
Computational Comparison of Differential Splicing Tools for Targeted RNA Long-Amplicon Sequencing (rLAS)



Abstract
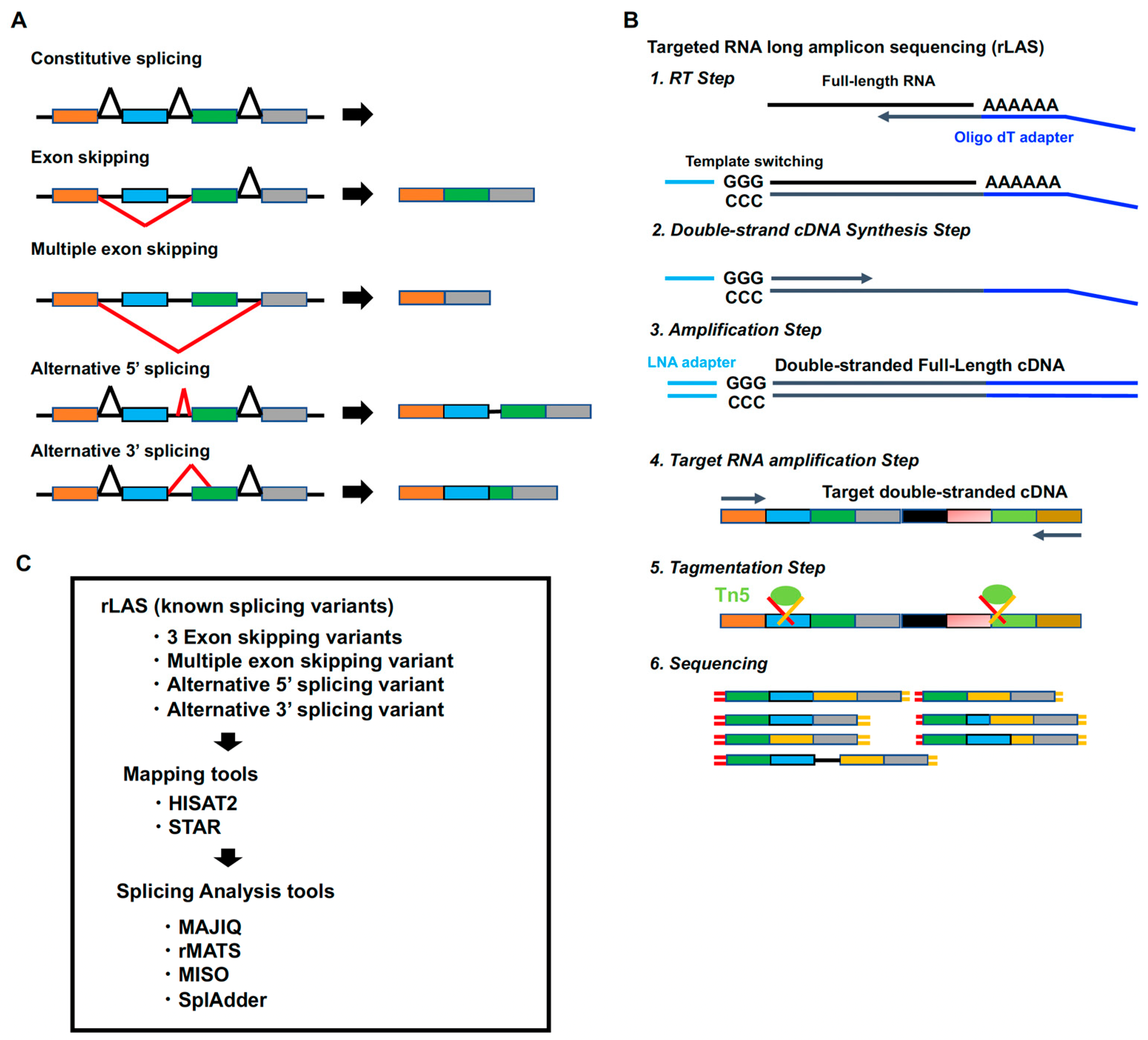
1. Introduction
2. Results
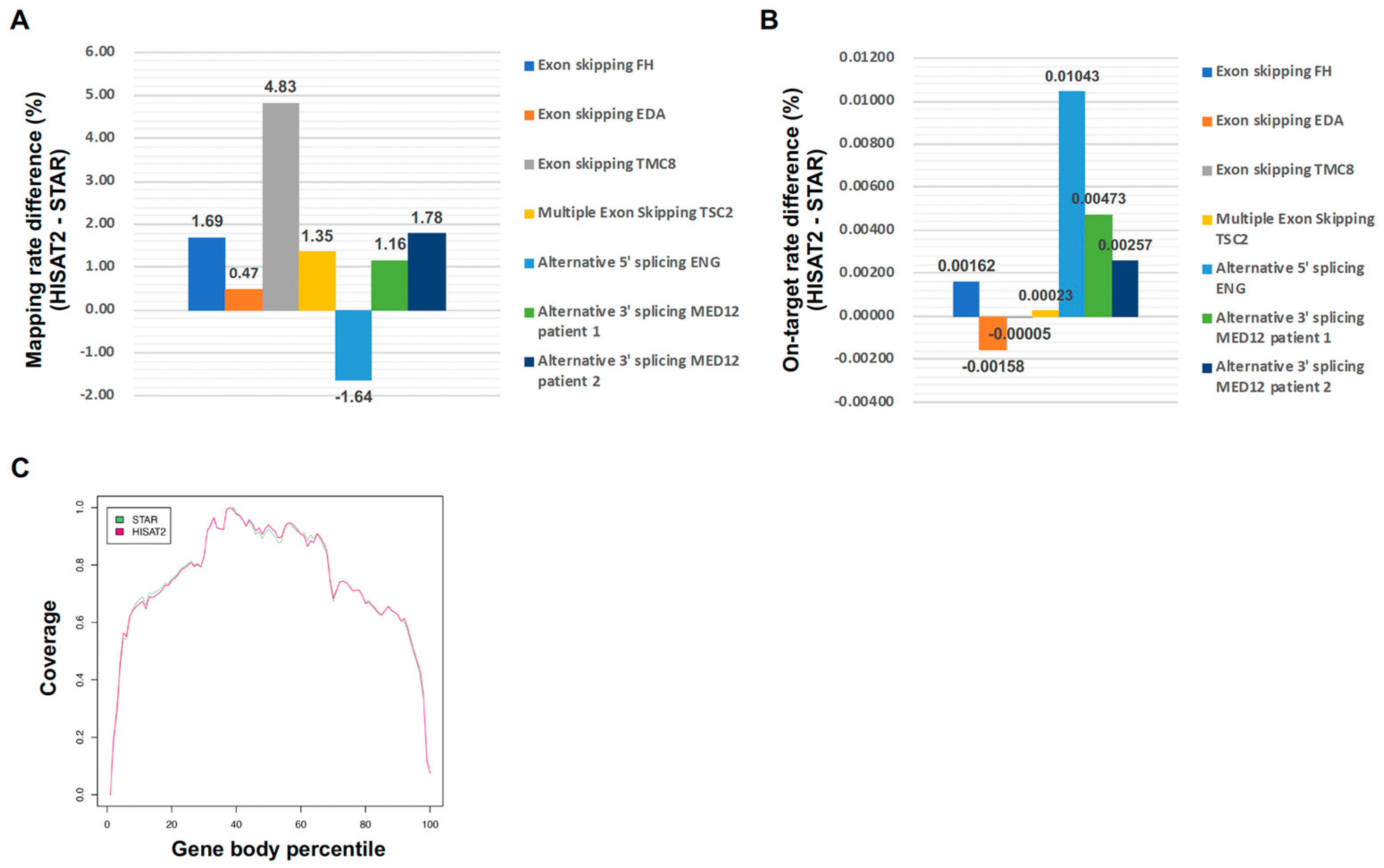
2.1. Comparison Between HISAT2 and STAR for Targeted RNA Long-Amplicon Sequencing (rLAS) Mapping
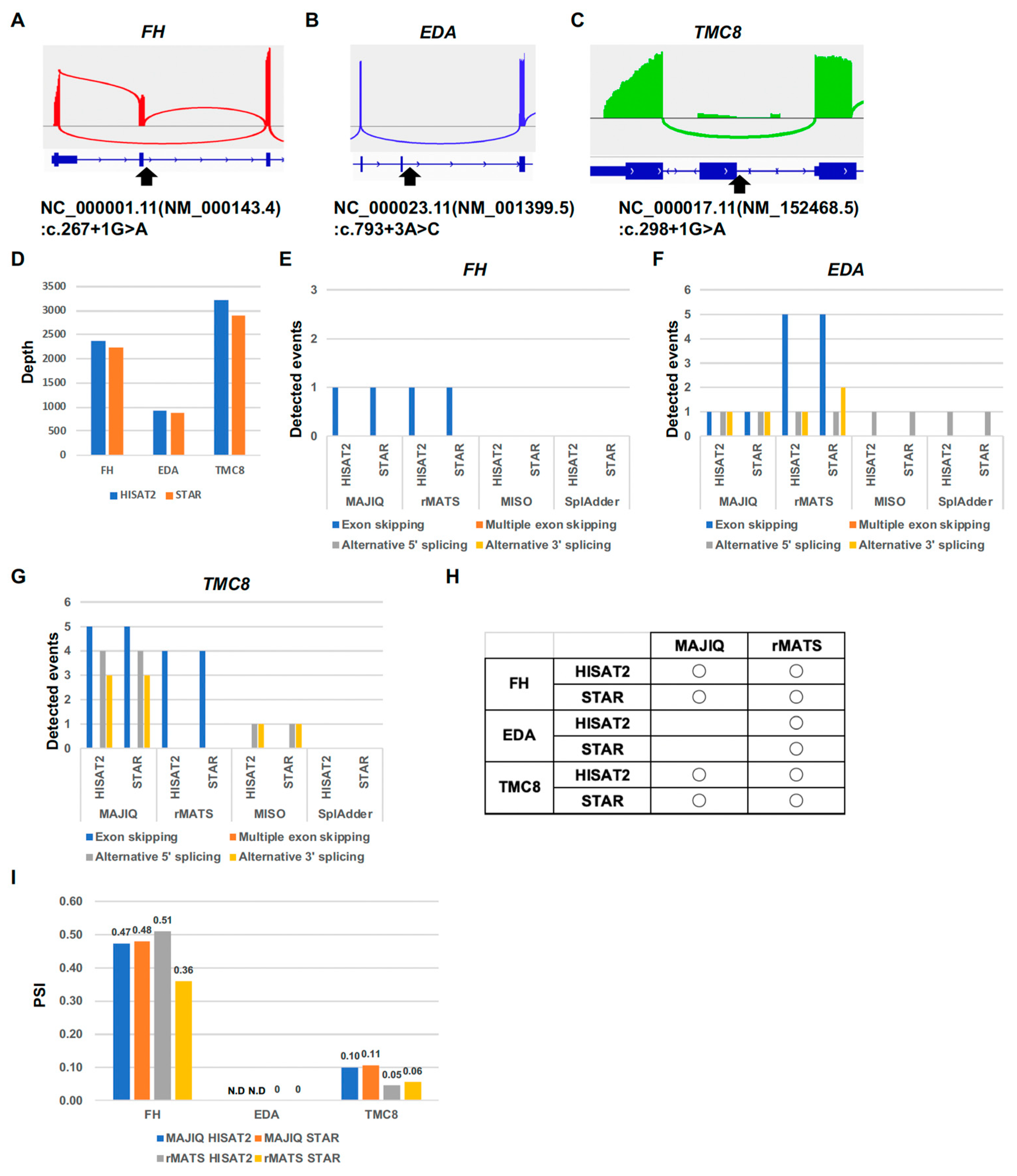
2.2. Computational Comparison of Targeted RNA Long-Amplicon Sequencing (rLAS) for Exon-Skipping Variants
2.3. Computational Comparison of Targeted RNA Long-Amplicon Sequencing (rLAS) for Multiple-Exon-Skipping Variant
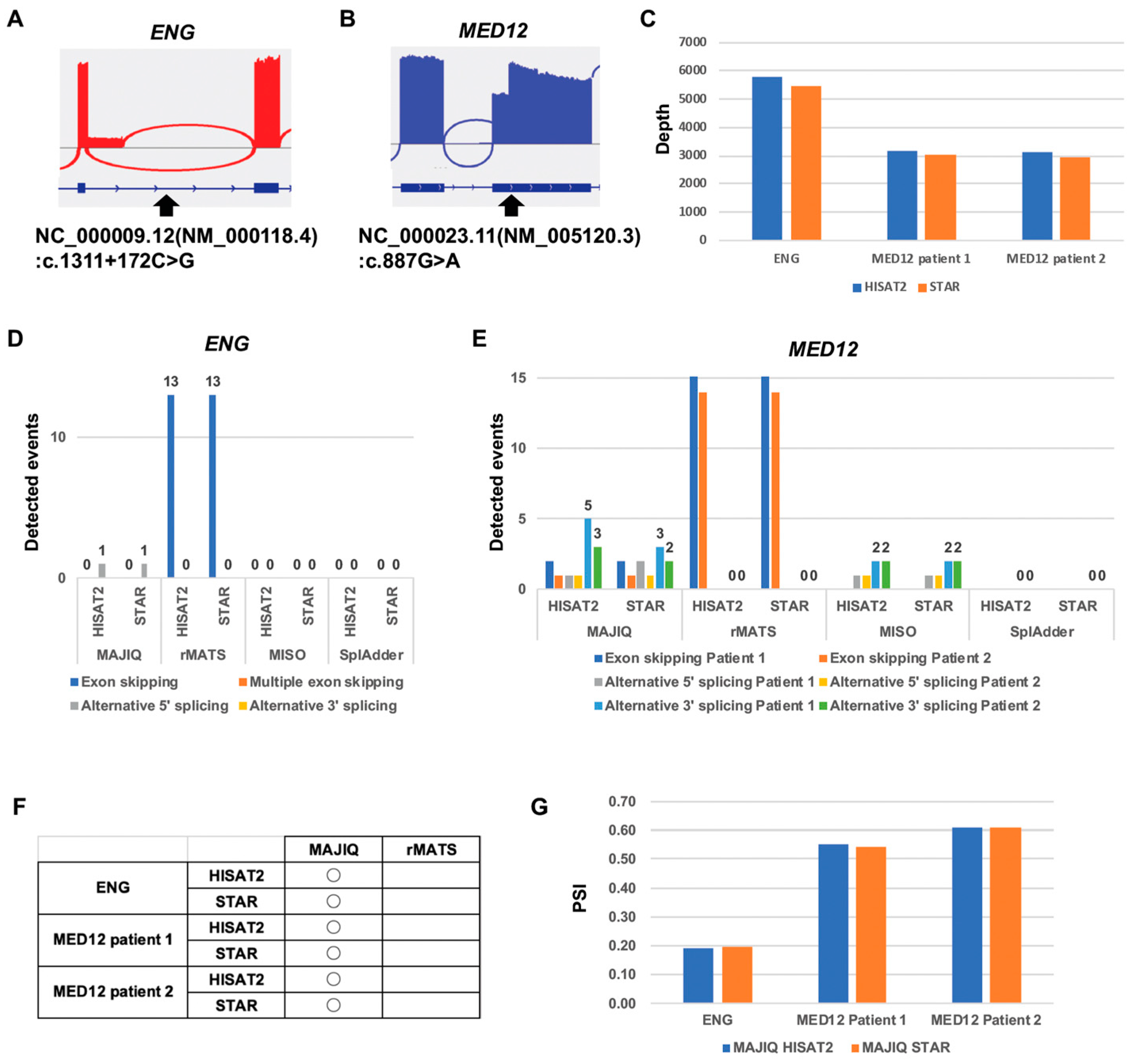
2.4. Computational Comparison of Targeted RNA Long-Amplicon Sequencing (rLAS) for the Alternative 5′ Splicing Variant and Alternative 3′ Splicing Variant
2.5. Computational Comparison of Targeted RNA Long-Amplicon Sequencing (rLAS) for Read Count Limitation
3. Discussion
4. Materials and Methods
4.1. Total RNA Extraction
4.2. Targeted RNA Long-Amplicon Sequecning (rLAS)
4.3. Library Preparation and Sequencing
4.4. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar]
- Martinez, N.M.; Pan, Q.; Cole, B.S.; Yarosh, C.A.; Babcock, G.A.; Heyd, F.; Zhu, W.; Ajith, S.; Blencowe, B.J.; Lynch, K.W. Alternative splicing networks regulated by signaling in human T cells. RNA 2012, 18, 1029–1040. [Google Scholar]
- Bhate, A.; Parker, D.J.; Bebee, T.W.; Ahn, J.; Arif, W.; Rashan, E.H.; Chorghade, S.; Chau, A.; Lee, J.H.; Anakk, S.; et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat. Commun. 2015, 6, 8768. [Google Scholar] [CrossRef]
- Llorian, M.; Gooding, C.; Bellora, N.; Hallegger, M.; Buckroyd, A.; Wang, X.; Rajgor, D.; Kayikci, M.; Feltham, J.; Ule, J.; et al. The alternative splicing program of differentiated smooth muscle cells involves concerted non-productive splicing of post-transcriptional regulators. Nucleic Acids Res. 2016, 44, 8933–8950. [Google Scholar] [CrossRef]
- Giudice, J.; Xia, Z.; Wang, E.T.; Scavuzzo, M.A.; Ward, A.J.; Kalsotra, A.; Wang, W.; Wehrens, X.H.; Burge, C.B.; Li, W.; et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 2014, 5, 3603. [Google Scholar] [CrossRef]
- Dillman, A.A.; Hauser, D.N.; Gibbs, J.R.; Nalls, M.A.; McCoy, M.K.; Rudenko, I.N.; Galter, D.; Cookson, M.R. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci. 2013, 16, 499–506. [Google Scholar] [CrossRef]
- Brinkman, B.M. Splice variants as cancer biomarkers. Clin. Biochem. 2004, 37, 584–594. [Google Scholar] [CrossRef]
- Srebrow, A.; Kornblihtt, A.R. The connection between splicing and cancer. J. Cell Sci. 2006, 119 Pt 13, 2635–2641. [Google Scholar] [CrossRef]
- Ura, H.; Niida, Y. Comparison of RNA-Sequencing Methods for Degraded RNA. Int. J. Mol. Sci. 2024, 25, 6143. [Google Scholar] [CrossRef]
- Togi, S.; Ura, H.; Niida, Y. Optimization and Validation of Multimodular, Long-Range PCR-Based Next-Generation Sequencing Assays for Comprehensive Detection of Mutation in Tuberous Sclerosis Complex. J. Mol. Diagn. JMD 2021, 23, 424–446. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Agirre, E.; Eyras, E. Methods to study splicing from high-throughput RNA sequencing data. Methods Mol. Biol. 2014, 1126, 357–397. [Google Scholar]
- Vaquero-Garcia, J.; Aicher, J.K.; Jewell, S.; Gazzara, M.R.; Radens, C.M.; Jha, A.; Norton, S.S.; Lahens, N.F.; Grant, G.R.; Barash, Y. RNA splicing analysis using heterogeneous and large RNA-seq datasets. Nat. Commun. 2023, 14, 1230. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Z.; Kutschera, E.; Adams, J.I.; Kadash-Edmondson, K.E.; Xing, Y. rMATS-turbo: An efficient and flexible computational tool for alternative splicing analysis of large-scale RNA-seq data. Nat. Protoc. 2024, 19, 1083–1104. [Google Scholar] [PubMed]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Rätsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar]
- Ura, H.; Togi, S.; Hatanaka, H.; Niida, Y. Establishment of a human induced pluripotent stem cell line, KMUGMCi005-A, from a patient with Epidermodysplasia verruciformis (EV) bearing homozygous splicing donor site mutation in the TMC8 gene. Stem. Cell. Res. 2022, 64, 102926. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Hatanaka, H.; Niida, Y. Establishment of a human induced pluripotent stem cell line, KMUGMCi010-A, from a patient with X-linked Ohdo syndrome bearing missense mutation in the MED12 gene. Stem Cell Res. 2024, 77, 103388. [Google Scholar] [CrossRef] [PubMed]
- Togi, S.; Ura, H.; Niida, Y. Qualitative and quantitative analysis of MED12 c.887G>A causing both missense and splicing variants in X-linked Ohdo syndrome. Am. J. Med. Genet. A 2024, 194, e63628. [Google Scholar] [PubMed]
- Simoneau, J.; Gosselin, R.; Scott, M.S. Factorial study of the RNA-seq computational workflow identifies biases as technical gene signatures. NAR Genom. Bioinform. 2020, 2, lqaa043. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar]
- Au, K.F.; Jiang, H.; Lin, L.; Xing, Y.; Wong, W.H. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010, 38, 4570–4578. [Google Scholar]
- Otto, C.; Stadler, P.F.; Hoffmann, S. Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinformatics 2014, 30, 1837–1843. [Google Scholar]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.J.; Ha, K.C.H.; Blencowe, B.J. Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol. Cell 2018, 72, 187–200.e6. [Google Scholar]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar]
- Ura, H.; Togi, S.; Niida, Y. Target-capture full-length double-strand cDNA sequencing for alternative splicing analysis. RNA Biol. 2021, 18, 1600–1607. [Google Scholar]
- Ura, H.; Togi, S.; Niida, Y. Target-capture full-length double-stranded cDNA long-read sequencing through Nanopore revealed novel intron retention in patient with tuberous sclerosis complex. Front. Genet. 2023, 14, 1256064. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant Type | Gene | dbSNP_ID | Reference Sequence | DNA Level (hg38) | cDNA Level | RNA Level | Protein Level |
|---|---|---|---|---|---|---|---|
| Exon skipping | FH | rs878853691 | NC_000001.11 (NM_000143.4) | g.241517181 C>T | c.267+1G>A | r.133_267del (exon 2 skip) | p.(Ala45_Pro89del) |
| EDA | NA | NC_000023.11 (NM_001399.5) | g.70030523 A>C | c.793+3A>C | r.742_793del (exon 6 skip) | p.(Pro248Ilefs * 15) | |
| TMC8 | rs1381151589 | NC_000017.11 (NM_152468.5) | g.78132031 G>A | c.298+1G>A | r.150_298del (exon 3 skip) | p.(Gln51Profs * 42) | |
| Multiple exon skipping | TSC2 | NA | NC_000016.10 (NM_000548.5) | g.2056989_ 2074645del | NA | r.775_2545del (exon 9 to 22 skip) | p.(Met260Trpfs * 44) |
| Alternative 5′ and 3′ splicing | ENG | NA | NC_000009.12 (NM_000118.4) | g.127819450 G>C | c.1311+172 C>G | r.1311_1312ins1311+1_1311+167 | p.(Lys438Valfs * 8) |
| MED12 * | rs1556334519 | NC_000023.11 (NM_005120.3) | g.71121602 G>A | c.887G>A | r.[887g>a;847_888del] | p.[Arg296Gln;Tyr283_Arg296del] |
| Gene | Size (kb) | Forward Primer | Reverse Primer |
|---|---|---|---|
| TSC1 | 8.5 | gtgctgtacgtccaagatgg | tagtgctttcagcgagaaaagg |
| TSC2 | 5.5 | gggaggggttttctggtg | ctgacaggcaataccgtccaag |
| ENG | 2.5 | acaagtcttgcagaaacagtcc | acaagtcttgcagaaacagtcc |
| TMC8 | 2.3 | tgcacagaggccatagccaa | gtaaggaggcctgaaggggc |
| MED12 | 6.7 | gtcgagagtttctaacgtgcc | gggaattaagaggaaagggtgg |
| FH | 1.7 | cagaaattctacccaagctccc | acttgtttaatccatcttagacctagc |
| EDA | 1.2 | tcaagagagtgggtgtctcc | caacaccaatacacctcactcc |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ura, H.; Hatanaka, H.; Togi, S.; Niida, Y. Computational Comparison of Differential Splicing Tools for Targeted RNA Long-Amplicon Sequencing (rLAS). Int. J. Mol. Sci. 2025, 26, 3220. https://doi.org/10.3390/ijms26073220
Ura H, Hatanaka H, Togi S, Niida Y. Computational Comparison of Differential Splicing Tools for Targeted RNA Long-Amplicon Sequencing (rLAS). International Journal of Molecular Sciences. 2025; 26(7):3220. https://doi.org/10.3390/ijms26073220
Chicago/Turabian StyleUra, Hiroki, Hisayo Hatanaka, Sumihito Togi, and Yo Niida. 2025. "Computational Comparison of Differential Splicing Tools for Targeted RNA Long-Amplicon Sequencing (rLAS)" International Journal of Molecular Sciences 26, no. 7: 3220. https://doi.org/10.3390/ijms26073220
APA StyleUra, H., Hatanaka, H., Togi, S., & Niida, Y. (2025). Computational Comparison of Differential Splicing Tools for Targeted RNA Long-Amplicon Sequencing (rLAS). International Journal of Molecular Sciences, 26(7), 3220. https://doi.org/10.3390/ijms26073220