3.2. Chemistry
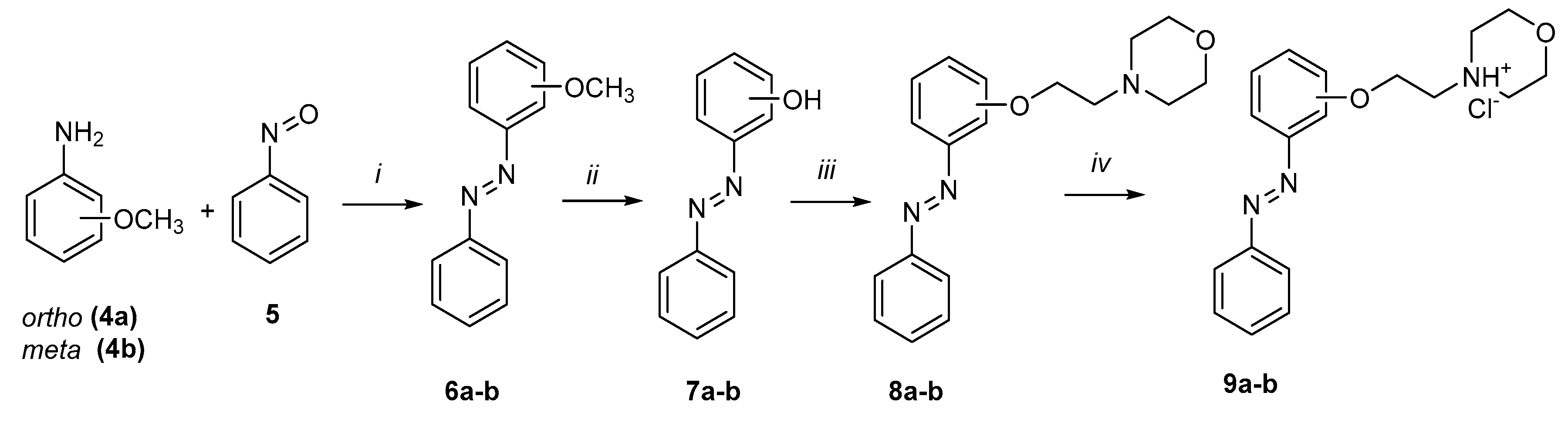
3.2.1. General Method for the Synthesis of Compounds 6a–b
To a solution of nitrosobenzene (5) in 100 mL of methanol, compound 4a or 4b (depending on the reaction) and 2 mL of acetic acid were added. The reaction mixture was stirred at RT for 72 h. After completion of the reaction (monitored by TLC), an equal volume of a 5% aqueous K2CO3 solution was added to the reaction mixture, followed by extraction with ethyl acetate from water. The organic layers were combined, washed with brine, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure. The product was purified by column chromatography (SiO2, hexane/EA).
Synthesis of o-methoxyazobenzene (6a)
Starting with compound 4a (1.085 g, 8.810 mmol) and compound 5 (0.788 g, 7.357 mmol), the yield of compound 6a was 30% (468 mg, yellow-orange amorphous solid).
1H NMR spectrum of compound 6a (300 MHz, CDCl3) δ, ppm: 7.92 (m, 2H, 2×CH), 7.67 (m, 1H, 1×CH), 7.58–7.38 (m, 4H, 4×CH), 7.10 (m, 1H, 1×CH), 7.03 (m, 1H, 1×CH), 4.03 (s, 3H, -OCH3).
13C NMR spectrum of compound 6a (75 MHz, CDCl3) δ, ppm: 157.1, 153.3, 142.4, 132.6, 130.9, 129.1, 123.1, 120.9, 117.1, 112.8, 56.4.
MS (ESI+) m/z: [M+H]+, calculated for (C13H13N2O)+ 213.1, found 213.1.
Synthesis of m-methoxyazobenzene (6b)
Starting with compound 4b (1.278 g, 10.378 mmol) and compound 5 (0.927 g, 8.655 mmol), the yield of compound 6b was 37% (679 mg, red-brown crystals).
1H NMR spectrum of compound 6b (300 MHz, CDCl3) δ, ppm: 7.93 (m, 2H, 2×CH), 7.58–7.46 (m, 6H, 6×CH), 7.06 (m, 1H, 1×CH), 3.91 (s, 3H, -OCH3).
13C NMR spectrum of compound 6b (75 MHz, CDCl3) δ, ppm: 160.4, 154.0, 152.7, 131.2, 129.9, 129.2, 123.0, 118.0, 117.3, 105.8, 55.6.
MS (ESI+) m/z: [M+H]+, calculated for (C13H13N2O)+ 213.1, found 213.0.
3.2.2. General Method for the Synthesis of Compounds 7a–b
A solution of compound 6a–b in DCM (5 mL) was cooled to −78 °C. Then, a BBr3 solution in DCM (1:7, v/v) was added to the mixture with stirring, and the reaction was left at RT for 16 h. After the reaction was complete (monitored by TLC), the mixture was poured onto ice, followed by extraction with DCM from water. The combined organic layers were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The product was purified by column chromatography (SiO2, hexane/EA).
Synthesis of o-hydroxyazobenzene (7a)
Starting from compound 6a (0.100 g, 0.471 mmol) and BBr3 (0.235 g, 0.938 mmol), the yield of compound 7a was 58% (54 mg, red-orange crystals).
1H NMR spectrum of compound 7a (300 MHz, DMSO-d6) δ, ppm: 11.18 (s, 1H, -OH), 7.98 (m, 2H, 2×CH), 7.76 (m, 1H, 1×CH), 7.65–7.48 (m, 3H, 3×CH), 7.43 (m, 1H, 1×CH), 7.12–6.95 (m, 2H, 2×CH).
13C NMR spectrum of compound 7a (75 MHz, DMSO-d6) δ, ppm: 154.4, 151.4, 138.3, 133.6, 131.3, 129.4, 123.3, 122.6, 119.8, 118.2.
MS (ESI+) m/z: [M+H]+, calculated for (C12H11N2O)+ 199.1, found 199.0.
Synthesis of m-hydroxyazobenzene (7b)
Starting from compound 6b (0.084 g, 0.396 mmol) and BBr3 (0.191 g, 0.762 mmol), the yield of compound 7b was 48% (38 mg, yellow-orange amorphous solid).
1H NMR spectrum of compound 7b (300 MHz, CDCl3) δ, ppm: 7.96–7.85 (m, 2H, 2×CH), 7.56–7.49 (m, 4H, 4×CH), 7.44–7.35 (m, 2H, 2×CH), 6.99 (m, 1H, 1×CH), 5.83 (s, 1H, -OH).
13C NMR spectrum of compound 7b (75 MHz, CDCl3) δ, ppm: 156.6, 154.0, 152.6, 131.1, 130.2, 129.2, 123.0, 118.4, 117.3, 108.0.
MS (ESI+) m/z: [M+H]+, calculated for (C12H11N2O)+ 199.1, found 199.1.
3.2.3. General Method for the Synthesis of Compounds 8a–b
To a solution of 7a–b in 10 mL of acetonitrile, N-(2-chloroethyl)-morpholine hydrochloride, K2CO3, and KI were added. The reaction was stirred under reflux for 6 h. After completion of the reaction (monitored by TLC), the solvent was removed under reduced pressure, and then the mixture was dissolved in ethyl acetate and washed with water. The organic layers were combined, washed with brine, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure. The product was purified by column chromatography (SiO2, EA/MeOH = 15:1).
Synthesis of 2-(2-(N-morpholino)-ethoxy)-azobenzene (8a)
Starting from compound 7a (0.185 g, 0.933 mmol), N-(2-chloroethyl)-morpholine hydrochloride (0.260 g, 1.397 mmol), K2CO3 (0.386 g, 2.793 mmol), and KI (0.185 g, 1.114 mmol), the yield of compound 8a was 82% (238 mg, orange amorphous solid).
1H NMR spectrum of compound 8a (300 MHz, CDCl3) δ, ppm: 7.90 (m, 2H, 2×CH), 7.67 (m, 1H, 1×CH), 7.53–7.39 (m, 4H, 4×CH), 7.08 (m, 2H, 2×CH), 4.35 (t, J = 5.5 Hz, 2H, 1×CH2), 3.71 (m, 4H, 2×CH2), 2.91 (t, J = 5.5 Hz, 2H, 1×CH2), 2.68 (m, 4H, 2×CH2).
13C NMR spectrum of compound 8a (75 MHz, CDCl3) δ, ppm: 156.4, 153.2, 142.8, 132.5, 131.0, 129.2, 123.0, 121.4, 117.1, 114.7, 68.5, 67.1, 57.7, 54.5.
MS (ESI+) m/z: [M+H]+, calculated for (C18H22N3O2)+ 312.2, found 312.3.
Synthesis of 3-(2-(N-morpholino)-ethoxy)-azobenzene (8b)
Starting from compound 7b (0.097 g, 0.489 mmol), N-(2-chloroethyl)-morpholine hydrochloride (0.137 g, 0.736 mmol), K2CO3 (0.203 g, 1.469 mmol), and KI (0.081 g, 0.488 mmol), the yield of compound 8b was 70% (106 mg, orange amorphous solid).
1H NMR spectrum of compound 8b (300 MHz, CDCl3) δ, ppm: 7.91 (m, 2H, 2×CH), 7.61–7.39 (m, 6H, 6×CH), 7.05 (m, 1H, 1×CH), 4.20 (t, J = 5.7 Hz, 2H, 1×CH2), 3.84–3.63 (m, 4H, 2×CH2), 2.84 (t, J = 5.7 Hz, 2H, 1×CH2), 2.60 (m, 4H, 2×CH2).
13C NMR spectrum of compound 8b (75 MHz, CDCl3) δ, ppm: 159.4, 153.9, 152.6, 131.2, 129.9, 129.2, 123.0, 118.3, 117.7, 106.3, 66.9, 65.9, 57.6, 54.1.
MS (ESI+) m/z: [M+H]+, calculated for (C18H22N3O2)+ 312.2, found 312.2.
3.2.4. General Method for the Synthesis of Compounds 9a–b
To a solution of compounds 8a–b in 15 mL of Et2O, a solution of HCl/Et2O (2.4 M) was added with stirring at 0–5 °C. The reaction mixture was stirred for 30 min at RT, after which the product was concentrated under reduced pressure.
Synthesis of the 2-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (9a)
Starting from compound 8a (0.219 g, 0.703 mmol) and HCl/Et2O (590 μL, 2.4 M), the yield of compound 9a was quantitative (244 mg, orange crystals).
1H NMR spectrum of compound 9a (300 MHz, DMSO-d6) δ, ppm: 11.87 (s, 1H, HCl), 7.83 (m, 2H, 2×CH), 7.67–7.51 (m, 5H, 5×CH), 7.36 (m, 1H, 1×CH), 7.13 (t, J = 7.7 Hz, 1H, 1××CH), 4.70 (t, J = 4.8 Hz, 2H, 1×CH2), 3.86 (m, 4H, 2×CH2), 3.62 (m, 4H, 2×CH2), 3.32 (m, 2H, 1×CH2).
13C NMR spectrum of compound 9a (75 MHz, DMSO-d6) δ, ppm: 155.2, 152.5, 141.6, 133.2, 131.5, 129.6, 122.5, 121.8, 116.5, 115.1, 64.4, 63.3, 55.0, 52.0.
MS (ESI+) m/z: [M-−HCl+H]+, calculated for (C18H22N3O2)+ 312.2, found 312.2.
Synthesis of the 3-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (9b)
Starting from compound 8b (0.125 g, 0.401 mmol) and HCl/Et2O (340 μL, 2.4 M), the yield of compound 9b was quantitative (139 mg, light yellow crystals).
1H NMR spectrum of compound 9b (300 MHz, DMSO-d6) δ, ppm: 11.61 (s, 1H, HCl), 7.90 (m, 2H, 2×CH), 7.65–7.52 (m, 5H, 5×CH), 7.48 (s, 1H, 1×CH), 7.23 (m, 1H, 1×CH), 4.57 (t, J = 5.1 Hz, 2H, 1×CH2), 4.04–3.77 (m, 4H, 2×CH2), 3.56 (m, 4H, 2×CH2), 3.22 (m, 2H, 1×CH2).
13C NMR spectrum of compound 9b (75 MHz, DMSO-d6) δ, ppm: 158.4, 153.1, 151.8, 131.8, 130.5, 129.6, 122.6, 118.3, 117.2, 106.9, 63.2, 62.5, 54.8, 51.7.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H22N3O2)+ 312.2, found 312.2.
3.2.5. Synthesis of 4-Hydroxyazobenzene (11)
Compound
11 was obtained using the procedure described earlier [
20].
1H NMR spectrum of compound 11 (300 MHz, DMSO-d6) δ, ppm: 10.31 (s, 1H, -OH); 7.81 (m, 4H, 4×CH); 7.52 (m, 3H, 3×CH); 6.95 (m, 2H, 2×CH).
13C NMR spectrum of compound 11 (75 MHz, DMSO-d6) δ, ppm: 161.0, 152.1, 145.2, 130.5, 129.4, 124.9, 122.1, 116.0.
MS (ESI+) m/z: [M+H]+, calculated for (C12H11N2O)+ 199.1, found 199.1.
3.2.6. Synthesis of 4-(1-Chloro-2-Ethoxy)-Azobenzene (12)
To a solution of compound 11 (1.000 g, 5.045 mmol) in acetonitrile (25 mL), K2CO3 (2.090 g, 15.123 mmol) and 1-bromo-2-chloroethane (1.260 mL, 2.171 g, 15.138 mmol) were added with stirring. The reaction mixture was stirred under reflux for 24 h and then extracted with ethyl acetate from water. The organic layer was washed with and dried over Na2SO4 (anhydrous). The solvent was removed under reduced pressure. After purification by chromatography (SiO2, hexane/EA = 5:1), the yield of compound 12 was 68% (894 mg, orange crystals).
1H NMR spectrum of compound 12 (300 MHz, CDCl3) δ, ppm: 7.97–7.86 (m, 4H, 4×CH), 7.56–7.42 (m, 3H, 3×CH), 7.03 (m, 2H, 2×CH), 4.30 (t, J = 5.8 Hz, 2H, 1×CH2), 3.85 (t, J = 5.8 Hz, 2H, 1×CH2).
13C NMR spectrum of compound 12 (75 MHz, CDCl3) δ, ppm: 160.7, 152.8, 147.5, 130.6, 129.2, 124.9, 122.7, 115.0, 68.3, 41.8.
3.2.7. Synthesis of 4-(2-(4-Methyl-1-Piperazino)-Ethoxy)-Azobenzene (13)
To a solution of compound 12 (516 mg, 1.979 mmol) in acetonitrile (15 mL), K2CO3 (819 mg, 5.926 mmol), 1-methylpiperazine (220 μL, 199 mg, 1.987 mmol), and KI (329 mg, 1.982 mmol) were added. The reaction mixture was stirred under reflux for 24 h and then extracted with ethyl acetate from water. The organic layer was washed with brine and dried over Na2SO4 (anhydrous). The solvent was removed under reduced pressure. After purification by chromatography (SiO2, DCM/MeOH/NH4OH = 90:10:1), the yield of compound 13 was 52% (333 mg, amorphous yellow solid).
1H NMR spectrum of compound 13 (300 MHz, DMSO-d6) δ, ppm: 7.85 (m, 4H, 4×CH), 7.65–7.46 (m, 3H, 3×CH), 7.13 (m, 2H, 2×CH), 4.17 (t, J = 5.8 Hz, 2H, 1×CH2), 2.71 (t, J = 5.8 Hz, 2H, 1×CH2), 2.46 (m, 4H, 2×CH2), 2.32 (m, 4H, 2×CH2), 2.14 (s, 3H, -CH3).
13C NMR spectrum of compound 13 (75 MHz, DMSO-d6) δ, ppm: 161.3, 152.0, 146.1, 130.8, 129.3, 124.6, 122.3, 115.1, 66.1, 56.5, 54.7, 53.0, 45.8.
MS (ESI+) m/z: [M+H]+, calculated for (C19H25N4O)+ 325.2, found 325.2.
3.2.8. Synthesis of the 4-(2-(4-Methyl-1-Piperazino)-Ethoxy)-Azobenzene Dihydrochloride (14)
To a solution of compound 13 (191 mg, 0.588 mmol) in diethyl ether (32 mL), a solution of HCl/Et2O (1.000 mL, 2.4 M) was added with stirring at 0–5 °C. The resulting mixture was stirred at RT for 30 min. The solvent was removed under reduced pressure. The yield of compound 14 was quantitative (233 mg, orange crystals).
1H NMR spectrum of compound 14 (300 MHz, DMSO-d6) δ, ppm: 12.06 (br s, HCl), 7.87 (m, 2H, 2×CH), 7.85 (m, 2H, 2×CH), 7.62–7.48 (m, 3H, 3×CH), 7.22 (m, 2H, 2×CH), 4.55 (m, 2H, 1×CH2), 3.91–3.55 (m, 10H, 5×CH2), 2.83 (s, 3H, -CH3).
13C NMR spectrum of compound 14 (75 MHz, D2O) δ (ppm): 159.8, 151.8, 146.6, 131.2, 129.4, 124.5, 122.1, 115.0, 61.6, 55.5, 50.1, 49.0, 42.8.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C19H25N4O)+ 325.2, found 325.2.
3.2.9. Synthesis of 4-(2-(N,N-Diethylamino)-Ethoxy)-Azobenzene (15)
To a solution of compound 11 (500 mg, 2.522 mmol) in acetonitrile (15 mL), K2CO3 (1047 mg, 7.576 mmol), 2-chloro-N,N-diethylethan-1-amine hydrochloride (1086 mg, 6.311 mmol), and KI (419 mg, 2.524 mmol) were added. The reaction mixture was stirred and heated for 3 h, after which it was extracted with ethyl acetate from water. The organic layer was washed with brine and dried using anhydrous Na2SO4. The solvent was removed under reduced pressure. After purification by column chromatography (SiO2, DCM/MeOH/NH4OH = from 200:10:1 to 25:10:1), compound 15 was obtained with an 18% yield (135 mg, amorphous orange solid).
1H NMR spectrum of compound 15 (300 MHz, DMSO-d6) δ (ppm): 7.94–7.78 (m, 4H, 4×CH), 7.63–7.46 (m, 3H, 3×CH), 7.13 (m, 2H, 2×CH), 4.12 (t, J = 6.1 Hz, 2H, 1×CH2), 2.80 (t, J = 6.1 Hz, 2H, 1×CH2), 2.56 (q, J = 7.1 Hz, 4H, 2×CH2), 0.98 (t, J = 7.1 Hz, 6H, 2×CH3).
13C NMR spectrum of compound 15 (75 MHz, DMSO-d6) δ (ppm): 161.3, 152.0, 146.1, 130.7, 129.3, 124.5, 122.2, 115.0, 66.9, 51.2, 47.0, 11.9.
MS (ESI+) m/z: [M+H]+, calculated for (C18H24N3O)+ 298.2, found 298.2.
3.2.10. Synthesis of 4-(2-(N,N-Diethylamino)Ethoxy)Azobenzene Hydrochloride (16)
To a solution of compound 15 (100 mg, 0.336 mmol) in diethyl ether (10 mL), a solution of HCl/Et2O (400 μL, 2.4 M) was added under stirring at 0–5 °C. The resulting mixture was stirred at RT for 30 min. The solvent was removed under reduced pressure. Compound 16 (112 mg, orange crystals) was obtained in quantitative yield.
1H NMR spectrum of compound 16 (300 MHz, DMSO-d6) δ, ppm: 11.01 (s, 1H, HCl), 7.93 (m, 2H, 2×CH), 7.85 (m, 2H, 2×CH), 7.64–7.49 (m, 3H, 3×CH), 7.20 (m, 2H, 2×CH), 4.54 (t, J = 5.1 Hz, 2H, 1×CH2), 3.53 (q, J = 5.0 Hz, 2H, 1×CH2), 3.21 (m, 4H, 2×CH2), 1.28 (t, J = 7.2 Hz, 6H, 2×CH3).
13C NMR spectrum of compound 16 (75 MHz, DMSO-d6) δ, ppm: 160.2, 151.9, 146.6, 130.9, 129.3, 124.5, 122.2, 115.3, 62.7, 49.5, 46.9, 8.4.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H24N3O)+ 298.2, found 298.2.
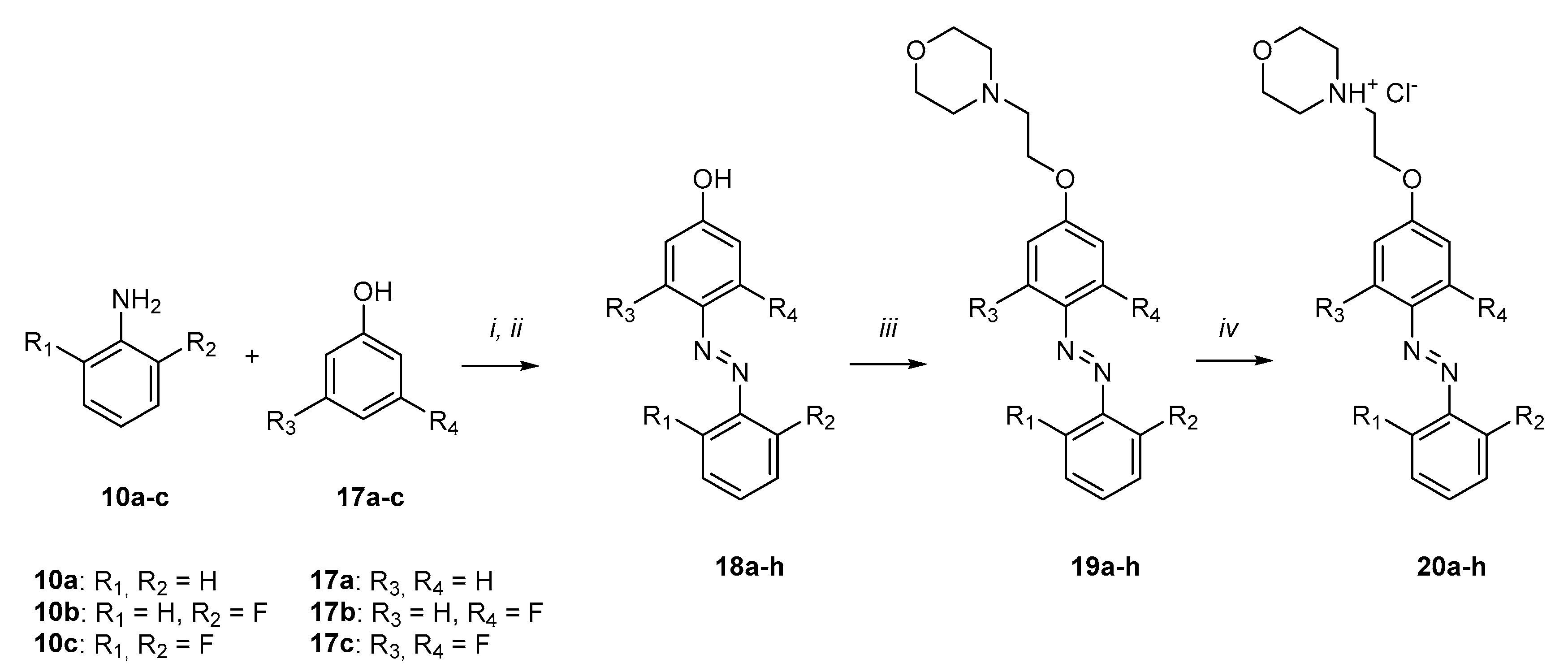
3.2.11. General Procedure for the Synthesis of Compounds 18a–h
To solution of 10a–c in an aqueous HCl (12% w/w), a solution of NaNO2 in water was added dropwise under stirring and cooling (0–5 °C). The reaction mixture was stirred for 30 min at 0–5 °C. Then, a solution of 17a–c in aqueous NaOH (14% w/w) was added dropwise to the reaction mixture under stirring over 10 min. The reaction was then stirred at 0–5 °C for 1 h. Upon completion, the pH of the reaction mixture was adjusted to 5–6, and the resulting precipitate was filtered and dried. The crude product was purified by column chromatography (SiO2, hexane/EA = 5:1).
Synthesis of 2-fluoro-4-hydroxyazobenzene (18a).
Starting from compound 10a (1.000 g, 10.738 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.741 g, 10.740 mmol) in water (10 mL), and compound 17b (1.204 g, 10.740 mmol) in 14% aqueous NaOH (40 mL), compound 18a was obtained with a yield of 65% (1.509 g, orange crystals).
1H NMR spectrum of compound 18a (300 MHz, DMSO-d6) δ, ppm: 10.79 (s, 1H, -OH), 7.86–7.76 (m, 2H, 2×CH), 7.70 (t, J = 8.9 Hz, 1H, 1×CH), 7.64–7.44 (m, 3H, 3×CH), 6.87–6.70 (m, 2H, 2×CH).
13C NMR spectrum of compound 18a (75 MHz, DMSO-d6) δ, ppm: 162.6 (d, J = 12.1 Hz), 159.4 (d, J = 255.9 Hz), 152.3, 133.1 (d, J = 6.8 Hz), 130.8, 129.3, 122.2, 118.4 (d, J = 2.2 Hz), 112.4 (d, J = 2.4 Hz), 103.3 (d, J = 21.8 Hz).
MS (ESI−) m/z: [M−H]− calculated for (C12H8FN2O)− 215.1, found: 215.1.
Synthesis of 2,6-difluoro-4-hydroxyazobenzene (18b).
Starting from compound 10a (1.000 g, 10.738 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.741 g, 10.741 mmol) in water (10 mL), and compound 17c (1.397 g, 10.739 mmol) in 14% aqueous NaOH (40 mL), compound 18b was obtained with a yield of 75% (1.886 g, orange crystals).
1H NMR spectrum of compound 18b (300 MHz, acetone-d6) δ, ppm: 9.83 (s, 1H, -OH), 7.85 (m, 2H, 2×CH), 7.62–7.46 (m, 3H, 3×CH), 6.68 (m, 2H, 2×CH).
13C NMR spectrum of compound 18b (75 MHz, acetone-d6) δ, ppm: 161.5 (t, J = 14.9 Hz), 158.4 (dd, J = 258.5, 7.6 Hz), 154.4, 132.0, 130.1, 125.3 (t, J = 9.6 Hz), 123.1, 101.07 (dd, J = 23.4, 3.0 Hz).
MS (ESI−) m/z: [M−H]− calculated for (C12H7F2N2O)− 233.0, found: 233.1.
Synthesis of 2′-fluoro-4-hydroxyazobenzene (18c).
Starting from compound 10b (1.000 g, 8.999 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.621 g, 9.001 mmol) in water (10 mL), and compound 17a (0.847 g, 9.000 mmol) in 14% aqueous NaOH (40 mL), compound 18c was obtained with a yield of 77% (1.498 g, orange crystals).
1H NMR spectrum of compound 18c (300 MHz, DMSO-d6) δ, ppm: 10.43 (s, 1H, -OH), 7.82 (m, 2H, 2×CH), 7.66 (m, 1H, 1×CH), 7.55–7.35 (m, 2H, 2×CH), 7.29 (m, 1H, 1×CH), 6.96 (m, 2H, 2×CH).
13C NMR spectrum of compound 18c (75 MHz, DMSO-d6) δ, ppm: 161.8, 159.3 (d, J = 254.3 Hz), 146.1, 140.4 (d, J = 6.8 Hz), 132.7 (d, J = 8.2 Hz), 125.7, 125.3 (d, J = 3.6 Hz), 117.9, 117.5 (d, J = 19.6 Hz), 116.5.
MS (ESI−) m/z: [M−H]− calculated for (C12H8FN2O)− 215.1, found: 215.0.
Synthesis of 2,2′-difluoro-4-hydroxyazobenzene (18d).
Starting from compound 10b (1.000 g, 8.999 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.621 g, 9.001 mmol) in water (10 mL), and compound 17b (1.009 g, 9.001 mmol) in 14% aqueous NaOH (40 mL), compound 18d was obtained with a yield of 73% (1.539 g, orange crystals).
1H NMR spectrum of compound 18d (300 MHz, DMSO-d6) δ, ppm: 10.87 (s, 1H, -OH), 7.73–7.59 (m, 2H, 2×CH), 7.58–7.49 (m, 1H, 1×CH), 7.43 (m, 1H, 1×CH), 7.30 (m, 1H, 1×CH), 6.85–6.70 (m, 1H, 2×CH).
13C NMR spectrum of compound 18d (75 MHz, DMSO-d6) δ, ppm: 163.2 (d, J = 12.3 Hz), 161.8 (d, J = 178.4 Hz), 158.5 (d, J = 176.5 Hz), 140.2 (d, J = 6.9 Hz), 133.5 (d, J = 6.8 Hz), 132.6 (d, J = 8.3 Hz), 124.9 (d, J = 3.7 Hz), 118.6 (d, J = 1.9 Hz), 117.5, 117.1 (d, J = 19.6 Hz), 112.6 (d, J = 2.4 Hz), 103.4 (d, J = 21.8 Hz).
MS (ESI−) m/z: [M−H]−, calculated for (C12H7F2N2O)− 233.0, found: 233.1.
Synthesis of 2,2′,6-trifluoro-4-hydroxyazobenzene (18e).
Starting from compound 10b (1.000 g, 8.999 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.621 g, 9.001 mmol) in water (10 mL), and compound 17c (1.171 g, 9.001 mmol) in 14% aqueous NaOH (40 mL), compound 18e was obtained with a yield of 70% (1.589 g, orange crystals).
1H NMR spectrum of compound 18e (300 MHz, acetone-d6) δ, ppm: 10.00 (s, 1H, OH), 7.68 (m, 1H, 1×CH), 7.62–7.52 (m, 1H, 1×CH), 7.44–7.26 (m, 2H, 2×CH), 6.67 (m, 2H, 2×CH).
13C NMR spectrum of compound 18e (75 MHz, acetone-d6) δ, ppm: 162.4 (t, J = 14.8 Hz), 160.6 (d, J = 255.1 Hz), 158.9 (dd, J = 259.3, 8.0 Hz), 142.3 (d, J = 7.1 Hz), 133.8 (d, J = 8.3 Hz), 125.5 (m), 117.9, 117.9 (d, J = 19.6 Hz), 101.1 (dd, J = 23.2, 3.1 Hz).
MS (ESI−) m/z: [M−H]−, calculated for (C12H6F3N2O)− 251.0, found: 251.0.
Synthesis of 2′,6′-difluoro-4-hydroxyazobenzene (18f).
Starting from compound 10c (1.000 g, 7.745 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.534 g, 7.740 mmol) in water (10 mL), and compound 17a (0.729 g, 7.746 mmol) in 14% aqueous NaOH (40 mL), compound 18f was obtained with a yield of 68% (1.233 g, orange crystals).
1H NMR spectrum of compound 18f (300 MHz, CDCl3) δ, ppm: 7.88 (m, 2H, 2×CH), 7.26 (m, 1H, 1×CH), 7.03 (m, 2H, 2×CH), 6.93 (m, 2H, 2×CH), 5.76 (s, 1H, OH).
13C NMR spectrum of compound 18f (75 MHz, CDCl3) δ, ppm: 159.5, 155.8 (dd, J = 257.8, 4.5 Hz), 147.8, 131.5 (t, J = 10.7 Hz), 129.8 (t, J = 10.4 Hz), 125.4, 116.1, 112.6 (m).
MS (ESI−) m/z: [M−H]−, calculated for (C12H7F2N2O)− 233.0, found: 233.1.
Synthesis of 2,2′,6′-trifluoro-4-hydroxyazobenzene (18g).
Starting from compound 10c (1.000 g, 7.745 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.534 g, 7.740 mmol) in water (10 mL), and compound 17b (0.868 g, 7.743 mmol) in 14% aqueous NaOH (40 mL), compound 18g was obtained with a yield of 60% (1.171 g, orange crystals).
1H NMR spectrum of compound 18g (300 MHz, acetone-d6) δ, ppm: 11.02 (s, 1H, -OH), 7.63 (t, J = 8.8 Hz, 1H, 1×CH), 7.47 (m, 1H, 1×CH), 7.32–7.18 (m, 2H, 2×CH), 6.86–6.72 (m, 2H, 2×CH).
MS (ESI−) m/z: [M−H]−, calculated for (C12H6F3N2O)− 251.0, found: 251.1.
Synthesis of 2,2′,6,6′-tetrafluoro-4-hydroxyazobenzene (18h).
Starting from compound 10c (1.000 g, 7.745 mmol) in 12% aqueous HCl (40 mL), NaNO2 (0.534 g, 7.740 mmol) in water (10 mL), and compound 17c (1.007 g, 7.741 mmol) in 14% aqueous NaOH (40 mL), compound 18h was obtained with a yield of 36% (753 mg, orange crystals).
1H NMR spectrum of compound 18h (300 MHz, acetone-d6) δ, ppm: 10.07 (s, 1H, -OH), 7.59–7.41 (m, 1H, 1×CH), 7.35–7.13 (m, 2H, 2×CH), 6.75–6.62 (m, 2H, 2×CH).
13C NMR spectrum of compound 18h (75 MHz, acetone-d6) δ, ppm: 162.9 (t, J = 15.0 Hz), 158.4 (dd, J = 260.4, 7.4 Hz), 156.0 (dd, J = 257.1, 4.5 Hz), 132.7 (t, J = 10.6 Hz), 131.8 (t, J = 10.4 Hz), 125.8 (t, J = 9.4 Hz), 113.5 (m), 101.1 (dd, J = 23.0, 3.0 Hz).
MS (ESI+) m/z: [M+H]+, calculated for (C12H7F4N2O)+ 271.0, found: 271.0.
3.2.12. General Procedure for the Preparation of Compounds 19a–h
A solution of compound 18a–f in acetonitrile was treated with K2CO3, KI, and N-(2-chloroethyl)-morpholine hydrochloride. The reaction mixture was stirred for 6 h under reflux, and the progress of the reaction was monitored by TLC. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The product was extracted with ethyl acetate from water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The reaction products 19a–f were purified by column chromatography (SiO2, EA/MeOH = 5:1).
Preparation of 2-fluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19a).
Starting from compound 18a (212 mg, 0.980 mmol) in acetonitrile (15 mL), K2CO3 (406 mg, 2.938 mmol), KI (163 mg, 0.982 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (274 mg, 1.472 mmol), the yield of compound 19a was 61% (197 mg, orange amorphous solid).
1H NMR spectrum of compound 19a (300 MHz, DMSO-d6) δ, ppm: 7.92–7.79 (m, 2H, 2×CH), 7.74 (t, J = 9.0 Hz, 1H, 1×CH), 7.63–7.47 (m, 3H, 3×CH), 7.13 (dd, J = 13.1, 2.6 Hz, 1H, 1×CH), 6.91 (m, 1H, 1×CH), 4.20 (t, J = 5.7 Hz, 2H, 1×CH2), 3.58 (m, 4H, 2×CH2), 2.71 (t, J = 5.6 Hz, 2H, 1×CH2), 2.47 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19a (75 MHz, DMSO-d6) δ, ppm: 162.7 (d, J = 5.5 Hz), 161.0 (d, J = 273.0 Hz), 152.2, 133.9 (d, J = 7.0 Hz), 131.2, 129.4, 122.4, 118.2 (d, J = 2.1 Hz), 112.1 (d, J = 2.8 Hz), 102.7 (d, J = 23.5 Hz), 66.4, 66.2, 56.8, 53.6.
MS (ESI+) m/z: [M+H]+, calculated for (C18H21FN3O2)+ 330.2, found 330.1.
Preparation of 2,6-difluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19b).
Starting from compound 18b (500 mg, 2.135 mmol) in acetonitrile (15 mL), K2CO3 (885 mg, 6.404 mmol), KI (354 mg, 2.132 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (596 mg, 3.203 mmol), the yield of compound 19b was 64% (474 mg, orange amorphous solid).
1H NMR spectrum of compound 19b (300 MHz, acetone-d6) δ, ppm: 7.87 (m, 2H, 2×CH), 7.63–7.45 (m, 3H, 3×CH), 6.80 (m, 2H, 2×CH), 4.24 (t, J = 5.7 Hz, 2H, 1×CH2), 3.60 (m, 4H, 2×CH2), 2.77 (t, J = 5.7 Hz, 2H, 1×CH2), 2.52 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19b (75 MHz, acetone-d6) δ, ppm: 162.3 (t, J = 14.2 Hz), 158.2 (dd, J = 258.3, 7.5 Hz), 154.4, 132.2, 130.1, 125.8 (t, J = 9.8 Hz), 123.2, 100.4 (dd, J = 24.4, 3.0 Hz), 68.1, 67.4, 57.9, 54.9.
MS (ESI+) m/z: [M+H]+, calculated for (C18H20F2N3O2)+ 348.1, found 348.2.
Preparation of 2′-fluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19c).
Starting from compound 18c (594 mg, 2.747 mmol) in acetonitrile (30 mL), K2CO3 (1.139 g, 8.242 mmol), KI (456 mg, 2.747 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (767 mg, 4.122 mmol), the yield of compound 19c was 70% (633 mg, orange amorphous solid).
1H NMR spectrum of compound 19c (300 MHz, CDCl3) δ, ppm: 7.94 (m, 2H, 2×CH); 7.73 (m, 1H, 1×CH); 7.45–7.35 (m, 1H, 1×CH); 7.30–7.18 (m, 2H, 2×CH); 7.02 (m, 2H, 2×CH); 4.20 (t, J = 5.7 Hz, 2H, 1×CH2); 3.75 (m, 4H, 2×CH2); 2.84 (t, J = 5.7 Hz, 2H, 1×CH2); 2.60 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19c (75 MHz, CDCl3) δ, ppm: 161.6, 159.9 (d, J = 257.2 Hz), 147.5, 140.9 (d, J = 6.9 Hz), 131.8 (d, J = 8.1 Hz), 125.2, 124.3 (d, J = 3.8 Hz), 117.9, 117.02 (d, J = 20.1 Hz), 114.9, 66.9, 66.2, 57.6, 54.2.
MS (ESI+) m/z: [M+H]+, calculated for (C18H21FN3O2)+ 330.2, found 330.3.
Synthesis of 2,2′-difluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19d).
Starting from compound 18d (1.217 g, 5.196 mmol) in acetonitrile (45 mL), with K2CO3 (2.154 g, 15.586 mmol), KI (863 mg, 5.199 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (1.450 g, 7.792 mmol), compound 19d was obtained in 57% yield (1.029 g, orange amorphous solid).
1H NMR spectrum of compound 19d (300 MHz, DMSO-d6) δ, ppm: 7.76–7.53 (m, 3H, 3×CH), 7.52–7.42 (m, 1H, 1×CH), 7.32 (m, 1H, 1×CH), 7.14 (dd, J = 13.0, 2.6 Hz, 1H, 1×CH), 6.92 (dd, J = 9.1, 2.6 Hz, 1H, 1×CH), 4.21 (t, J = 5.6 Hz, 2H, 1×CH2), 3.58 (m, 4H, 2×CH2), 2.71 (t, J = 5.6 Hz, 2H, 1×CH2), 2.47 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19d (75 MHz, DMSO-d6) δ, ppm: 163.3 (d, J = 11.3 Hz), 161.2 (d, J = 257.1 Hz), 159.2 (d, J = 255.5 Hz), 140.1 (d, J = 6.7 Hz), 134.3 (d, J = 6.8 Hz), 133.2 (d, J = 8.4 Hz), 125.0 (d, J = 3.6 Hz), 118.4 (d, J = 1.9 Hz), 117.5, 117.3 (d, J = 19.5 Hz), 112.3 (d, J = 2.6 Hz), 102.7 (d, J = 23.3 Hz), 66.4, 66.2, 56.8, 53.6.
MS (ESI+) m/z: [M+H]+, calculated for (C18H20F2N3O2)+ 348.1, found 348.2.
Synthesis of 2,2′,6-trifluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19e).
Starting from compound 18e (500 mg, 1.982 mmol) in acetonitrile (15 mL), with K2CO3 (822 mg, 5.948 mmol), KI (329 mg, 1.982 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (553 mg, 2.972 mmol), compound 19e was obtained in 51% yield (369 mg, orange amorphous solid).
1H NMR spectrum of compound 19e (300 MHz, acetone-d6) δ, ppm: 7.69 (m, 1H, 1×CH), 7.59 (m, 1H, 1×CH), 7.45–7.25 (m, 2H, 2×CH), 6.84 (m, 2H, 2×CH), 4.30 (t, J = 5.8 Hz, 2H, 1×CH2), 3.62 (m, 4H, 2×CH2), 2.80 (t, J = 5.7 Hz, 2H, 1×CH2), 2.54 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19e (75 MHz, CDCl3) δ, ppm: 161.2 (t, J = 13.4 Hz), 158.3 (dd, J = 259.4, 7.4 Hz), 151.8 (d, J = 249.6 Hz), 142.3 (d, J = 6.9 Hz), 131.1 (d, J = 7.7 Hz), 125.5 (t, J = 3.7 Hz), 121.8, 117.9, 117.3 (d, J = 19.8 Hz), 99.8 (dd, J = 24.7, 2.9 Hz), 67.9, 67.4, 57.9, 54.8.
MS (ESI+) m/z: [M+H]+, calculated for (C18H19F3N3O2)+ 366.1, found 366.2.
Synthesis of 2′,6′-difluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19f).
Starting from compound 18f (347 mg, 1.482 mmol) in acetonitrile (20 mL), with K2CO3 (614 mg, 4.443 mmol), KI (246 mg, 1.482 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (413 mg, 2.219 mmol), the yield of compound 19f was 56% (288 mg, orange amorphous solid).
1H NMR spectrum of compound 19f (300 MHz, CDCl3) δ, ppm: 7.92 (m, 2H, 2×CH), 7.34–7.20 (m, 1H, 1×CH), 7.07–7.00 (m, 4H, 4×CH), 4.20 (t, J = 5.7 Hz, 2H, 1×CH2), 3.77–3.72 (m, 4H, 2×CH2), 2.84 (t, J = 5.7 Hz, 2H, 1×CH2), 2.63–2.57 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19f (75 MHz, CDCl3) δ, ppm: 162.0, 155.7 (dd, J = 257.9, 4.6 Hz), 147.8, 131.5 (t, low intensity), 129.6 (t, J = 10.2 Hz), 125.1, 114.9, 112.5 (m), 66.9, 66.2, 57.5, 54.1.
MS (ESI+) m/z: [M+H]+, calculated for (C18H20F2N3O2)+ 348.1, found: 348.2.
Synthesis of 2,2′,6′-trifluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19g).
Starting from compound 18g (946 mg, 3.751 mmol) in acetonitrile (45 mL), with K2CO3 (1.555 g, 11.252 mmol), KI (623 mg, 3.753 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (1.047 g, 5.627 mmol), the yield of compound 19g was 77% (1.055 g, orange amorphous solid).
1H NMR spectrum of compound 19g (300 MHz, DMSO-d6) δ, ppm: 7.68 (t, J = 8.9 Hz, 1H, 1×CH), 7.57–7.46 (m, 1H, 1×CH), 7.29 (m, 2H, 2×CH), 7.14 (m, 1H, 1×CH), 6.92 (m, 1H, 1×CH), 4.22 (t, J = 5.6 Hz, 2H, 1×CH2), 3.58 (m, 4H, 2×CH2), 2.71 (t, J = 5.6 Hz, 2H, 1×CH2), 2.47 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19g (75 MHz, DMSO-d6) δ, ppm: 163.8 (d, J = 11.5 Hz), 161.3 (d, J = 258.0 Hz), 154.9 (dd, J = 256.9, 4.5 Hz), 134.7 (d, J = 7.0 Hz), 131.4 (t, J = 10.4 Hz), 130.6 (t, J = 10.3 Hz), 117.8 (d, J = 1.7 Hz), 113.0 (m), 112.3 (d, J = 2.6 Hz), 102.7 (d, J = 23.4 Hz), 66.5, 66.2, 56.7, 53.6.
MS (ESI+) m/z: [M+H]+, calculated for (C18H19F3N3O2)+ 366.1, found: 366.1.
Synthesis of 2,2′,6,6′-tetrafluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene (19h).
Starting from compound 18h (573 mg, 2.121 mmol) in acetonitrile (30 mL), with K2CO3 (880 mg, 6.368 mmol), KI (352 mg, 2.120 mmol), and N-(2-chloroethyl)-morpholine hydrochloride (592 mg, 3.181 mmol), the yield of compound 19h was 76% (618 mg, orange amorphous solid).
1H NMR spectrum of compound 19h (300 MHz, DMSO-d6) δ, ppm: 7.48–7.36 (m, 1H), 7.19 (t, J = 8.6 Hz, 2H, 2×CH), 6.86 (m, 2H, 2×CH), 4.09 (t, J = 5.6 Hz, 2H, 1×CH2), 3.53 (m, 4H, 2×CH2), 2.63 (t, J = 5.6 Hz, 2H, 1×CH2), 2.41 (m, 4H, 2×CH2).
13C NMR spectrum of compound 19h (75 MHz, acetone-d6) δ, ppm: 163.5 (t, J = 14.4 Hz), 158.2 (dd, J = 260.2, 7.3 Hz), 156.1 (dd, J = 257.7, 4.4 Hz), 132.7 (t, J = 10.6 Hz), 132.1 (t, J = 10.4 Hz), 126.5 (t, J = 9.7 Hz), 113.6 (m), 100.5 (dd, J = 24.1, 3.2 Hz), 68.3, 67.5, 57.9, 54.9.
MS (ESI+) m/z: [M+H]+, calculated for (C18H18F4N3O2)+ 384.1, found: 384.2.
3.2.13. General Method for the Synthesis of Compounds 20a–h
To the solutions of compounds 19a–f in diethyl ether, a solution of HCl/Et2O (2.4 M) was added dropwise with stirring at 0–5 °C. The reaction mixture was then stirred for 30 min at RT. The product was concentrated under reduced pressure.
Synthesis of 2-fluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20a).
Starting from compound 19a (128 mg, 0.389 mmol) in Et2O (20 mL) and HCl/Et2O (328 µL, 2.4 M), the yield of compound 20a was quantitative (142 mg, light orange crystals).
1H NMR spectrum of compound 20a (300 MHz, DMSO-d6) δ, ppm: 11.81 (s, 1H, HCl), 7.89–7.82 (m, 2H, 2×CH), 7.79 (t, J = 8.9 Hz, 1H, 1×CH), 7.63–7.53 (m, 3H, 3×CH), 7.23 (dd, J = 12.8, 2.6 Hz, 1H, 1×CH), 7.00 (m, 1H, 1×CH), 4.60 (t, J = 5.0 Hz, 2H, 1×CH2), 4.01–3.82 (m, 4H, 2×CH2), 3.65–3.44 (m, 4H, 2×CH2), 3.22 (s, 2H, 1×CH2).
13C NMR spectrum of compound 20a (75 MHz, DMSO-d6) δ, ppm: 161.6 (d, J = 11.2 Hz), 160.8 (d, J = 256.5 Hz), 152.1, 134.4 (d, J = 7.1 Hz), 131.5, 129.5, 122.6, 118.4 (d, J = 1.2 Hz), 112.2 (d, J = 2.7 Hz), 103.2 (d, J = 23.7 Hz), 63.3, 63.2, 54.5, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H21FN3O2)+ 330.2, found 330.2.
Synthesis of 2,6-difluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20b).
Starting from compound 19b (160 mg, 0.461 mmol) in Et2O (10 mL) and HCl/Et2O (389 µL, 2.4 M), the yield of compound 20b was quantitative (176 mg, light orange crystals).
1H NMR spectrum of compound 20b (300 MHz, DMSO-d6) δ, ppm: 11.90 (s, 1H, HCl), 7.86–7.74 (m, 2H, 2×CH), 7.66–7.51 (m, 3H, 3×CH), 7.08 (m, 2H, 2×CH), 4.61 (t, J = 5.0 Hz, 2H, 1×CH2), 3.99–3.88 (m, 4H, 2×CH2), 3.64–3.43 (m, 4H, 2×CH2), 3.21 (s, 2H, 1×CH2).
13C NMR spectrum of compound 20b (75 MHz, DMSO-d6) δ, ppm: 160.0 (t, J = 14.4 Hz), 156.5 (dd, J = 257.7, 6.9 Hz), 152.7, 131.9, 129.5, 129.0 (t, J = 22.8 Hz), 122.3, 100.3 (d, J = 24.6 Hz), 63.6, 63.1, 54.3, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H20F2N3O2)+ 348.1, found 348.1.
Synthesis of 2′-fluoro-4-(2-(N- morpholino)-ethoxy)-azobenzene hydrochloride (20c).
Starting from compound 19c (249 mg, 0.756 mmol) in Et2O (14 mL) and HCl/Et2O (638 µL, 2.4 M), the yield of compound 20c was quantitative (276 mg, light orange crystals).
1H NMR spectrum of compound 20c (300 MHz, DMSO-d6) δ, ppm: 12.05 (s, 1H, HCl), 7.91 (m, 2H, 2×CH), 7.68 (m, 1H, 1×CH), 7.56 (m, 1H, 1×CH), 7.46 (m, 1H, 1×CH), 7.31 (m, 1H, 1×CH), 7.21 (m, 2H, 2×CH), 4.61 (m, 2H, 1×CH2), 3.93 (m, 4H, 2×CH2), 3.73–3.40 (m, 4H, 2×CH2), 3.25 (s, 2H, 1×CH2).
13C NMR spectrum of compound 20c (75 MHz, DMSO-d6) δ, ppm: 160.6, 159.6 (d, J = 254.2 Hz), 146.9, 139.8 (d, J = 6.7 Hz), 132.8 (d, J = 8.3 Hz), 124.9, 117.4, 117.2 (d, J = 20.5 Hz), 115.4, 63.1, 62.8, 54.6, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H21FN3O2)+ 330.2, found 330.2.
Synthesis of 2,2′-difluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20d).
Starting from compound 19d (392 mg, 1.128 mmol) in Et2O (10 mL) and HCl/Et2O (952 µL, 2.4 M), the yield of compound 20d was quantitative (433 mg, light orange crystals).
1H NMR spectrum of compound 20d (300 MHz, DMSO-d6) δ, ppm: 11.97 (s, 1H, HCl), 7.75 (t, J = 8.9 Hz, 1H, 1×CH), 7.66 (m, 1H, 1×CH), 7.58 (m, 1H, 1×CH), 7.47 (m, 1H, 1×CH), 7.33 (m, 1H, 1×CH), 7.23 (dd, J = 12.8, 2.6 Hz, 1H, 1×CH), 7.00 (dd, J = 9.2, 2.6 Hz, 1H, 1×CH), 4.62 (t, J = 4.9 Hz, 2H, 1×CH2), 3.99–3.84 (m, 4H, 2×CH2), 3.66–3.44 (m, 4H, 2×CH2), 3.23 (m, 2H, 1×CH2).
13C NMR spectrum of compound 20d (75 MHz, DMSO-d6) δ, ppm: 162.0 (d, J = 11.3 Hz), 160.9 (d, J = 257.4 Hz), 159.1 (d, J = 255.7 Hz), 140.0 (d, J = 6.8 Hz), 134.7 (d, J = 6.9 Hz), 133.2 (d, J = 8.5 Hz), 125.0 (d, J = 3.8 Hz), 118.5 (d, J = 1.7 Hz), 117.5, 117.2 (d, J = 19.5 Hz), 112.3 (d, J = 2.7 Hz), 103.2 (d, J = 23.5 Hz), 63.2, 63.1, 54.4, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H20F2N3O2)+ 348.1, found 348.2.
Synthesis of 2,2′,6-trifluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20e).
Starting from compound 19e (126 mg, 0.345 mmol) in Et2O (10 mL) and HCl/Et2O (291 µL, 2.4 M), the yield of compound 20e was quantitative (138 mg, light orange crystals).
1H NMR spectrum of compound 20e (300 MHz, DMSO-d6) δ, ppm: 12.04 (s, 1H, HCl), 7.61 (m, 2H, 2×CH), 7.48 (m, 1H, 1×CH), 7.33 (m, 1H, 1×CH), 7.09 (m, 2H, 2×CH), 4.63 (t, J = 4.9 Hz, 2H, 1×CH2), 4.01–3.89 (m, 4H, 2×CH2), 3.64–3.48 (m, 4H, 2×CH2), 3.22 (m, 2H, 1×CH2).
13C NMR spectrum of compound 20e (75 MHz, DMSO-d6) δ, ppm: 160.5 (t, J = 14.6 Hz), 159.2 (d, J = 256.5 Hz), 156.7 (dd, J = 258.9, 7.2 Hz), 140.6 (d, J = 6.8 Hz), 133.8 (d, J = 8.5 Hz), 125.1 (d, J = 4.1 Hz), 117.4 (d, J = 19.5 Hz), 116.8, 100.4 (m), 63.7, 63.1, 54.3, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H19F3N3O2)+ 366.1, found 366.1.
Synthesis of 2′,6′-difluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20f).
Starting from compound 19f (219 mg, 0.630 mmol) in Et2O (12 mL) and HCl/Et2O (532 µL, 2.4 M), the yield of compound 20f was quantitative (241 mg, light orange crystals).
1H NMR spectrum of compound 20f (300 MHz, DMSO-d6) δ, ppm: 12.00 (s, 1H, OH), 7.90 (m, 2H, 2×CH), 7.51 (m, 1H, 1×CH), 7.37–7.16 (m, 4H, 4×CH), 4.62 (m, 2H, 1×CH2), 3.93 (m, 4H, 2×CH2), 3.69–3.45 (m, 4H, 2×CH2), 3.24 (m, 2H, 1×CH2).
13C NMR spectrum of compound 20f (75 MHz, DMSO-d6) δ, ppm: 161.0, 153.1 (dd, J = 255.9, 3.4 Hz), 147.3, 131.0 (t, J = 10.4 Hz), 130.5 (t, J = 10.6 Hz), 124.8, 115.5, 113.0 (d, J = 20.5 Hz), 63.2, 62.8, 54.6, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H20F2N3O2)+ 348.1, found 348.2.
Synthesis of 2,2′,6′-trifluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20g).
Starting from compound 19g (273 mg, 0.747 mmol) in Et2O (15 mL) and HCl/Et2O (630 µL, 2.4 M), the yield of compound 20g was quantitative (300 mg, light orange crystals).
1H NMR spectrum of compound 20g (300 MHz, DMSO-d6) δ, ppm: 11.98 (s, 1H, HCl), 7.72 (t, J = 8.9 Hz, 1H, 1×CH), 7.64–7.46 (m, 1H, 1×CH), 7.37–7.20 (m, 3H, 3×CH), 7.01 (m, 1H, 1×CH), 4.63 (t, J = 4.9 Hz, 2H, 1×CH2), 4.02–3.83 (m, 4H, 2×CH2), 3.66–3.46 (m, 4H, 2×CH2), 3.22 (m, 2H, 1×CH2).
13C NMR spectrum of compound 20g (75 MHz, DMSO-d6) δ, ppm: 162.61 (d, J = 11.4 Hz), 161.12 (d, J = 258.1 Hz), 154.9 (dd, J = 257.1, 4.2 Hz), 135.2 (d, J = 6.9 Hz), 131.6 (t, J = 10.5 Hz), 130.3 (t, J = 9.7 Hz), 118.0 (m), 113.1 (m), 112.5 (d, J = 2.9 Hz), 103.2 (d, J = 23.5 Hz), 63.4, 63.1, 54.5, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H19F3N3O2)+ 366.1, found 366.1.
Synthesis of 2,2′,6,6′-tetrafluoro-4-(2-(N-morpholino)-ethoxy)-azobenzene hydrochloride (20h).
Starting from compound 19h (96 mg, 0.250 mmol) in Et2O (7 mL) and HCl/Et2O (211 µL, 2.4 M), the yield of compound 20h was quantitative (105 mg, light orange crystals).
1H NMR spectrum of compound 20h (300 MHz, DMSO-d6) δ, ppm: 11.69 (s, 1H, HCl), 7.48–7.39 (m, 1H, 1×CH), 7.21 (m, 2H, 2×CH), 6.95 (m, 2H, 2×CH), 4.47 (t, J = 4.9 Hz, 2H, 1×CH2), 3.90 (m, 4H, 2×CH2), 3.46 (m, 4H, 2×CH2), 3.17 (m, 2H, 1×CH2).
13C NMR spectrum of compound 20h (75 MHz, DMSO-d6) δ, ppm: 159.4 (t, J = 13.8 Hz), 151.9 (dd, J = 249.6, 8.5 Hz), 150.9 (dd, J = 250.9, 5.2 Hz), 132.2 (t, J = 11.0 Hz), 131.1 (t, J = 9.9 Hz), 125.4 (t, J = 17.2 Hz), 112.8 (dd, J = 20.3, 2.9 Hz), 99.8 (dd, J = 24.5, 2.9 Hz), 63.5, 63.1, 54.4, 51.6.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C18H18F4N3O2)+ 384.1, found 384.1.
3.2.14. General Procedure for the Synthesis of 4-Hydroxyphenylazothiazoles (22a–b)
A concentrated aqueous solution of HCl was added to a solution of 21a–b in water with stirring at 0–5 °C. The resulting mixture was stirred at 0–5 °C for 15 min. Subsequently, an aqueous solution of NaNO2 was added dropwise to the mixture over 10 min while maintaining the temperature at 0–5 °C, followed by stirring for an additional 30 min under cooling. A solution of phenol in 16% aqueous NaOH was then added dropwise to the diazonium salt solution over 10 min. The reaction mixture was stirred for 1 h at 0–5 °C, after which the pH was adjusted to 5. The precipitated product was filtered and dried. The products were purified by column chromatography.
Synthesis of 2-(4-hydroxyphenylazo)-thiazole (22a).
Starting from 2-aminothiazole (21a) (5.400 g, 53.924 mmol) dissolved in 30 mL of water, HCl (36.6%, 16 mL), NaNO2 (3.720 g, 53.921 mmol) in 18 mL of water, and a solution of phenol (5.075 g, 53.926 mmol) in 16% aqueous NaOH (40 mL), the reaction product was purified by chromatography (SiO2, EA/MeOH = 50:1). The yield of compound 22a was 38% (4.205 g, brown crystals).
1H NMR spectrum of compound 22a (300 MHz, DMSO-d6) δ, ppm: 10.72 (s, 1H, -OH), 8.04 (d, J = 3.4 Hz, 1H, 1×CH), 7.86 (m, 2H, 2×CH), 7.81 (d, J = 3.4 Hz, 1H, 1×CH), 6.98 (m, 2H, 2×CH).
13C NMR spectrum of compound 22a (75 MHz, DMSO-d6) δ, ppm: 176.8, 162.9, 144.3, 143.7, 126.2, 121.9, 116.5.
MS (ESI+) m/z: [M+H]+, calculated for (C9H8N3OS)+ 206.0, found 206.0.
Synthesis of 2-(4-hydroxyphenylazo)-5-methylthiazole (22b).
Starting from 2-amino-5-methylthiazole (21b) (500 mg, 4.379 mmol) dissolved in 5 mL of water, HCl (36.6%, 5 mL), NaNO2 (302 mg, 4.377 mmol) in water (5 mL), and a solution of phenol (412 mg, 4.378 mmol) in 16% aqueous NaOH (15 mL), the reaction product was purified by chromatography (SiO2, EA/MeOH = 50:1). The yield of compound 22b was 83% (796 mg, brown crystals).
1H NMR spectrum of compound 22b (300 MHz, DMSO-d6) δ, ppm: 10.65 (s, 1H, -OH), 7.81 (m, 2H, 2×CH), 7.76 (m, 1H, 1×CH), 6.97 (m, 2H, 2×CH), 2.49 (s, 3H, 1×CH3).
13C NMR spectrum of compound 22b (75 MHz, DMSO-d6) δ, ppm: 174.7, 162.6, 144.3, 141.8, 136.4, 126.0, 116.5, 12.5.
MS (ESI+) m/z: [M+H]+ calculated for (C10H10N3OS)+ 220.0, found 220.0.
3.2.15. General Procedure for the Preparation of Compounds 23a–b and 24a–b
To a solution of compound 22a–b in DMF, K2CO3, N-(2-chloroethyl)-morpholine hydrochloride and KI were added under stirring. The reaction mixture was heated under reflux with stirring for 3 h, followed by extraction with ethyl acetate from water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure. The products were purified by column chromatography.
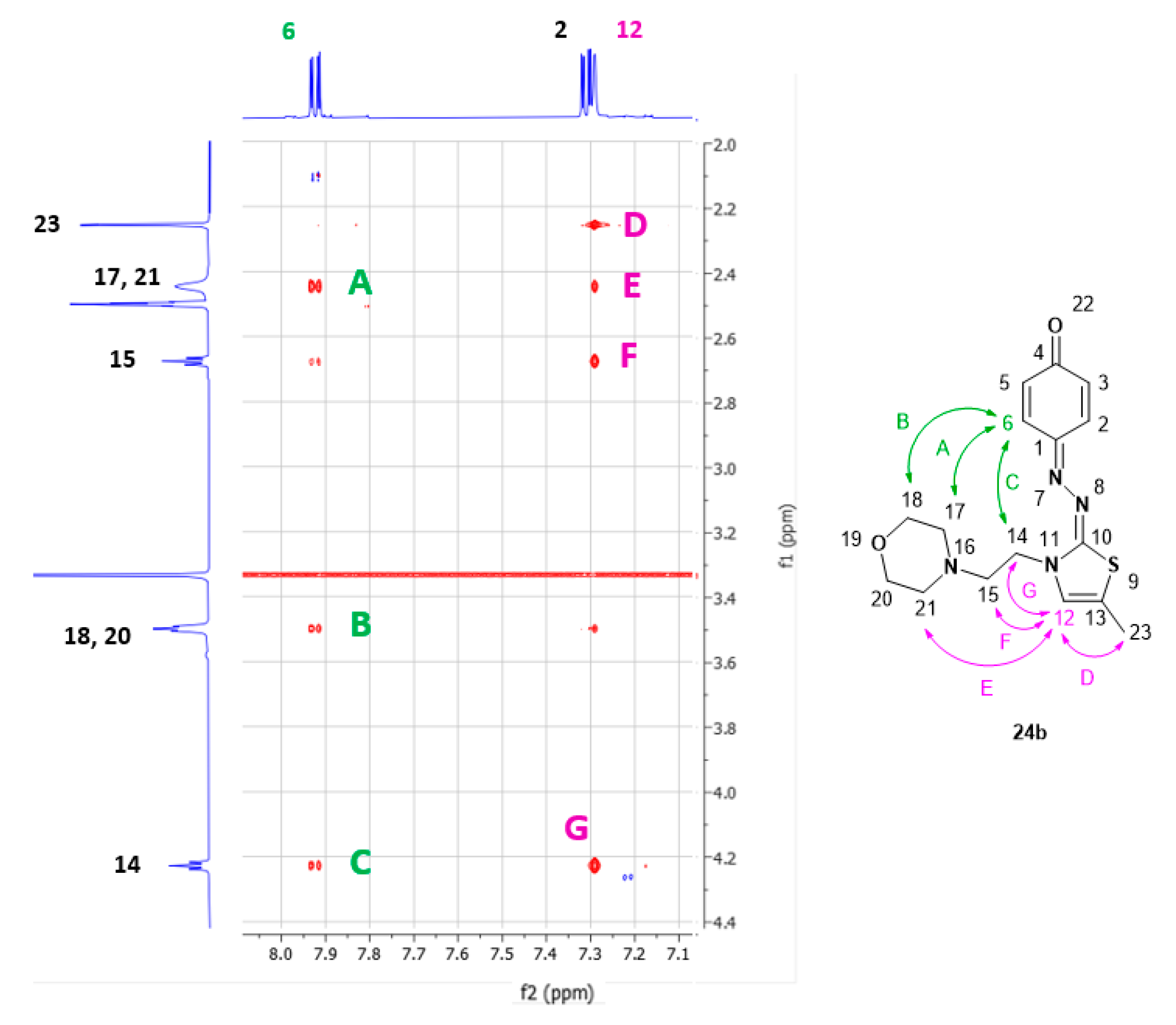
Preparation of 2-(4-(2-(N-morpholino)-ethoxy)-phenylazo)-thiazole (23a) and 4-((3-(2-(N-morpholino)-ethyl)-thiazol-2(3H)-ylidene)-hydrazono)-cyclohexa-2,5-dien-1-one (24a).
Starting from compound 22a (1000 mg, 4.873 mmol) in DMF (30 mL), K2CO3 (2022 mg, 14.631 mmol), N-(2-chloroethyl)-morpholine hydrochloride (1361 mg, 7.314 mmol), and KI (810 mg, 4.879 mmol) after purification by chromatography (SiO2, EA/MeOH = 5:1), the yield of compound 23a was 19% (295 mg, orange crystals), and the yield of compound 24a was 5% (78 mg, violet amorphous solid).
1H NMR spectrum of compound 23a (600 MHz, DMSO-d6) δ, ppm: 8.08 (d, J = 3.3 Hz, 1H, 1×CH), 7.94 (m, 2H, 2×CH), 7.86 (d, J = 3.3 Hz, 1H, 1×CH), 7.18 (m, 2H, 2×CH), 4.24 (t, J = 5.7 Hz, 2H, 1×CH2), 3.58 (m, 4H, 2×CH2), 2.73 (t, J = 5.7 Hz, 2H, 1×CH2), 2.48 (m, 4H, 2×CH2).
13C NMR spectrum of compound 23a (150 MHz, DMSO-d6) δ, ppm: 176.5, 162.8, 145.2, 143.8, 125.7, 122.2, 115.6, 66.1, 66.0, 56.8, 53.5.
MS (ESI+) m/z: [M+H]+, calculated for (C15H19N4O2S)+ 319.1, found 319.0.
1H NMR spectrum of compound 24a (600 MHz, DMSO-d6) δ, ppm: 7.95 (m, 1H), 7.53 (d, J = 4.5 Hz, 1H), 7.34 (m, 1H), 6.98 (d, J = 4.5 Hz, 1H), 6.37 (m, 2H), 4.28 (t, J = 6.2 Hz, 2H, 1×CH2), 3.50 (m, 4H, 2×CH2), 2.70 (t, J = 6.2 Hz, 2H, 1×CH2), 2.45 (m, 4H, 2×CH2).
13C NMR spectrum of compound 24a (150 MHz, DMSO-d6) δ, ppm: 186.5, 174.2, 145.5, 139.9, 131.1, 128.9, 126.9, 124.8, 107.8, 66.1, 56.2, 53.1, 44.9.
MS (ESI+) m/z: [M+H]+, calculated for (C15H19N4O2S)+ 319.1, found 319.0.
Preparation of 2-(4-(2-(N-morpholino)-ethoxy)-phenylazo)-5-methylthiazole (23b) and 4-((5-methyl-3-(2-(N-morpholino)-ethyl)-thiazol-2(3H)-ylidene)-hydrazono)-cyclohexa-2,5-dien-1-one (24b).
Starting from compound 22b (144 mg, 0.657 mmol) in DMF (5 mL), K2CO3 (272 mg, 1.968 mmol), N-(2-chloroethyl)-morpholine hydrochloride (183 mg, 0.983 mmol), and KI (109 mg, 0.657 mmol) were used. After purification by chromatography (SiO2, DCM/MeOH/NH4OH = 200:10:1), the yield of compound 23b was 62% (135 mg, orange crystals), and the yield of compound 24b was 5% (11 mg, violet amorphous solid).
1H NMR spectrum of compound 23b (600 MHz, DMSO-d6) δ, ppm: 7.89 (m, 2H, 2×CH), 7.80 (m, 1H, 1×CH), 7.17 (m, 2H, 2×CH), 4.23 (t, J = 5.7 Hz, 2H, 1×CH2), 3.58 (m, 4H, 2×CH2), 2.73 (t, J = 5.7 Hz, 2H, 1×CH2), 2.51 (s, 3H, 1×CH3), 2.48 (m, 4H, 2×CH2).
13C NMR spectrum of compound 23b (150 MHz, DMSO-d6) δ, ppm: 174.4, 162.5, 145.2, 142.0, 136.9, 125.5, 115.5, 66.1, 66.0, 56.8, 53.5, 12.4.
MS (ESI+) m/z: [M+H]+, calculated for (C16H21N4O2S)+ 333.1, found 333.1.
1H NMR spectrum of compound 24b (600 MHz, DMSO-d6) δ, ppm: 7.92 (m, 1H, 1×CH), 7.31 (m, 1H, 1×CH), 7.29 (m, 1H, 1×CH), 6.35 (m, 2H, 2×CH), 4.23 (t, J = 6.2 Hz, 2H, 1×CH2), 3.50 (m, 4H, 2×CH2), 2.68 (t, J = 6.2 Hz, 2H, 1×CH2), 2.45 (m, 4H, 2×CH2), 2.26 (d, J = 1.4 Hz, 3H, 1×CH3).
13C NMR spectrum of compound 24b (150 MHz, DMSO-d6) δ, ppm: 186.4, 173.6, 145.2, 139.9, 128.8, 127.4, 126.8, 124.7, 120.1, 66.2, 56.4, 53.1, 44.7, 12.7
MS (ESI+) m/z: [M+H]+, calculated for (C16H21N4O2S)+ 333.1, found 333.1.
3.2.16. General Procedure for the Preparation of Compounds 25a–b
To the solutions of compounds 23a–b in Et2O, a solution of HCl/Et2O (2.4 M) was added dropwise with stirring at 0–5 °C. The resulting mixture was stirred at RT for 30 min. The product was concentrated under reduced pressure.
Synthesis of 2-(4-(2-(N-morpholino)-ethoxy)-phenylazo)-thiazole hydrochloride (25a).
Starting from compound 23a (80 mg, 0.251 mmol) in Et2O (10 mL) and HCl/Et2O (500 μL, 2.4 M), compound 25a was obtained quantitatively (89 mg, light-orange crystals).
1H NMR spectrum of compound 25a (300 MHz, DMSO-d6) δ, ppm: 11.59 (s, 1H, HCl), 8.10 (d, J = 3.3 Hz, 1H, 1×CH), 7.99 (m, 2H, 2×CH), 7.90 (d, J = 3.3 Hz, 1H, 1×CH), 7.25 (m, 2H, 2×CH), 4.61 (t, J = 5.0 Hz, 2H, 1×CH2), 4.01–3.70 (m, 6H, 3×CH2), 3.64–3.46 (m, 4H, 2×CH2).
13C NMR spectrum of compound 25a (75 MHz, DMSO-d6) δ, ppm: 176.4, 161.7, 145.7, 144.0, 125.8, 122.6, 115.8, 63.1, 62.8, 54.6, 51.6.
MS (ESI+) m/z: [M−HCl+H]+ calculated for (C15H19N4O2S)+ 319.1, found 319.1.
Synthesis of 2-(4-(2-(N-morpholino)-ethoxy)-phenylazo)-5-methylthiazole hydrochloride (25b).
Starting from compound 23b (100 mg, 0.301 mmol) in Et2O (20 mL) and HCl/Et2O (400 μL, 2.4 M), compound 25b was obtained quantitatively (110 mg, light-orange crystals).
1H NMR spectrum of compound 25b (300 MHz, DMSO-d6) δ, ppm: 11.29 (s, 1H, HCl), 7.95 (m, 2H, 2×CH), 7.83 (d, J = 1.2 Hz, 1H, 1×CH), 7.23 (m, 2H, 2×CH), 4.58 (t, J = 4.9 Hz, 2H, 1×CH2), 4.04–3.74 (m, 4H, 2×CH2), 3.55 (m, 4H, 2×CH2), 3.22 (m, 2H, 1×CH2), 2.52 (m, 3H, 1×CH3).
13C NMR spectrum of compound 25b (75 MHz, DMSO-d6) δ, ppm: 161.4, 145.8, 142.2, 137.2, 125.5, 122.5, 115.8, 63.1, 62.7, 54.7, 51.7, 12.4.
MS (ESI+) m/z: [M−HCl+H]+, calculated for (C16H21N4O2S)+ 333.1, found 333.1.
3.2.17. Preparation of Micellar Solutions of Compounds
A solution of corresponding compound (50 mg) in dichloromethane (2 mL) was added dropwise to a freshly prepared 4% aqueous solution of Kolliphor® ELP (5 mL) heated to 45 °C with continuous argon bubbling. Bubbling was continued until strong foaming started. A clear orange solution with a concentration of 1.0% was obtained, which was subsequently filtered in turn through a 0.45 µm PTFE filter and then through a 0.22 µm PTFE filter.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}