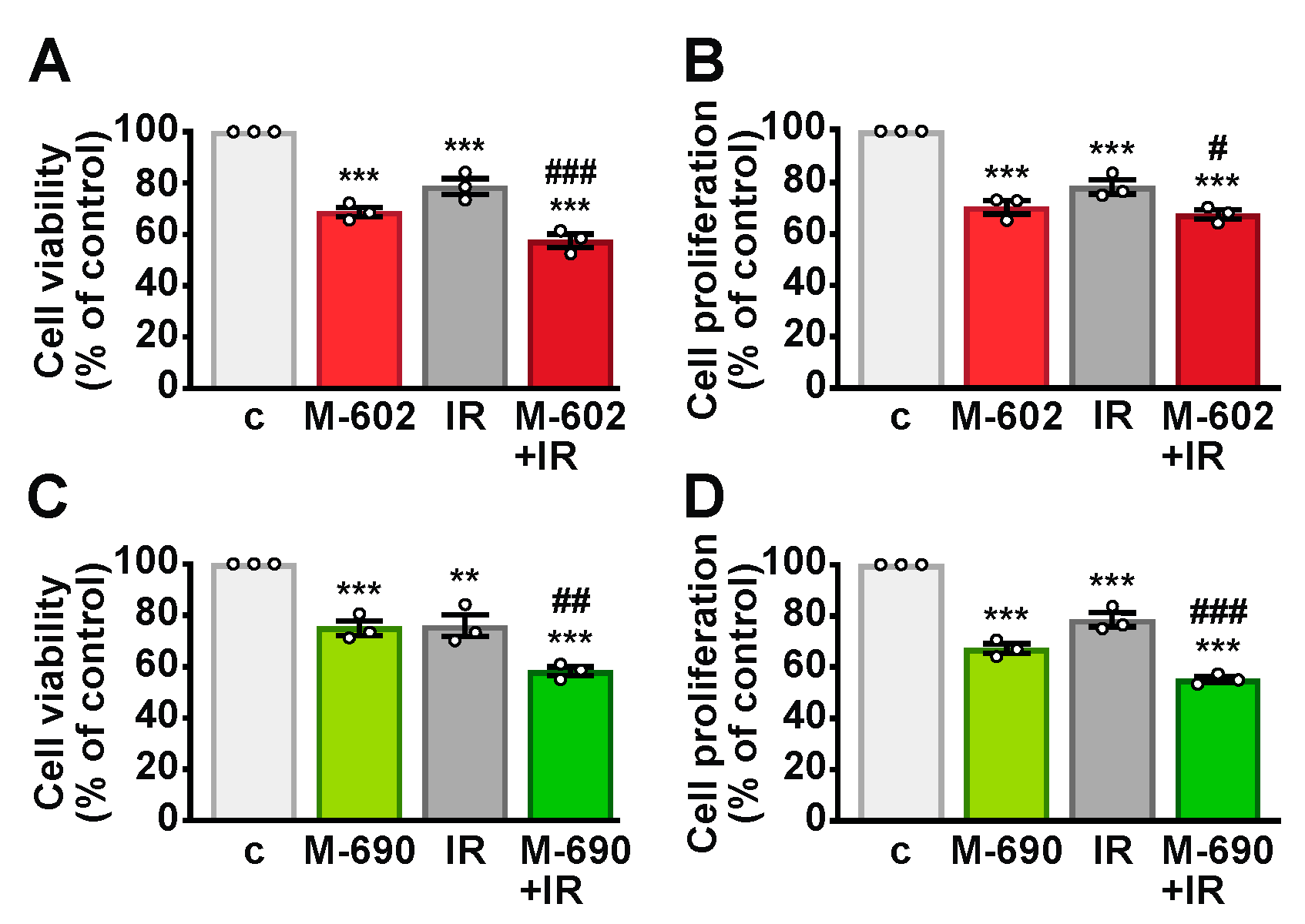
2.1. MIA-602 and MIA-690 Reduce Viability and Proliferation in NSCLC Cells
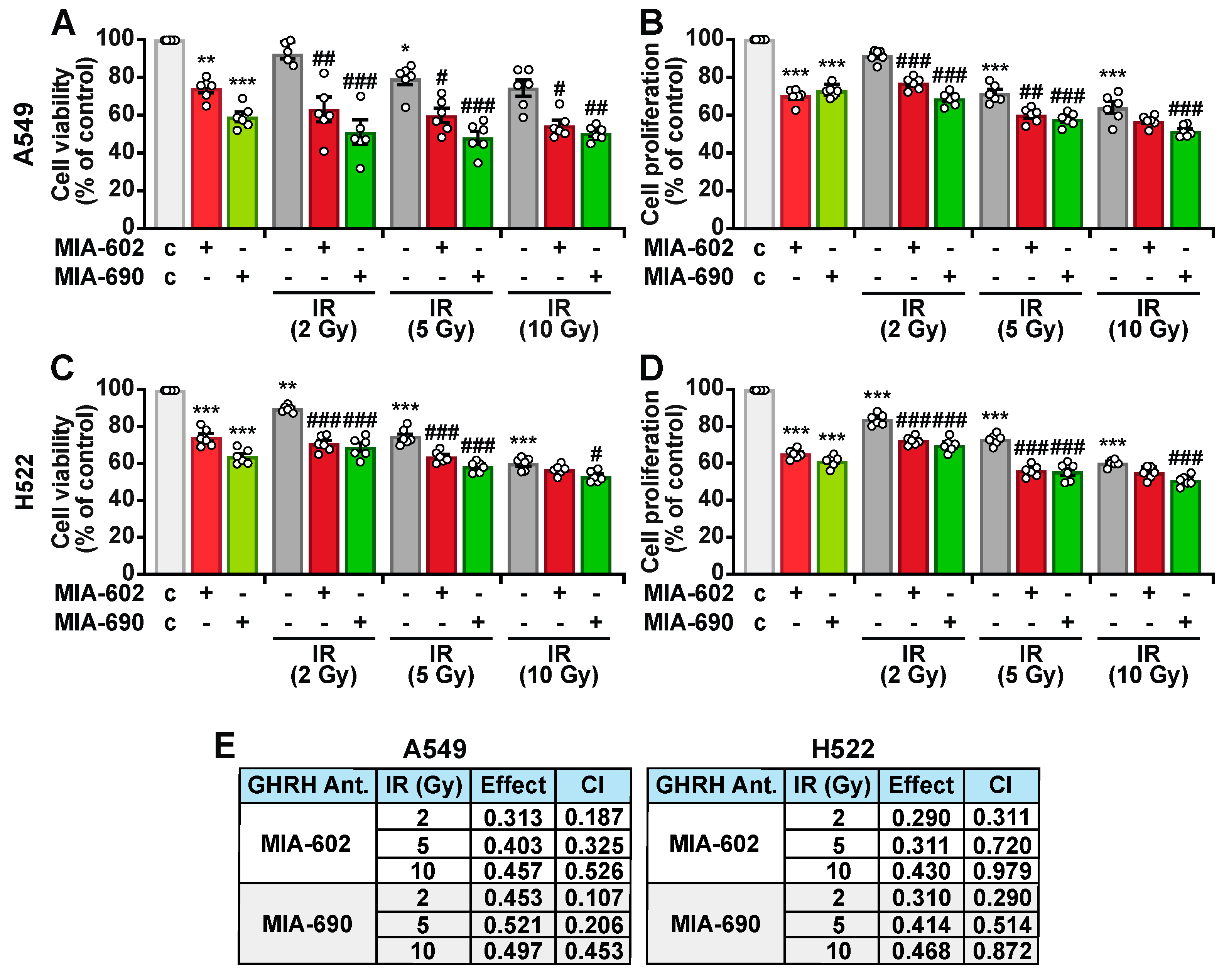
2.2. MIA-602 and MIA-690 Increase the Sensitivity of NSCLC Cells to Ionizing Radiation
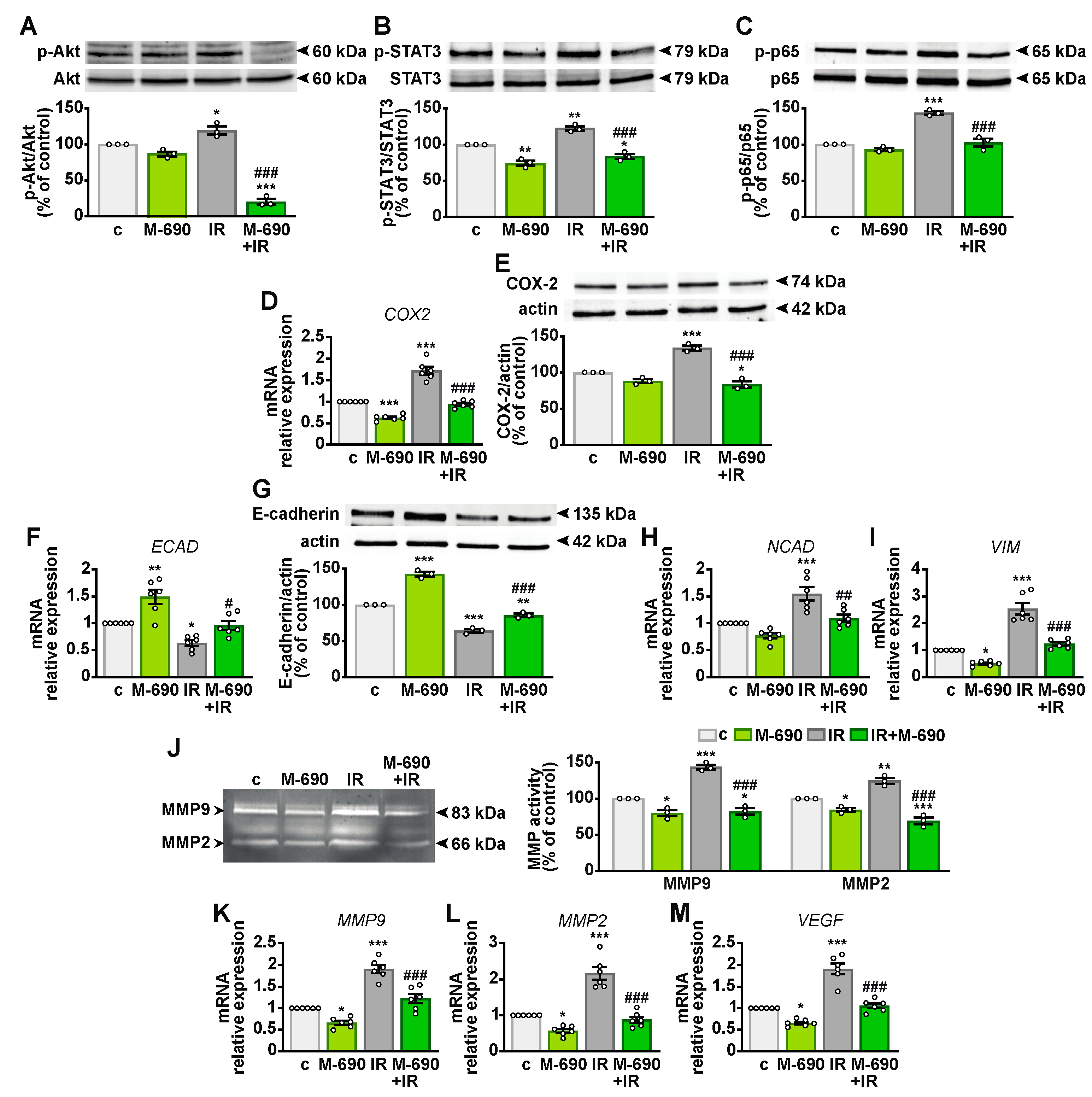
2.3. MIA-690 Reduces the Expression of GHRH Receptors and IGF1 in A549 Cells Exposed to IR
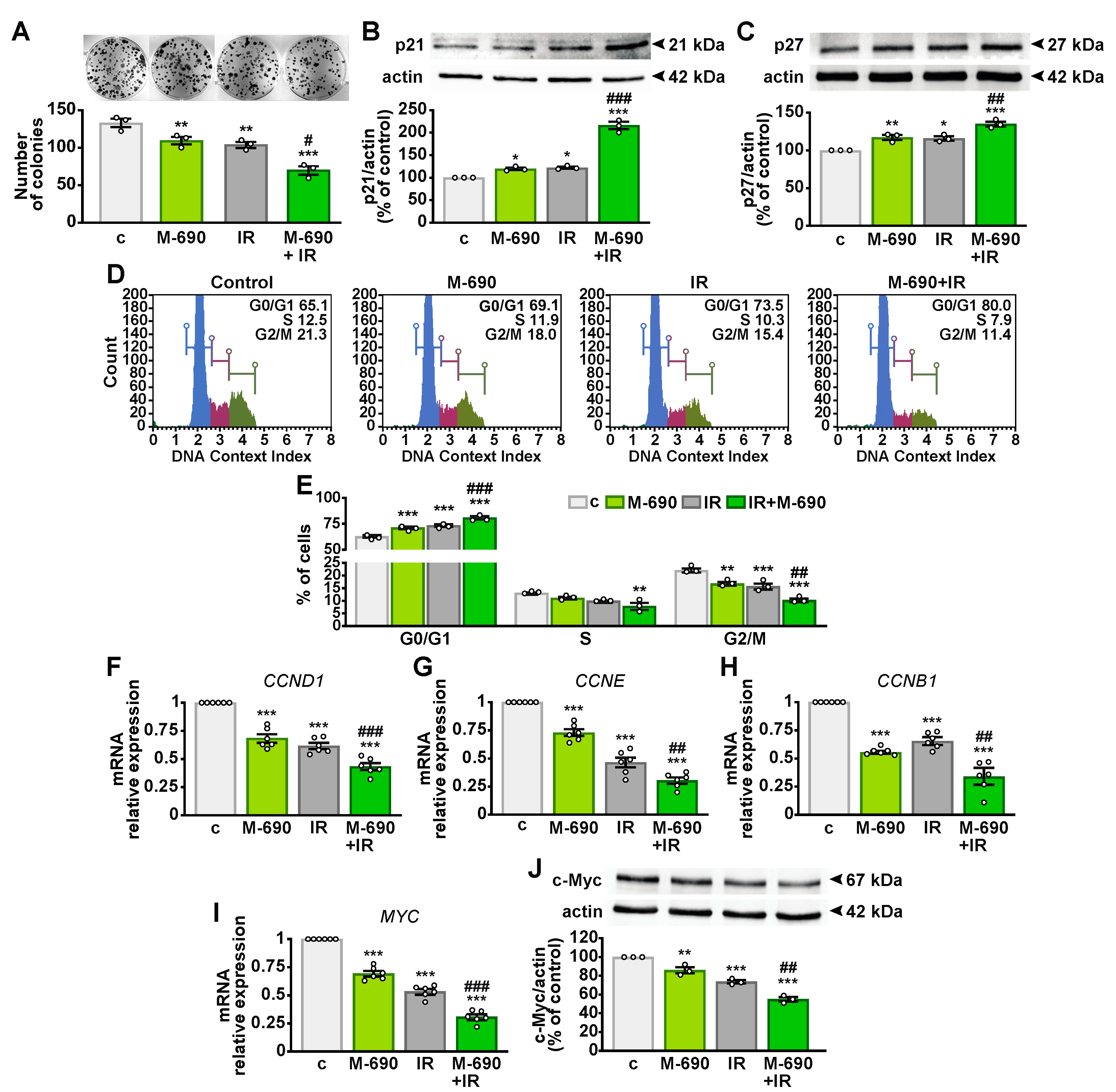
2.4. MIA-690 Inhibits Cell Growth and Regulates the Cell Cycle in Irradiated NSCLC Cells
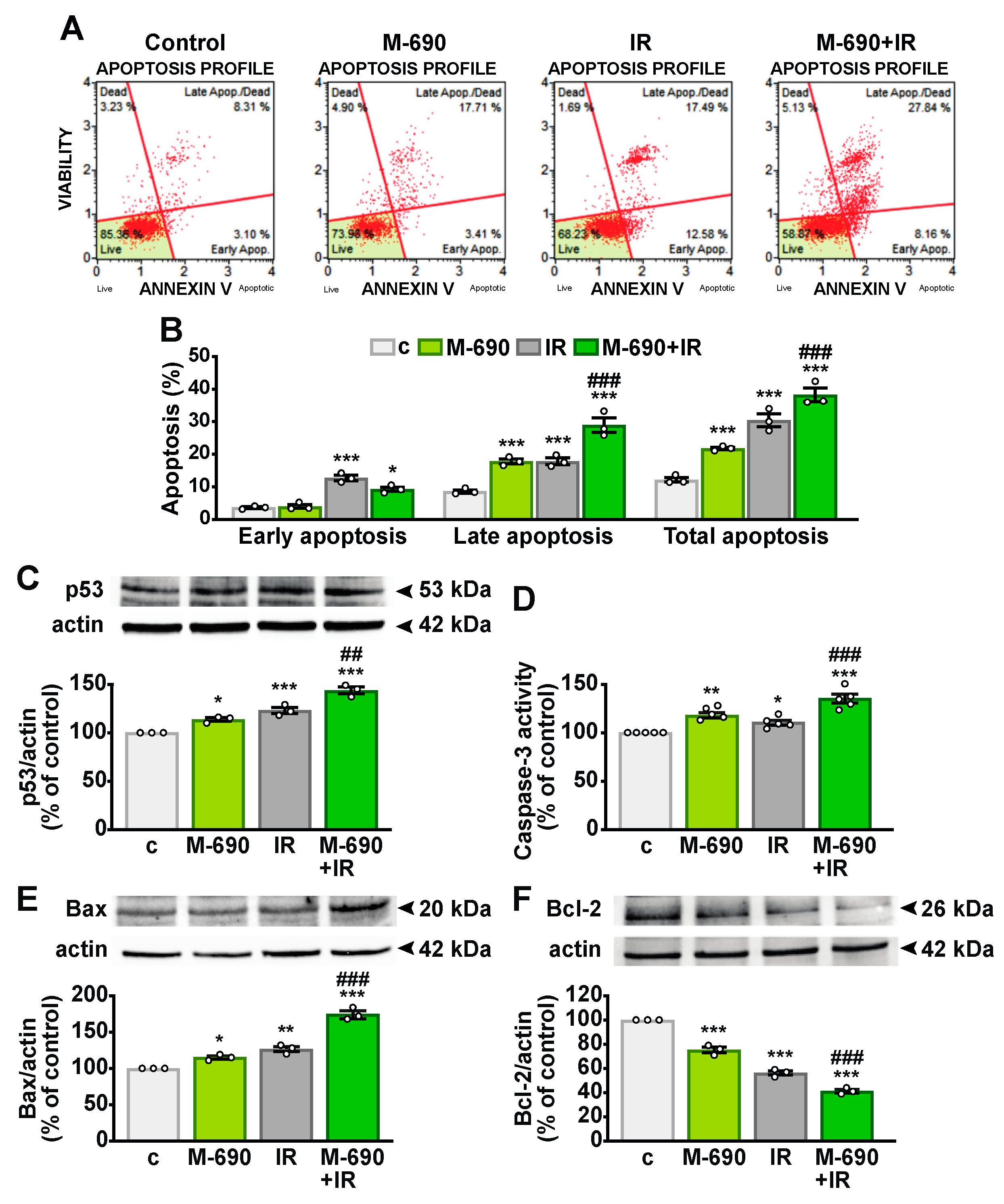
2.5. MIA-690 Sensitizes IR-Induced Apoptosis in NSCLC Cells
2.6. MIA-690 Mitigates Radioresistance in NSCLC Cells
2.7. MIA-690 and MIA-602 Enhance the Sensitivity of Primary NSCLC Cells to IR
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatments
4.3. Isolation of Primary Lung Cancer Cells
4.4. Cell Viability and Proliferation
4.5. Caspase-3 Activity
4.6. Colony Formation
4.7. Cell Cycle Analysis
4.8. Annexin V Analysis
4.9. Real-Time PCR
4.10. Western Blot Analysis
4.11. Gelatin Zymography
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients with Oncogenic Driver Molecular Alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.G.; Yuan, H.; Deng, W.; Li, J.; Huang, Y.; Kim, B.Y.S.; Story, M.D.; Jiang, W. The Reciprocity between Radiotherapy and Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhang, L.Y.; He, J.Z.; Miao, Z.M.; Li, Y.Y.; Zhang, Y.M.; Liu, Z.W.; Zhang, S.Z.; Chen, Y.; Zhou, G.C.; et al. Review: Mechanisms and perspective treatment of radioresistance in non-small cell lung cancer. Front. Immunol. 2023, 14, 1133899. [Google Scholar] [CrossRef]
- Schally, A.V.; Zhang, X.; Cai, R.; Hare, J.M.; Granata, R.; Bartoli, M. Actions and Potential Therapeutic Applications of Growth Hormone-Releasing Hormone Agonists. Endocrinology 2019, 160, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Granata, R.; Leone, S.; Zhang, X.; Gesmundo, I.; Steenblock, C.; Cai, R.; Sha, W.; Ghigo, E.; Hare, J.M.; Bornstein, S.R.; et al. Growth hormone-releasing hormone and its analogues in health and disease. Nat. Rev. Endocrinol. 2024, 21, 180–195. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, R.; Jia, H.; Yang, K.; Ren, F.; Zhou, M.S. GHRH and its analogues in central nervous system diseases. Rev. Endocr. Metab. Disord. 2024. [Google Scholar] [CrossRef]
- Dulce, R.A.; Hatzistergos, K.E.; Kanashiro-Takeuchi, R.M.; Takeuchi, L.M.; Balkan, W.; Hare, J.M. Growth hormone-releasing hormone signaling and manifestations within the cardiovascular system. Rev. Endocr. Metab. Disord. 2025. [Google Scholar] [CrossRef]
- Steenblock, C.; Bornstein, S.R. GHRH in diabetes and metabolism. Rev. Endocr. Metab. Disord. 2024. [Google Scholar] [CrossRef]
- Siejka, A.; Lawnicka, H.; Fakir, S.; Barabutis, N. Growth hormone—Releasing hormone in the immune system. Rev. Endocr. Metab. Disord. 2024. [Google Scholar] [CrossRef]
- Halmos, G.; Szabo, Z.; Dobos, N.; Juhasz, E.; Schally, A.V. Growth hormone-releasing hormone receptor (GHRH-R) and its signaling. Rev. Endocr. Metab. Disord. 2025. [Google Scholar] [CrossRef]
- Hohla, F.; Schally, A.V.; Szepeshazi, K.; Varga, J.L.; Buchholz, S.; Koster, F.; Heinrich, E.; Halmos, G.; Rick, F.G.; Kannadka, C.; et al. Synergistic inhibition of growth of lung carcinomas by antagonists of growth hormone-releasing hormone in combination with docetaxel. Proc. Natl. Acad. Sci. USA 2006, 103, 14513–14518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cui, T.; Cai, R.; Wangpaichitr, M.; Mirsaeidi, M.; Schally, A.V.; Jackson, R.M. Growth Hormone-Releasing Hormone in Lung Physiology and Pulmonary Disease. Cells 2020, 9, 2331. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Vidaurre, I.; Cai, R.; Sha, W.; Schally, A.V. Inhibition of experimental small-cell and non-small-cell lung cancers by novel antagonists of growth hormone-releasing hormone. Int. J. Cancer 2018, 142, 2394–2404. [Google Scholar] [CrossRef]
- Siejka, A.; Barabutis, N.; Schally, A.V. GHRH antagonist inhibits focal adhesion kinase (FAK) and decreases expression of vascular endothelial growth factor (VEGF) in human lung cancer cells in vitro. Peptides 2012, 37, 63–68. [Google Scholar] [CrossRef]
- Zarandi, M.; Cai, R.; Kovacs, M.; Popovics, P.; Szalontay, L.; Cui, T.; Sha, W.; Jaszberenyi, M.; Varga, J.; Zhang, X.; et al. Synthesis and structure-activity studies on novel analogs of human growth hormone releasing hormone (GHRH) with enhanced inhibitory activities on tumor growth. Peptides 2017, 89, 60–70. [Google Scholar] [CrossRef]
- Schally, A.V.; Cai, R.; Zhang, X.; Sha, W.; Wangpaichitr, M. The development of growth hormone-releasing hormone analogs: Therapeutic advances in cancer, regenerative medicine, and metabolic disorders. Rev. Endocr. Metab. Disord. 2024. [Google Scholar] [CrossRef]
- Recinella, L.; Chiavaroli, A.; Veschi, S.; Di Valerio, V.; Lattanzio, R.; Orlando, G.; Ferrante, C.; Gesmundo, I.; Granata, R.; Cai, R.; et al. Antagonist of growth hormone-releasing hormone MIA-690 attenuates the progression and inhibits growth of colorectal cancer in mice. Biomed. Pharmacother. 2022, 146, 112554. [Google Scholar] [CrossRef]
- Gesmundo, I.; Pedrolli, F.; Cai, R.; Sha, W.; Schally, A.V.; Granata, R. Growth hormone-releasing hormone and cancer. Rev. Endocr. Metab. Disord. 2024. [Google Scholar] [CrossRef]
- Munoz-Moreno, L.; Roman, I.D.; Bajo, A.M. GHRH and the prostate. Rev. Endocr. Metab. Disord. 2024. [Google Scholar] [CrossRef]
- Gesmundo, I.; Pedrolli, F.; Vitale, N.; Bertoldo, A.; Orlando, G.; Banfi, D.; Granato, G.; Kasarla, R.; Balzola, F.; Deaglio, S.; et al. Antagonist of Growth Hormone-Releasing Hormone Potentiates the Antitumor Effect of Pemetrexed and Cisplatin in Pleural Mesothelioma. Int. J. Mol. Sci. 2022, 23, 1248. [Google Scholar] [CrossRef]
- Villanova, T.; Gesmundo, I.; Audrito, V.; Vitale, N.; Silvagno, F.; Musuraca, C.; Righi, L.; Libener, R.; Riganti, C.; Bironzo, P.; et al. Antagonists of growth hormone-releasing hormone (GHRH) inhibit the growth of human malignant pleural mesothelioma. Proc. Natl. Acad. Sci. USA 2019, 116, 2226–2231. [Google Scholar] [CrossRef]
- Granato, G.; Gesmundo, I.; Pedrolli, F.; Kasarla, R.; Begani, L.; Banfi, D.; Bruno, S.; Lopatina, T.; Brizzi, M.F.; Cai, R.; et al. Growth hormone-releasing hormone antagonist MIA-602 inhibits inflammation induced by SARS-CoV-2 spike protein and bacterial lipopolysaccharide synergism in macrophages and human peripheral blood mononuclear cells. Front. Immunol. 2023, 14, 1231363. [Google Scholar] [CrossRef]
- Brix, N.; Samaga, D.; Belka, C.; Zitzelsberger, H.; Lauber, K. Analysis of clonogenic growth in vitro. Nat. Protoc. 2021, 16, 4963–4991. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Qiao, L.; Chen, Y.; Liang, N.; Xie, J.; Deng, G.; Chen, F.; Wang, X.; Liu, F.; Li, Y.; Zhang, J. Targeting Epithelial-to-Mesenchymal Transition in Radioresistance: Crosslinked Mechanisms and Strategies. Front. Oncol. 2022, 12, 775238. [Google Scholar] [CrossRef]
- Vilalta, M.; Rafat, M.; Graves, E.E. Effects of radiation on metastasis and tumor cell migration. Cell Mol. Life Sci. 2016, 73, 2999–3007. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Havt, A.; Schally, A.V.; Halmos, G.; Varga, J.L.; Toller, G.L.; Horvath, J.E.; Szepeshazi, K.; Koster, F.; Kovitz, K.; Groot, K.; et al. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 17424–17429. [Google Scholar] [CrossRef]
- Barabutis, N.; Siejka, A.; Schally, A.V. Growth hormone releasing hormone induces the expression of nitric oxide synthase. J. Cell Mol. Med. 2011, 15, 1148–1155. [Google Scholar] [CrossRef]
- Farkas, R.; Pozsgai, E.; Schally, A.V.; Szigeti, A.; Szigeti, E.; Laszlo, Z.; Papp, A.; Gomori, E.; Mangel, L.; Horvath, P.O.; et al. Possible predictors of histopathological response to neoadjuvant chemoradiotherapy for rectal cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Perez, R.; Block, N.L.; Rick, F.G. Potentiating effects of GHRH analogs on the response to chemotherapy. Cell Cycle 2015, 14, 699–704. [Google Scholar] [CrossRef]
- Gesmundo, I.; Granato, G.; Fuentes-Fayos, A.C.; Alvarez, C.V.; Dieguez, C.; Zatelli, M.C.; Congiusta, N.; Banfi, D.; Prencipe, N.; Leone, S.; et al. Antagonists of Growth Hormone-Releasing Hormone Inhibit the Growth of Pituitary Adenoma Cells by Hampering Oncogenic Pathways and Promoting Apoptotic Signaling. Cancers 2021, 13, 3950. [Google Scholar] [CrossRef]
- Abdel-Wahab, M.; Schally, A.V.; Rick, F.G.; Szalontay, L.; Block, N.L.; Jorda, M.; Mahmoud, O.; Markoe, A.; Shi, Y.-F.; Reiner, T.; et al. Antagonists of growth hormone releasing hormone (GHRH) given before whole body radiation lead to modulation of radiation response and organ-specific changes in the expression of angiogenesis. J. Radiat. Oncol. 2012, 1, 389–396. [Google Scholar] [CrossRef]
- Chen, K.; Shang, Z.; Dai, A.L.; Dai, P.L. Novel PI3K/Akt/mTOR pathway inhibitors plus radiotherapy: Strategy for non-small cell lung cancer with mutant RAS gene. Life Sci. 2020, 255, 117816. [Google Scholar] [CrossRef] [PubMed]
- Boumelha, J.; de Castro, A.; Bah, N.; Cha, H.; de Carne Trecesson, S.; Rana, S.; Tomaschko, M.; Anastasiou, P.; Mugarza, E.; Moore, C.; et al. CRISPR-Cas9 Screening Identifies KRAS-Induced COX2 as a Driver of Immunotherapy Resistance in Lung Cancer. Cancer Res. 2024, 84, 2231–2246. [Google Scholar] [CrossRef]
- Csernus, V.J.; Schally, A.V.; Kiaris, H.; Armatis, P. Inhibition of growth, production of insulin-like growth factor-II (IGF-II), and expression of IGF-II mRNA of human cancer cell lines by antagonistic analogs of growth hormone-releasing hormone in vitro. Proc. Natl. Acad. Sci. USA 1999, 96, 3098–3103. [Google Scholar] [CrossRef]
- Szereday, Z.; Schally, A.V.; Varga, J.L.; Kanashiro, C.A.; Hebert, F.; Armatis, P.; Groot, K.; Szepeshazi, K.; Halmos, G.; Busto, R. Antagonists of growth hormone-releasing hormone inhibit the proliferation of experimental non-small cell lung carcinoma. Cancer Res. 2003, 63, 7913–7919. [Google Scholar]
- Pohlman, A.W.; Moudgalya, H.; Jordano, L.; Lobato, G.C.; Gerard, D.; Liptay, M.J.; Seder, C.W.; Borgia, J.A. The role of IGF-pathway biomarkers in determining risks, screening, and prognosis in lung cancer. Oncotarget 2022, 13, 393–407. [Google Scholar] [CrossRef]
- Szalontay, L.; Benveniste, R.J.; Schally, A.V.; Vidaurre, I.; Nadji, M.; Zarandi, M.; Block, N.L.; Kovacs, M. Inhibitory effects of GHRH antagonists on human GH-secreting adenoma tissue. Neuroendocrinology 2012, 96, 81–88. [Google Scholar] [CrossRef]
- Qin, Y.J.; Chan, S.O.; Chong, K.K.; Li, B.F.; Ng, T.K.; Yip, Y.W.; Chen, H.; Zhang, M.; Block, N.L.; Cheung, H.S.; et al. Antagonist of GH-releasing hormone receptors alleviates experimental ocular inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, 18303–18308. [Google Scholar] [CrossRef] [PubMed]
- Kiaris, H.; Chatzistamou, I.; Schally, A.V.; Halmos, G.; Varga, J.L.; Koutselini, H.; Kalofoutis, A. Ligand-dependent and -independent effects of splice variant 1 of growth hormone-releasing hormone receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 9512–9517. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Ke, X.; Wang, L.; Yao, Z.; Guo, Y.; Zhang, X.; Chen, Y.; Pang, C.P.; Schally, A.V.; Zhang, H. Splice variant of growth hormone-releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target. Proc. Natl. Acad. Sci. USA 2020, 117, 6726–6732. [Google Scholar] [CrossRef]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Szalontay, L.; Schally, A.V.; Popovics, P.; Vidaurre, I.; Krishan, A.; Zarandi, M.; Cai, R.Z.; Klukovits, A.; Block, N.L.; Rick, F.G. Novel GHRH antagonists suppress the growth of human malignant melanoma by restoring nuclear p27 function. Cell Cycle 2014, 13, 2790–2797. [Google Scholar] [CrossRef]
- Volakaki, A.A.; Lafkas, D.; Kassi, E.; Schally, A.V.; Papavassiliou, A.G.; Kiaris, H. Essential role of p21/waf1 in the mediation of the anti-proliferative effects of GHRH antagonist JMR-132. J. Mol. Endocrinol. 2008, 41, 389–392. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef]
- Zhang, L.; Ludden, C.M.; Cullen, A.J.; Tew, K.D.; Branco de Barros, A.L.; Townsend, D.M. Nuclear factor kappa B expression in non-small cell lung cancer. Biomed. Pharmacother. 2023, 167, 115459. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-kappaB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Kobayashi, A.; Hiroyama, Y.; Mamiya, T.; Oikawa, M.; Konishi, T. The COX-2/PGE2 Response Pathway Upregulates Radioresistance in A549 Human Lung Cancer Cells through Radiation-Induced Bystander Signaling. Biology 2023, 12, 1368. [Google Scholar] [CrossRef]
- Parakh, S.; Ernst, M.; Poh, A.R. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef] [PubMed]
- Sinkevicius, K.W.; Bellaria, K.J.; Barrios, J.; Pessina, P.; Gupta, M.; Brainson, C.F.; Bronson, R.T.; Kim, C.F. E-Cadherin Loss Accelerates Tumor Progression and Metastasis in a Mouse Model of Lung Adenocarcinoma. Am. J. Respir. Cell Mol. Biol. 2018, 59, 237–245. [Google Scholar] [CrossRef]
- Bellyei, S.; Schally, A.V.; Zarandi, M.; Varga, J.L.; Vidaurre, I.; Pozsgai, E. GHRH antagonists reduce the invasive and metastatic potential of human cancer cell lines in vitro. Cancer Lett. 2010, 293, 31–40. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gesmundo, I.; Pedrolli, F.; Giglioli, F.R.; Jazaj, F.; Granato, G.; Bertoldo, A.; Bistolfi, F.; Gregorc, V.; Sapino, A.; Righi, L.; et al. Growth Hormone-Releasing Hormone Antagonists Increase Radiosensitivity in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2025, 26, 3267. https://doi.org/10.3390/ijms26073267
Gesmundo I, Pedrolli F, Giglioli FR, Jazaj F, Granato G, Bertoldo A, Bistolfi F, Gregorc V, Sapino A, Righi L, et al. Growth Hormone-Releasing Hormone Antagonists Increase Radiosensitivity in Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2025; 26(7):3267. https://doi.org/10.3390/ijms26073267
Chicago/Turabian StyleGesmundo, Iacopo, Francesca Pedrolli, Francesca Romana Giglioli, Florian Jazaj, Giuseppina Granato, Alessia Bertoldo, Federica Bistolfi, Vanesa Gregorc, Anna Sapino, Luisella Righi, and et al. 2025. "Growth Hormone-Releasing Hormone Antagonists Increase Radiosensitivity in Non-Small Cell Lung Cancer Cells" International Journal of Molecular Sciences 26, no. 7: 3267. https://doi.org/10.3390/ijms26073267
APA StyleGesmundo, I., Pedrolli, F., Giglioli, F. R., Jazaj, F., Granato, G., Bertoldo, A., Bistolfi, F., Gregorc, V., Sapino, A., Righi, L., Cai, R., Sha, W., Wangpaichitr, M., Papotti, M., Ghigo, E., Ricardi, U., Schally, A. V., & Granata, R. (2025). Growth Hormone-Releasing Hormone Antagonists Increase Radiosensitivity in Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences, 26(7), 3267. https://doi.org/10.3390/ijms26073267