
Retinal Organoids: Innovative Tools for Understanding Retinal Degeneration

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Pioneering Research: From the Beginning of Retinal Organoids
3. Generating Retinal Organoids: Growing and Differentiation Methods

{kind=link}
{kind=link}
{kind=link}
| Stage 1: Neuroectoderm Induction | |
| Molecule | Role |
| Noggin [18] | Inhibits BMP signaling, promoting neuroectoderm specification. |
| SB431542 [31] | TGF-β pathway inhibitor, enhances neuronal differentiation. |
| LDN-193189 [31] | BMP inhibitor, promotes differentiation into neural progenitors. |
| BMP4 (Bone Morphogenetic Protein 4) [32] | Induces neural ectoderm formation and promotes early retinal differentiation. |
| Stage 2: Retinal Specification | |
| Molecule | Role |
| IGF-1 (Insulin-like Growth Factor 1) [32] | Promotes the differentiation of retinal progenitors. |
| bFGF (Basic Fibroblast Growth Factor, FGF2) [33] | Supports neural and early retinal progenitor proliferation, initiating differentiation toward retinal fate. |
| IWR-1 (Inhibitor of Wnt Response 1) [22] | Inhibits Wnt signaling, facilitating retinal cell differentiation and organized neuroretinal epithelium development. |
| Dkk-1 [34] | Wnt inhibitor, promotes differentiation into retinal progenitors. |
| Activin A [22,35] | Enhances spheroid formation by inducing the expression of early retinal development genes such as PAX6, supporting retinal progenitor proliferation. |
| Stage 3: Optic Vesicle and Neuroretina Formation | |
| Molecule | Role |
| Retinoic Acid (RA) [22,35] | Potent inducer of differentiation and retinal layer formation, promoting progenitor specification via CRX and PAX6 expression. However, it could delay maturation. |
| Sonic hedgehog agonist SAG [22,35] | Regulates retinal progenitor proliferation and retinal pigment epithelium (RPE) organization. |
| EGF (Epidermal Growth Factor) [36] | Supports proliferation and survival of retinal progenitor cells and it is useful to isolate and maintain Müller cells. |
| Wnt3a [22] | Prevents premature differentiation of retinal progenitors and maintains stem cell pluripotency. |
| CHIR99021 (GSK-3β inhibitor) [22] | Activates Wnt signaling, promoting early optic neuroepithelium growth and epithelial differentiation with MITF positive cells. |
| BMP4 [32] | Regulates RPE specification. |
| Stage 4: Retinal Differentiation and Maturation | |
| Molecule | Role |
| B27, non-essential amino acid solution (NEAA) and N2 supplements [33] | Essential for maintaining optimal stem cell growth and differentiation conditions, supporting the survival and structural organization of retinal organoids. |
| DAPT (Notch inhibitor) [22] | Inhibits Notch signaling, promoting retinal progenitor differentiation and precise stratification, mimicking human retina organization. |
| BDNF (Brain-Derived Neurotrophic Factor) and CNTF (Ciliary Neurotrophic Factor) [37] | Neurotrophic factors that support retinal cell survival and maturation, facilitating photoreceptor layer development and Müller glia differentiation. |
| SU5402 [38] | FGF inhibitor, promotes photoreceptor maturation and RPE differentiation. |
| Stage 5: Advanced Maturation and Functional Photoreceptors | |
| Molecule | Role |
| Taurine [39] | Sulfonated amino acid critical for photoreceptor development, neuroprotection, and calcium homeostasis. Enhances neurotrophic factors’ effects and accelerates retinal organoid maturation and stratification. |
| 9-cis-Retinal [40] | Opsin cofactor, essential for functional photoreceptor maturation. |
| DAPT (Notch inhibitor) [22] | Inhibits Notch signaling, promoting retinal progenitor differentiation and precise stratification, mimicking human retina organization. |
4. Metabolic Changes in Retinal Organoids
4.1. Oxygen Gradient in the Retina and Its Importance in Retinal Development and Function
4.2. Influence of Hypoxia on Retinal Progenitor and Ganglion Cells in Retinal Organoids
4.3. Challenges in Oxygenation and Metabolism in Retinal Organoid Cultures
4.4. Key Metabolic Challenges in Retinal Organoids
- Oxygenation Limitations: Unlike the highly vascularized in vivo retina, retinal organoids depend on the passive diffusion of oxygen and nutrients. As they grow, they may develop hypoxic regions that affect cellular differentiation and photoreceptor viability.
- Impact on Cellular Maturation: Oxygen availability regulates key retinal processes, such as the differentiation of ganglion and photoreceptor cells. Low oxygen levels can activate HIFs, which may influence gene expression and cellular metabolism.
- Metabolic Differences from the Native Retina: In physiological conditions, photoreceptors consume large amounts of oxygen and depend on oxidative phosphorylation. If organoids fail to replicate this environment, they may have an altered function and may not serve as accurate disease models.
- Strategies to Improve Oxygenation: Novel approaches, such as metabolic supplementation, bioreactor-based culture systems, or vascularized organoid engineering, could enhance oxygenation and make these models more comparable to the native retina.
5. The Cellular Mechanisms of Neurodegeneration in the Retina and the Utilization of Retinal Organoids for Research
5.1. Apoptosis
5.2. Autophagy
5.3. Ciliogenesis and Ciliopathies in the Retina
6. Disease Modeling with Retinal Organoids
| Treatment | Disease | Treatment Studies | |
|---|---|---|---|
| CRISPR-Cas9-mediated correction | RP |
| [97] |
| [104] | ||
| |||
| LCA |
| [105] | |
| [83] | ||
| AAV-mediated gene repair | RP |
| [96] |
| [85] | ||
| LCA |
| [106] | |
| X-linked RP |
| [89] | |
| [107] | ||
| XLRS |
| [71] | |
| [108] | ||
| Protein trans-splicing | - |
| [109] |
| QR-110 (RNA oligonucleotide) | LCA |
| [79] |
| Eupatilin | LCA |
| [110] |
| Reserpine | LCA |
| [48] |
| AA147 (Small molecule ATF6 agonist) | Achromatopsia |
| [111] |
| Stem-cell transplantation using retinal organoids | - |
| [99] |
| RP |
| [100] | |
| RP |
| [112] | |
| Direct transplantation of cultured retinal organoids tissue as a retinal sheet into animals and human retinas | RP |
| [102,103] |
| - |
| [113] | |
| Age-related macular degeneration |
| [72] | |
7. Advantages and Limitations of Organoids as Models for Studying Retinal Neurodegeneration
8. Future Perspectives and Challenges
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | Recombinant Adeno-Associated Virus |
| AAV2 | AAV serotype 2 |
| AAV5 | AAV serotype 5 |
| AAV2-7m8 | AAV serotype 2-7m8 |
| ABCA4 | ATP-binding cassette subfamily 4 |
| AIPL1 | Aryl Hydrocarbon Receptor Interacting Protein-like 1 |
| ASCs | Adult Stem Cells |
| ATF6 | Activating Transcription Factor 6 |
| AMD | Age-Related Macular Degeneration |
| Cas9 | CRISPR-associated protein 9 |
| CEP290 | Centrosomal protein 290 |
| CRB | Crumbs homolog |
| CRB1 | Crumbs homolog 1 |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CRX | Cone–rod homeobox |
| ECM | Extracellular Matrix |
| ESCs | Embryonic Stem Cells |
| EBs | Embryoid Bodies |
| hESCs | Human Embryonic Stem Cells |
| hiPSCs | Human-Induced Pluripotent Stem Cells |
| HDAC6 | Histone Deacetylase 6 |
| HIFs | Hypoxia-inducible factors |
| INL | Inner Nuclear Layer |
| IPL | Inner Plexiform Layer |
| iPSCs | Induced Pluripotent Stem Cells |
| IRD | Inherited Retinal Diseases |
| JSRD | Joubert Syndrome and Related Disorders |
| LCA | Leber Congenital Amaurosi |
| mESCs | Mouse Embryonic Stem Cells |
| mRNA | Messenger RNA |
| MYO7A | Myosin VIIA |
| NR | Neural retina |
| NPHP5 | Nephrocystin-5 |
| OFD1 | Oral–Facial–Digital Syndrome 1 |
| ONL | Outer Nuclear Layer |
| OPA1 | Optic Atrophy 1 |
| OPL | Outer Plexiform Layer |
| ORF15 | Open Reading Frame 15 |
| OS | Outer Segment |
| OV | Optic Vesicle-like |
| PRGR | Retinitis Pigmentosa GTPase regulator |
| PRPF31 | Pre-mRNA Processing Factor 31 |
| PTC124 | Ataluren |
| QR110 | Sophora RNA oligonucleotide therapeutic designed to correct the splicing defect in the CEP290 |
| RB1 | Retinoblastoma 1 |
| RDD | Retinal degeneration diseases |
| rd1 | Retinal degeneration 1 |
| RGCs | Retinal Ganglion Cells |
| RPGRORF15 | Isoform of the RPGR gene |
| RNA | Ribonucleic Acid |
| RP | Retinitis Pigmentosa |
| RP2 | Retinitis Pigmentosa 2 |
| RPE | Retinal pigment epithelium |
| RS1 | Retinoschisin 1 |
| USH2A | Usher syndrome type 2A |
| USH1B | Usher syndrome type 1B |
| WT | Wild-Type |
| XLRS | X-linked Retinoschisis |
| 3D | Three dimensions(al) |
References
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [PubMed]
- Boye, S.E.; Boye, S.L.; Lewin, A.S.; Hauswirth, W.W. A comprehensive review of retinal gene therapy. Mol. Ther. 2013, 21, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Al-khersan, H.; Shah, K.P.; Jung, S.C.; Rodriguez, A.; Madduri, R.K.; Grassi, M.A. A novel MERTK mutation causing retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1613–1619. [Google Scholar] [CrossRef]
- Ashworth, K.E.; Weisbrod, J.; Ballios, B.G. Inherited Retinal Diseases and Retinal Organoids as Preclinical Cell Models for Inherited Retinal Disease Research. Genes 2024, 15, 705. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell. Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef]
- Kandoi, S.; Lamba, D.A. Retinal Organoids: A Human Model System for Development, Diseases, and Therapies. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2023; Volume 1415, pp. 549–554. [Google Scholar] [CrossRef]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Gao, M.L.; Deng, W.L.; Wu, K.C.; Sugita, S.; Mandai, M.; Takahashi, M. Stemming retinal regeneration with pluripotent stem cells. Prog. Retin. Eye Res. 2019, 69, 38–56. [Google Scholar] [CrossRef]
- Masland, R.H. The Neuronal Organization of the Retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O.L. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef]
- Cuenca, N.; Fernández-Sánchez, L.; Campello, L.; Maneu, V.; De la Villa, P.; Lax, P.; Pinilla, I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog. Retin. Eye Res. 2014, 43, 17–75. [Google Scholar] [CrossRef]
- Kruczek, K.; Swaroop, A. Pluripotent stem cell-derived retinal organoids for disease modeling and development of therapies. Stem Cells 2020, 38, 1206–1215. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vis. Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Slijkerman, R.W.N.; Song, F.; Astuti, G.D.N.; Huynen, M.A.; van Wijk, E.; Stieger, K.; Collin, R.W.J. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog. Retin. Eye Res. 2015, 48, 137–159. [Google Scholar] [CrossRef]
- Corrò, C.; Novellasdemunt, L.; Li, V.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Belova, L.; Lavrov, A.; Smirnikhina, S. Organoid transduction using recombinant adeno-associated viral vectors: Challenges and opportunities. BioEssays News Rev. Mol. Cell. Dev. Biol. 2022, 44, e2200055. [Google Scholar] [CrossRef]
- Bell, C.M.; Zack, D.J.; Berlinicke, C.A. Human Organoids for the Study of Retinal Development and Disease. Annu. Rev. Vis. Sci. 2020, 6, 91–114. [Google Scholar] [CrossRef] [PubMed]
- O’Hara-Wright, M.; Gonzalez-Cordero, A. Retinal organoids: A window into human retinal development. Development 2020, 147, dev189746. [Google Scholar] [CrossRef]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–58. [Google Scholar] [CrossRef]
- Meyer, J.S.; Howden, S.E.; Wallace, K.A.; Verhoeven, A.D.; Wright, L.S.; Capowski, E.E.; Pinilla, I.; Martin, J.M.; Tian, S.; Stewart, R.; et al. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 2011, 29, 1206–1218. [Google Scholar] [CrossRef]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Soon Park, T.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional Retinal Organoids Facilitate the Investigation of Retinal Ganglion Cell Development, Organization and Neurite Outgrowth from Human Pluripotent Stem Cells. Sci. Rep. 2018, 8, 14520. [Google Scholar] [CrossRef]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2019, 146, dev171686. [Google Scholar] [CrossRef]
- Rozanska, A.; Cerna-Chavez, R.; Queen, R.; Collin, J.; Zerti, D.; Dorgau, B.; Beh, C.S.; Davey, T.; Cochead, J.; Hussain, R.; et al. PRB-Depleted Pluripotent Stem Cell Retinal Organoids Recapitulate Cell State Transitions of Retinoblastoma Development and Suggest an Important Role for pRB in Retinal Cell Differentiation. Stem Cells Transl. Med. 2022, 11, 415–433. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Erkilic, N.; Damodar, K.; Boukhaddaoui, H.; Diakatou, M.; Garita-Hernandez, M.; Mamaeva, D.; Dubois, G.; Jazouli, Z.; Jimenez-Medina, C.; et al. Retinoic acid delays initial photoreceptor differentiation and results in a highly structured mature retinal organoid. Stem Cell Res. Ther. 2022, 13, 478. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Jin, Z.B. Retinal organoids as models for development and diseases. Cell Regen. 2021, 10, 33. [Google Scholar] [CrossRef]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.-A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef]
- Wagstaff, E.L.; Berzal, A.H.; Boon, C.J.F.; Quinn, P.M.J.; Ten Asbroek, A.L.M.A.; Bergen, A.A. The role of small molecules and their effect on the molecular mechanisms of early retinal organoid development. Int. J. Mol. Sci. 2021, 22, 7081. [Google Scholar] [CrossRef]
- Chichagova, V.; Hilgen, G.; Ghareeb, A.; Georgiou, M.; Carter, M.; Sernagor, E.; Lako, M.; Armstrong, L. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line- and method-dependent. Stem Cells 2019, 38, 195. [Google Scholar] [CrossRef]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zhong, X.; Li, K.; Xie, B.; Liu, Y.; Ye, M.; Li, K.; Xu, C.; Ge, J. An Optimized System for Effective Derivation of Three-Dimensional Retinal Tissue via Wnt Signaling Regulation. Stem Cells 2018, 36, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Onishi, A.; Matsuyama, T.; Masuda, T.; Ogino, Y.; Kageyama, M.; Takahashi, M.; Uchiumi, F. Rapid and efficient generation of mature retinal organoids derived from human pluripotent stem cells via optimized pharmacological modulation of Sonic hedgehog, activin A, and retinoic acid signal transduction. PLoS ONE 2024, 19, e0308743. [Google Scholar] [CrossRef]
- Eastlake, K.; Wang, W.; Jayaram, H.; Murray-Dunning, C.; Carr, A.J.F.; Ramsden, C.M.; Vugler, A.; Gore, K.; Clemo, N.; Stewart, M.; et al. Phenotypic and Functional Characterization of Müller Glia Isolated from Induced Pluripotent Stem Cell-Derived Retinal Organoids: Improvement of Retinal Ganglion Cell Function upon Transplantation. Stem Cells Transl. Med. 2019, 8, 775–784. [Google Scholar] [CrossRef]
- Wong, N.K.; Yip, S.P.; Huang, C.L. Establishing Functional Retina in a Dish: Progress and Promises of Induced Pluripotent Stem Cell-Based Retinal Neuron Differentiation. Int. J. Mol. Sci. 2023, 24, 13652. [Google Scholar] [CrossRef]
- Kuwahara, A.; Ozone, C.; Nakano, T.; Saito, K.; Eiraku, M.; Sasai, Y. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 2015, 6, 6286. [Google Scholar] [CrossRef]
- Wagstaff, E.L.; ten Asbroek, A.L.M.A.; ten Brink, J.B.; Jansonius, N.M.; Bergen, A.A.B. An alternative approach to produce versatile retinal organoids with accelerated ganglion cell development. Sci. Rep. 2021, 11, 1101. [Google Scholar] [CrossRef]
- Kelley, R.A.; Chen, H.Y.; Swaroop, A.; Li, T. Accelerated Development of Rod Photoreceptors in Retinal Organoids Derived from Human Pluripotent Stem Cells by Supplementation with 9-cis Retinal. STAR Protoc. 2020, 1, 100033. [Google Scholar] [CrossRef]
- Isla-Magrané, H.; Veiga, A.; García-Arumí, J.; Duarri, A. Multiocular organoids from human induced pluripotent stem cells displayed retinal, corneal, and retinal pigment epithelium lineages. Stem Cell Res. Ther. 2021, 12, 581. [Google Scholar] [CrossRef]
- Vincent, A.; Krishnakumar, S.; Parameswaran, S. Heterozygous RB1 mutation enhanced ATP production in human iPSC-derived retinal organoids. Mol. Biol. Rep. 2024, 51, 606. [Google Scholar] [CrossRef] [PubMed]
- Drabbe, E.; Pelaez, D.; Agarwal, A. Retinal organoid chip: Engineering a physiomimetic oxygen gradient for optimizing long term culture of human retinal organoids. Lab A Chip 2024, 25, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Du, J.L.; Gao, L.X.; Wang, T.; Ye, Z.; Li, H.Y.; Li, W.; Zeng, Q.; Xi, J.-F.; Yue, W.; Li, Z.-H. Influence of hypoxia on retinal progenitor and ganglion cells in human induced pluripotent stem cell-derived retinal organoids. Int. J. Ophthalmol. 2023, 16, 1574–1581. [Google Scholar] [CrossRef]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Phoida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Rep. 2018, 10, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.Y.; Woo, T.T.Y.; Wong, R.L.M.; Wong, D. Apoptosis and other cell death mechanisms after retinal detachment: Implications for photoreceptor rescue. Ophthalmologica 2011, 226, 10–17. [Google Scholar] [CrossRef]
- Moos, W.H.; Faller, D.V.; Glavas, I.P.; Harpp, D.N.; Kamperi, N.; Kanara, I.; Kodukula, K.; Mavrakis, A.N.; Pernokas, J.; Pernokas, M.; et al. Treatment and prevention of pathological mitochondrial dysfunction in retinal degeneration and in photoreceptor injury. Biochem. Pharmacol. 2022, 203, 115168. [Google Scholar] [CrossRef]
- Chen, H.Y.; Swaroop, M.; Papal, S.; Mondal, A.K.; Song, H.B.; Campello, L.; Tawa, G.J.; Regent, F.; Shimada, H.; Nagashima, K.; et al. Reserpine maintains photoreceptor survival in retinal ciliopathy by resolving proteostasis imbalance and ciliogenesis defects. eLife 2023, 12, e83205. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Liang, L.; Zhang, L.; Wang, J.; Chen, L.; Su, C.; Cao, J.; Yu, Q.; Deng, S.; Chan, H.F.; et al. Retinal organoids and microfluidic chip-based approaches to explore the retinitis pigmentosa with USH2A mutations. Front. Bioeng. Biotechnol. 2022, 10, 939774. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xu, G. Autophagy: A Role in the Apoptosis, Survival, Inflammation, and Development of the Retina. Ophthalmic Res. 2019, 61, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Loygorri, J.I.; Viedma-Poyatos, Á.; Gómez-Sintes, R.; Boya, P. Urolithin A promotes p62-dependent lysophagy to prevent acute retinal neurodegeneration. Mol. Neurodegener. 2024, 19, 49. [Google Scholar] [CrossRef]
- Watson, A.; Lako, M. Retinal organoids provide unique insights into molecular signatures of inherited retinal disease throughout retinogenesis. J. Anat. 2023, 243, 186–203. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Rusten, T.E.; Stenmark, H. P62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Díaz-Carretero, A.; Beau, I.; Condongo, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef]
- Yanamoto, Y.; Mizushima, N. Autophagy and Ciliogenesis. JMA J. 2021, 4, 207–215. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef]
- Lako, M.; Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Hilgen, G.; et al. Human iPSC-derived RPE and retinal organoids reveal impaired alternative splicing of genes involved in pre-mRNA splicing in PRPF31 autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1563. [Google Scholar]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin. Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef]
- Ma, S.; Xie, Y.; Wang, Q.; Fu, S.; Wu, H. Application of eye organoids in the study of eye diseases. Exp. Eye Res. 2024, 247, 110068. [Google Scholar] [CrossRef]
- Boon, N.; Wijnholds, J.; Pellissier, L.P. Research Models and Gene Augmentation Therapy for CRB1 Retinal Dystrophies. Front. Neurosci. 2020, 14, 860. [Google Scholar] [CrossRef]
- McDonald, A.; Wijnholds, J. Retinal Ciliopathies and Potential Gene Therapies: A Focus on Human iPSC-Derived Organoid Models. Int. J. Mol. Sci. 2024, 25, 2887. [Google Scholar] [CrossRef]
- Han, J.W.; Chang, H.S.; Park, S.C.; Yang, J.Y.; Kim, Y.J.; Kim, J.H.; Park, H.S.; Jeong, H.; Lee, J.; Yoon, C.K.; et al. Early Developmental Characteristics and Features of a Three-Dimensional Retinal Organoid Model of X-Linked Juvenile Retinoschisis. Int. J. Mol. Sci. 2024, 25, 8203. [Google Scholar] [CrossRef]
- Liang, Y.; Sun, X.; Duan, C.; Tang, S.; Chen, J. Application of patient-derived induced pluripotent stem cells and organoids in inherited retinal diseases. Stem Cell Res. Ther. 2023, 14, 340. [Google Scholar] [CrossRef]
- Maeda, T.; Mandai, M.; Sugita, S.; Kime, C.; Takahashi, M. Strategies of pluripotent stem cell-based therapy for retinal degeneration: Update and challenges. Trends Mol. Med. 2022, 28, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Singh, N. Inflammation and retinal degenerative diseases. Neural Regen. Res. 2023, 18, 513–518. [Google Scholar] [CrossRef]
- Kondkar, A.A.; Abu-Amero, K.K. Leber congenital amaurosis: Current genetic basis, scope for genetic testing and personalized medicine. Exp. Eye Res. 2019, 189, 107834. [Google Scholar] [CrossRef]
- Ghenciu, L.A.; Hațegan, O.A.; Stoicescu, E.R.; Iacob, R.; Șișu, A.M. Emerging Therapeutic Approaches and Genetic Insights in Stargardt Disease: A Comprehensive Review. Int. J. Mol. Sci. 2024, 25, 8859. [Google Scholar] [CrossRef] [PubMed]
- Zaw, K.; Carvalho, L.S.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D.; Chen, F.K.; McLenachan, S. Pathogenesis and Treatment of Usher Syndrome Type IIA. Asia-Pacific J. Ophthalmol. 2022, 11, 369–379. [Google Scholar] [CrossRef]
- Su, P.Y.; Lee, W.; Zernant, J.; Tsang, S.H.; Nagasaki, T.; Corneo, B.; Allikmets, R. Establishment of the iPSC line CUIMCi005-A from a patient with Stargardt disease for retinal organoid culture. Stem Cell Res. 2022, 65, 102973. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.J.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769. [Google Scholar] [CrossRef]
- Dulla, K.; Aguila, M.; Lane, A.; Jovanovic, K.; Parfitt, D.A.; Schulkens, I.; Chan, H.L.; Schmidt, I.; Beumer, W.; Vorthoren, L.; et al. Splice-Modulating Oligonucleotide QR-110 Restores CEP290 mRNA and Function in Human c.2991+1655A>G LCA10 Models. Mol. Therapy. Nucleic Acids 2018, 12, 730. [Google Scholar] [CrossRef]
- Shimada, H.; Lu, Q.; Insinna-Kettenhofen, C.; Nagashima, K.; English, M.A.; Semler, E.M.; Mahgerefteh, J.; Cideciyan, A.V.; Li, T.; Brooks, B.P.; et al. In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations. Cell Rep. 2017, 20, 384–396. [Google Scholar] [CrossRef]
- Lukovic, D.; Artero Castro, A.; Kaya, K.D.; Munezero, D.; Gieser, L.; Davó-Martínez, C.; Corton, M.; Cuenca, N.; Swaroop, A.; Ramamurthy, V.; et al. Retinal Organoids derived from hiPSCs of an AIPL1-LCA Patient Maintain Cytoarchitecture despite Reduced levels of Mutant AIPL1. Sci. Rep. 2020, 10, 5426. [Google Scholar] [CrossRef]
- Leung, A.; Sacristan-Reviriego, A.; Perdigão, P.R.L.; Sai, H.; Georgiou, M.; Kalitzeos, A.; Carr, A.J.F.; Coffey, P.J.; Muchaelides, M.; Bainbridge, J.; et al. Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital amaurosis. Stem Cell Rep. 2022, 17, 2187–2202. [Google Scholar] [CrossRef]
- Chirco, K.R.; Chew, S.; Moore, A.T.; Duncan, J.L.; Lamba, D.A. Allele-specific gene editing to rescue dominant CRX-associated LCA7 phenotypes in a retinal organoid model. Stem Cell Rep. 2021, 16, 2690. [Google Scholar] [CrossRef]
- Kruczek, K.; Qu, Z.; Gentry, J.; Fadl, B.R.; Gieser, L.; Hiriyanna, S.; Batz, Z.; Samant, M.; Samanta, A.; Chu, C.J.; et al. Gene Therapy of Dominant CRX-Leber Congenital Amaurosis using Patient Stem Cell-Derived Retinal Organoids. Stem Cell Rep. 2021, 16, 252–263. [Google Scholar] [CrossRef]
- Boon, N.; Lu, X.; Andriessen, C.A.; Moustakas, I.; Buck, T.M.; Freund, C.; Arendzen, C.H.; Bohringer, S.; Mei, H.; Wijnholds, J.; et al. AAV-mediated gene augmentation therapy of CRB1 patient-derived retinal organoids restores the histological and transcriptional retinal phenotype. Stem Cell Rep. 2023, 18, 1123. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated From the iPSCs of a Patient with the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell. Neurosci. 2019, 13, 463934. [Google Scholar] [CrossRef]
- Xu, J.; Yu, S.J.; Sun, S.; Li, Y.P.; Zhang, X.; Jin, K.; Jin, Z.B. Enhanced innate responses in microglia derived from retinoblastoma patient-specific iPSCs. Glia 2024, 72, 872–884. [Google Scholar] [CrossRef]
- Bocquet, B.; Borday, C.; Erkilic, N.; Mamaeva, D.; Donval, A.; Masson, C.; Parain, K.; Kaminska, K.; Quinodoz, M.; Perea-Romero, I. TBC1D32 variants disrupt retinal ciliogenesis and cause retinitis pigmentosa. JCI Insight 2023, 8, e169426. [Google Scholar] [CrossRef]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67. [Google Scholar] [CrossRef]
- Leong, Y.C.; Di Foggia, V.; Pramod, H.; Bitner-Glindzicz, M.; Patel, A.; Sowden, J.C. Molecular pathology of Usher 1B patient-derived retinal organoids at single cell resolution. Stem Cell Rep. 2022, 17, 2421. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Jimenez-Medina, C.; Erkilic, N.; Cappellino, L.; Lefevre, A.; Nagel-Wolfrum, K.; Wolfrum, U.; Wijk, E.V.; Roux, A.F.; Meunier, I.; et al. USH2A variants causing retinitis pigmentosa or Usher syndrome provoke differential retinal phenotypes in disease-specific organoids. Hum. Genet. Genom. Adv. 2023, 4, 100229. [Google Scholar] [CrossRef]
- Lei, Q.; Xiang, K.; Cheng, L.; Xiang, M. Human retinal organoids with an OPA1 mutation are defective in retinal ganglion cell differentiation and function. Stem Cell Rep. 2024, 19, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Ding, C.; Sun, X.; Mao, S.; Liang, Y.; Liu, X.; Ding, X.; Chen, J.; Tang, S. Retinal organoids with X-linked retinoschisis RS1 (E72K) mutation exhibit a photoreceptor developmental delay and are rescued by gene augmentation therapy. Stem Cell Res. Ther. 2024, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Wang, M.L.; Chen, S.J.; Kuo, J.C.; Wang, W.J.; Nhi Nguyen, P.N.; Wahlin, K.J.; Lu, J.F.; Tran, A.A.; Shi, M.; et al. Morphological and Molecular Defects in Human Three-Dimensional Retinal Organoid Model of X-Linked Juvenile Retinoschisis. Stem Cell Rep. 2019, 13, 906. [Google Scholar] [CrossRef]
- Chakrabarty, K.; Nayak, D.; Debnath, J.; Das, D.; Shetty, R.; Ghosh, A. Retinal organoids in disease modeling and drug discovery: Opportunities and challenges. Surv. Ophthalmol. 2024, 69, 179–189. [Google Scholar] [CrossRef]
- Sladen, P.E.; Naeem, A.; Adefila-Ideozu, T.; Vermeule, T.; Busson, S.L.; Michaelides, M.; Naylor, S.; Forbes, A.; Lane, A.; Georgiadis, A. AAV-RPGR Gene Therapy Rescues Opsin Mislocalisation in a Human Retinal Organoid Model of RPGR-Associated X-Linked Retinitis Pigmentosa. Int. J. Mol. Sci. 2024, 25, 1839. [Google Scholar] [CrossRef] [PubMed]
- da Costa, B.L.; Li, Y.; Levi, S.R.; Tsang, S.H.; Quinn, P.M.J. Generation of CRB1 RP Patient-Derived iPSCs and a CRISPR/Cas9-Mediated Homology-Directed Repair Strategy for the CRB1 c.2480G>T Mutation. Adv. Exp. Med. Biol. 2023, 1415, 571–576. [Google Scholar] [CrossRef]
- Xue, Y.; Lin, B.; Chen, J.T.; Tang, W.C.; Browne, A.W.; Seiler, M.J. The Prospects for Retinal Organoids in Treatment of Retinal Diseases. Asia-Pac. J. Ophthalmol. 2022, 11, 314. [Google Scholar] [CrossRef]
- Yu, C.T.; Kandoi, S.; Periasamy, R.; Reddy, L.V.K.; Follett, H.M.; Summerfelt, P.; Martinez, C.; Guillaume, C.; Bowie, O.; Connor, T.B.; et al. Human iPSC-derived photoreceptor transplantation in the cone dominant 13-lined ground squirrel. Stem Cell Rep. 2024, 19, 331–342. [Google Scholar] [CrossRef]
- Lin, B.; Singh, R.K.; Seiler, M.J.; Nasonkin, I.O. Survival and Functional Integration of Human Embryonic Stem Cell-Derived Retinal Organoids After Shipping and Transplantation into Retinal Degeneration Rats. Stem Cells Dev. 2024, 33, 201–213. [Google Scholar] [CrossRef]
- Ishikura, M.; Muraoka, Y.; Hirami, Y.; Tu, H.-Y.; Mandai, M. Adaptive Optics Optical Coherence Tomography Analysis of Induced Pluripotent Stem Cell-Derived Retinal Organoid Transplantation in Retinitis Pigmentosa. Cureus 2024, 16, e64962. [Google Scholar] [CrossRef]
- Zhang, C.J.; Jin, Z.B. Turning point of organoid transplantation: First-in-human trial of iPSC-derived retinal organoid grafting in patients with retinitis pigmentosa. Sci. China Life Sci. 2024, 67, 1082–1084. [Google Scholar] [CrossRef]
- Hirami, Y.; Mandai, M.; Sugita, S.; Maeda, A.; Maeda, T.; Yamamoto, M.; Uyama, H.; Yokota, S.; Fujihara, M.; Igeta, M.; et al. Safety and stable survival of stem-cell-derived retinal organoid for 2 years in patients with retinitis pigmentosa. Cell Stem Cell 2023, 30, 1585–1596.e6. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Afanasyeva, T.A.V.; Athanasiou, D.; Perdigao, P.R.L.; Whiting, K.R.; Duijkers, L.; Astuti, G.D.N.; Bennett, J.; Garanto, A.; Spuy, J.; Roepman, R.; et al. CRISPR-Cas9 correction of a nonsense mutation in LCA5 rescues lebercilin expression and localization in human retinal organoids. Mol. Therapy. Methods Clin. Dev. 2023, 29, 522. [Google Scholar] [CrossRef] [PubMed]
- Kruczek, K.; Qu, Z.; Welby, E.; Shimada, H.; Hiriyanna, S.; English, M.A.; Zein, W.M.; Brooks, B.P.; Swaroop, A. In vitro modeling and rescue of ciliopathy associated with IQCB1/NPHP5 mutations using patient-derived cells. Stem Cell Rep. 2022, 17, 2172. [Google Scholar] [CrossRef]
- West, E.L.; Majumder, P.; Naeem, A.; Fernando, M.; O’Hara-Wright, M.; Lanning, E.; Kloc, M.; Ribero, J.; Ovando-Roche, P.; Shum, I.O.; et al. Antioxidant and lipid supplementation improve the development of photoreceptor outer segments in pluripotent stem cell-derived retinal organoids. Stem Cell Rep. 2022, 17, 775. [Google Scholar] [CrossRef]
- Sai, H.; Ollington, B.; Rezek, F.O.; Chai, N.; Lane, A.; Georgiadis, T.; Bainbridge, J.; Michaelides, M.; Sacristan-Reviriego, A.; Perdigao, P.R.L.; et al. Effective AAV-mediated gene replacement therapy in retinal organoids modeling AIPL1-associated LCA4. Mol. Ther. Nucleic Acids 2024, 35, 102148. [Google Scholar] [CrossRef]
- Tornabene, P.; Trapani, I.; Minopoli, R.; Centrulo, M.; Lupo, M.; De Simone, S.; Tiberi, P.; Dell‘Aquila, F.; Marrocco, E.; Iodice, C.; et al. Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci. Transl. Med. 2019, 11, eaav4523. [Google Scholar] [CrossRef]
- Corral-Serrano, J.C.; Sladen, P.E.; Ottaviani, D.; Rezek, O.F.; Athanasiou, D.; Jovanovic, K.; Spuy, J.V.D.; Mansfield, B.C.; Cheetham, M.E. Eupatilin Improves Cilia Defects in Human CEP290 Ciliopathy Models. Cells 2023, 12, 1575. [Google Scholar] [CrossRef]
- Kroeger, H.; Grandjean, J.M.D.; Chiang, W.C.J.; Bindels, D.D.; Mastey, R.; Okalova, J.; Nguyen, A.; Powers, E.T.; Kelly, P.W.; Grimsey, N.J.; et al. ATF6 is essential for human cone photoreceptor development. Proc. Natl. Acad. Sci. USA 2021, 118, e2103196118. [Google Scholar] [CrossRef]
- Ribeiro, J.; Procyk, C.A.; West, E.L.; O’Hara-Wright, M.; Martins, M.F.; Khorasani, M.M.; Hare, A.; Basche, M.; Fernando, M.; Goh, D.; et al. Restoration of visual function in advanced disease after transplantation of purified human pluripotent stem cell-derived cone photoreceptors. Cell Rep. 2021, 35, 109022. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.B.; Lin, B.; Martinez-Camarillo, J.C.; Zhu, D.; McLelland, B.T.; Nistor, G.; Keirstead, H.S.; Humayun, M.S.; Seiler, M.J. Co-grafts of Human Embryonic Stem Cell Derived Retina Organoids and Retinal Pigment Epithelium for Retinal Reconstruction in Immunodeficient Retinal Degenerate Royal College of Surgeons Rats. Front. Neurosci. 2021, 15, 752958. [Google Scholar] [CrossRef]
- Garcia-Delgado, A.B.; Valdes-Sanchez, L.; Morillo-Sanchez, M.J.; Ponte-Zuñiga, B.; Diaz-Corrales, F.J.; de la Cerda, B. Dissecting the role of EYS in retinal degeneration: Clinical and molecular aspects and its implications for future therapy. Orphanet J. Rare Dis. 2021, 16, 222. [Google Scholar] [CrossRef]
- Aasen, D.M.; Vergara, M.N. New Drug Discovery Paradigms for Retinal Diseases: A Focus on Retinal Organoids. J. Ocul. Pharmacol. Ther. 2020, 36, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, Z.; Yuan, F.; Jin, K.; Xiang, M. Retinal organoid technology: Where are we now? Int. J. Mol. Sci. 2021, 22, 10244. [Google Scholar] [CrossRef]
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi- layer tissue models in a human retina-on-a-chip platform. eLife 2019, 8, e46188. [Google Scholar] [CrossRef] [PubMed]
- Vielle, A.; Park, Y.K.; Secora, C.; Vergara, M.N. Organoids for the Study of Retinal Development and Developmental Abnormalities. Front. Cell. Neurosci. 2021, 15, 667880. [Google Scholar] [CrossRef]
- Arik, Y.B.; Buijsman, W.; Loessberg-Zahl, J.; Cuartas-Vélez, C.; Veenstra, C.; Logtenberg, S.; Grobbink, A.M.; Bergveld, P.; Gagliardi, G.; Hollander, A.I.D.; et al. Microfluidic organ-on-a-chip model of the outer blood-retinal barrier with clinically relevant read-outs for tissue permeability and vascular structure. Lab A Chip 2021, 21, 272–283. [Google Scholar] [CrossRef]
- Wang, L.; Hiler, D.; Xu, B.; AlDiri, I.; Chen, X.; Zhou, X.; Griffiths, L.; Valentine, M.; Shirinifard, A.; Sablauer, A.; et al. Retinal Cell Type DNA Methylation and Histone Modifications Predict Reprogramming Efficiency and Retinogenesis in 3D Organoid Cultures. Cell Rep. 2018, 22, 2601–2614. [Google Scholar] [CrossRef]
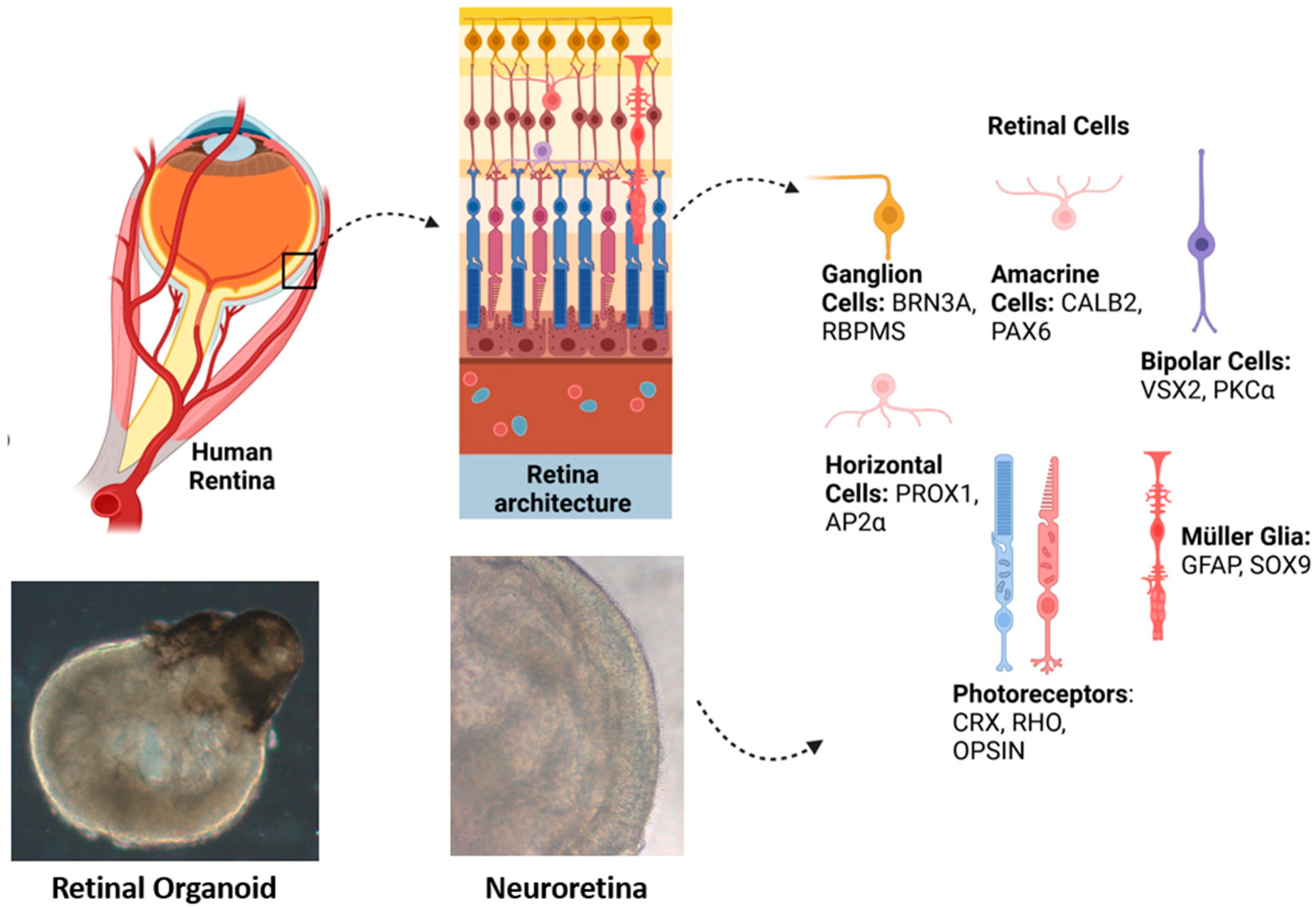
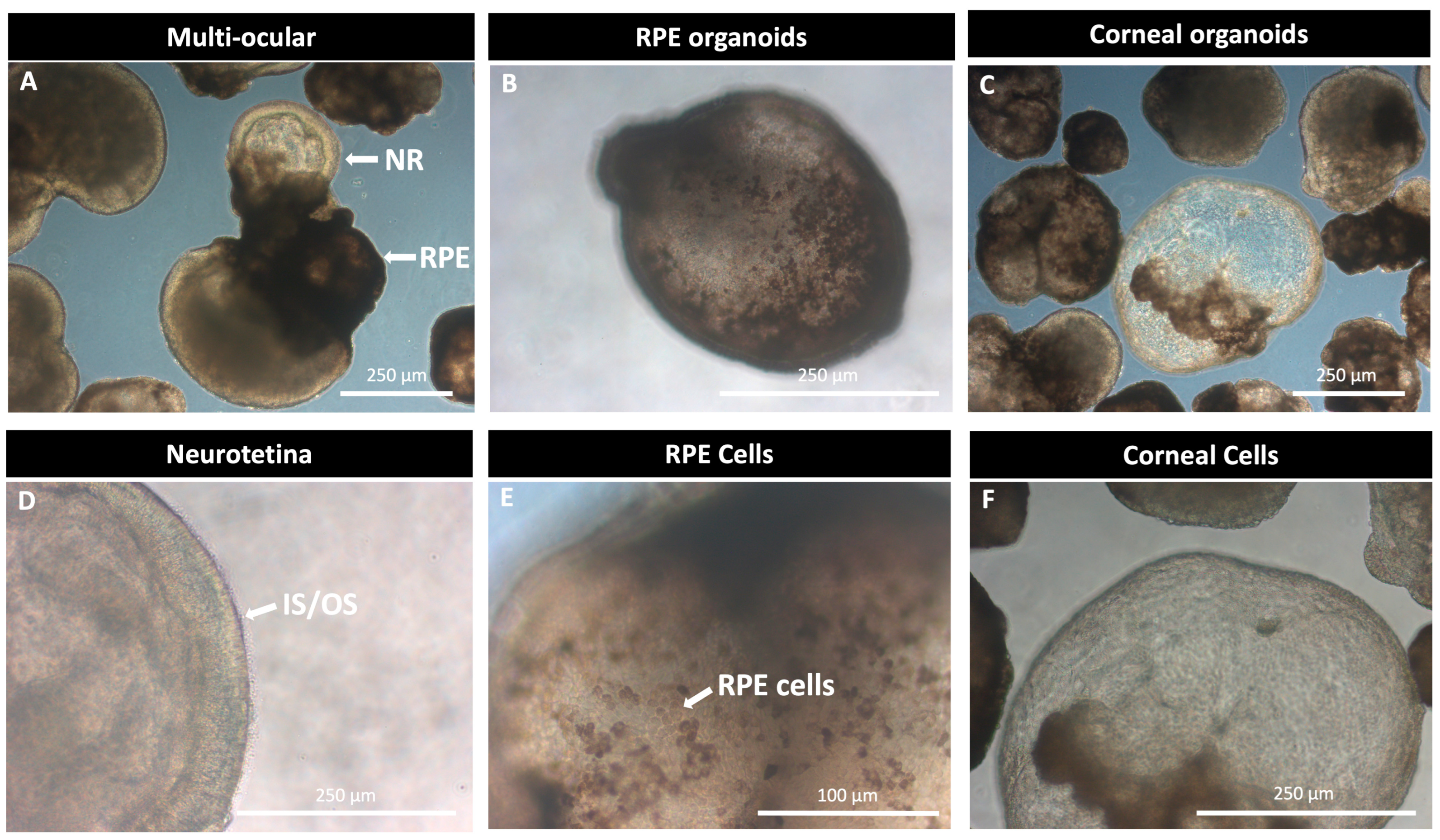
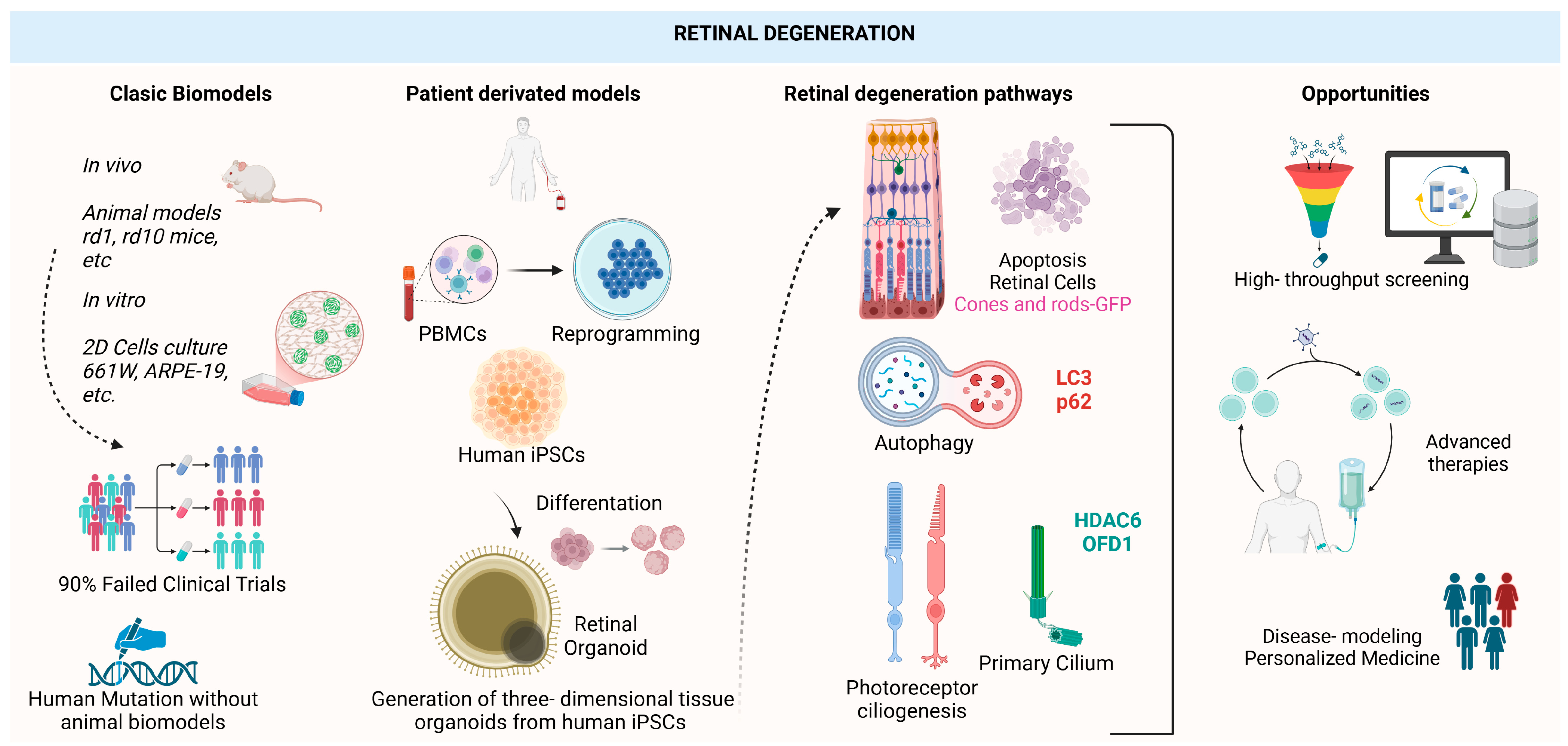
| Disease | Gene | Studies on iPSC Lines’ Retinal Organoids with Induced or Naïve Disease-Specific Mutations | Reference | |
|---|---|---|---|---|
| Stargardt disease | ABCA4 | Mutation expression | Su et al. (2022) | [77] |
| LCA | CEP290 | Retinogenesis, morphology, markers, and pathogenic processes | Parfitt et al. (2016) | [78] |
| Dulla et al. (2018) | [79] | |||
| Shimada et al. (2017) | [80] | |||
| AIPL1 (LCA 4) | Study of expression, therapy with the PTC124 drug | Lukovic et al. (2020) | [81] | |
| AIPL1 | Mutation expression | Leung et al. (2022) | [82] | |
| CRX | Development and opsin expression | Chirco et al. (2021) | [83] | |
| Kruczek et al. (2021) | [84] | |||
| JSRD | CEP290 | Retinogenesis, morphology, markers, and pathogenic processes | Shimada et al. (2017) | [80] |
| RP | CRB1 | Compound heterozygous mutations | Boon et al. (2023) | [85] |
| PRPF31 | Mutation expression | Buskin et al. (2018) | [50] | |
| USH2A | Mutation expression | Guo et al. (2019) | [86] | |
| TBC1D32 | Mutation expression | Xu et al. (2024) | [87] | |
| Late-onset RP | PDE6B | Mutation expression | Bocquet et al. (2023) | [88] |
| X-linked RP | RPGR (ORF15 region, and intron 11) | Mutation expression | McDonald et al. (2024) | [69] |
| RP2 | Mutation expression | Lane et al. (2020) | [89] | |
| RPGR | Knockout animal models’ mutation | Lane et al. (2020) | [89] | |
| Non-syndromic RP | USH2A | Mutation expression | Guo et al. (2019) | [86] |
| Usher syndrome type 1 | USH1B—MYO7A | Mutation expression | Leong et al. (2022) | [90] |
| Usher syndrome type 2 | USH2A | Mutation expression | Guo et al. (2019) | [86] |
| Sanjurjo-Soriano et al. (2023) | [91] | |||
| Autosomal dominant optic atrophy | OPA1 | Mutation expression | Lei et al. (2024) | [92] |
| XLRS | RS1 | Mutation expression | Duan et al. (2024) | [93] |
| Retinal development and expression of retinoschisin | Huang et al. (2019) | [94] | ||
| Retinoblastoma | RB1 | Cell-to-cell interactions | Xu et al. (2024) | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galindo-Cabello, N.; Caballano-Infantes, E.; Benites, G.; Pastor-Idoate, S.; Diaz-Corrales, F.J.; Usategui-Martín, R. Retinal Organoids: Innovative Tools for Understanding Retinal Degeneration. Int. J. Mol. Sci. 2025, 26, 3263. https://doi.org/10.3390/ijms26073263
Galindo-Cabello N, Caballano-Infantes E, Benites G, Pastor-Idoate S, Diaz-Corrales FJ, Usategui-Martín R. Retinal Organoids: Innovative Tools for Understanding Retinal Degeneration. International Journal of Molecular Sciences. 2025; 26(7):3263. https://doi.org/10.3390/ijms26073263
Chicago/Turabian StyleGalindo-Cabello, Nadia, Estefanía Caballano-Infantes, Gregorio Benites, Salvador Pastor-Idoate, Francisco J. Diaz-Corrales, and Ricardo Usategui-Martín. 2025. "Retinal Organoids: Innovative Tools for Understanding Retinal Degeneration" International Journal of Molecular Sciences 26, no. 7: 3263. https://doi.org/10.3390/ijms26073263
APA StyleGalindo-Cabello, N., Caballano-Infantes, E., Benites, G., Pastor-Idoate, S., Diaz-Corrales, F. J., & Usategui-Martín, R. (2025). Retinal Organoids: Innovative Tools for Understanding Retinal Degeneration. International Journal of Molecular Sciences, 26(7), 3263. https://doi.org/10.3390/ijms26073263