
Strategies for Altering Delivery Technologies to Optimize CAR Therapy


Abstract
1. Introduction
2. Main
2.1. In Vitro Production Mode
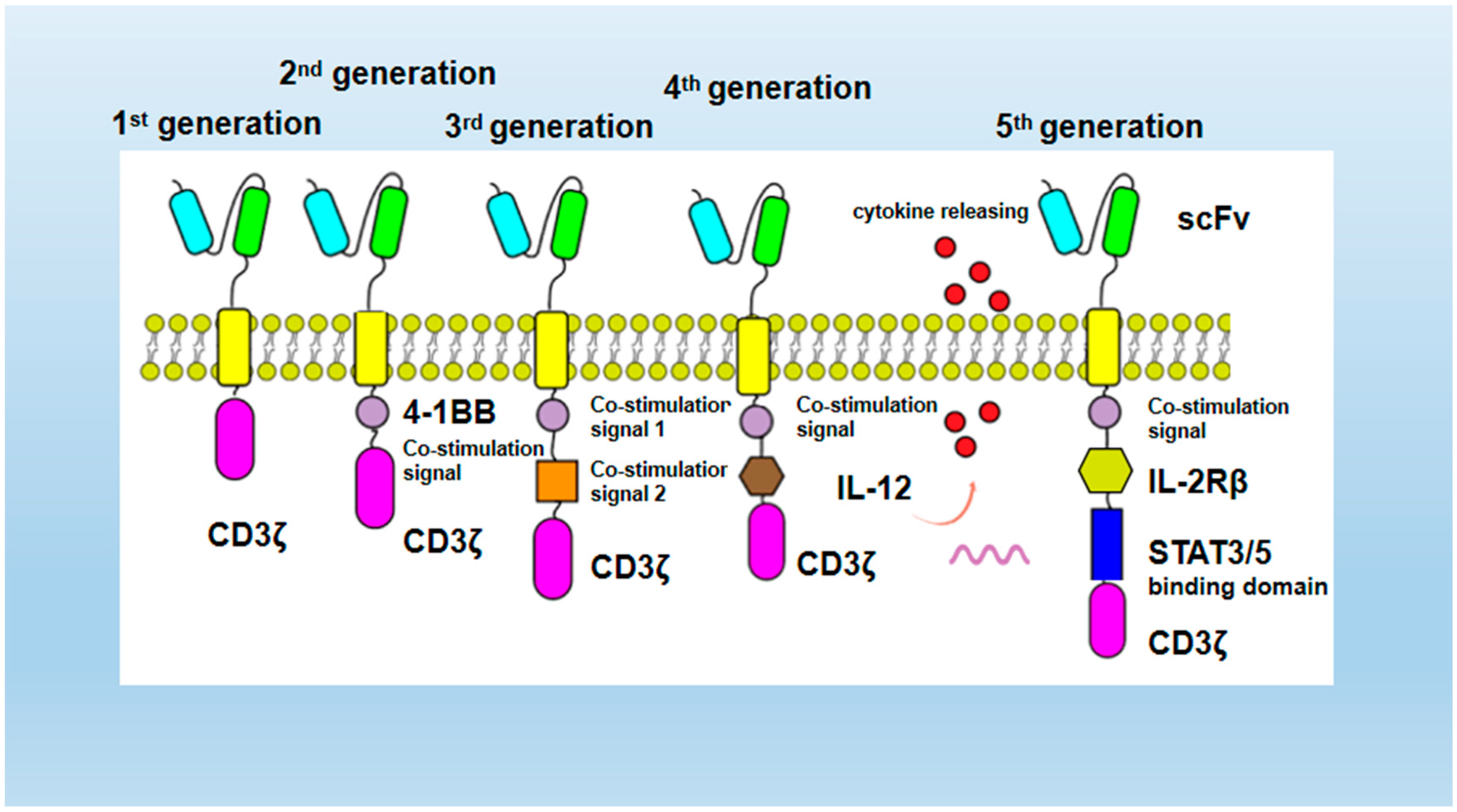
2.1.1. CAR Structure Optimization
2.1.2. Intravenous Delivery
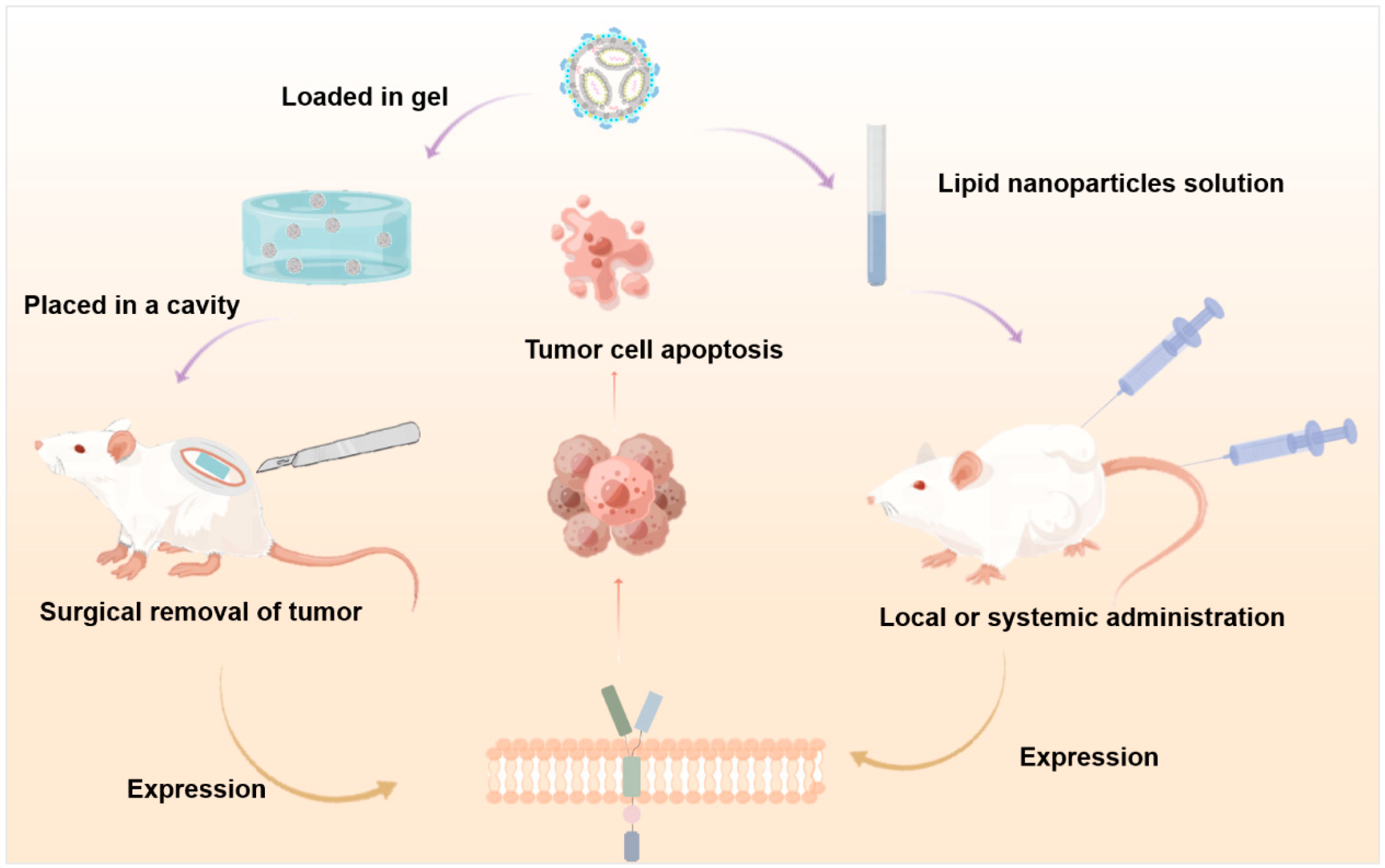
2.1.3. Local Drug Delivery
2.2. In Vivo Production Mode
2.2.1. Viral Vector
Lentivirus
Adeno-Associated Virus
2.2.2. Nonviral Vector
Transposon
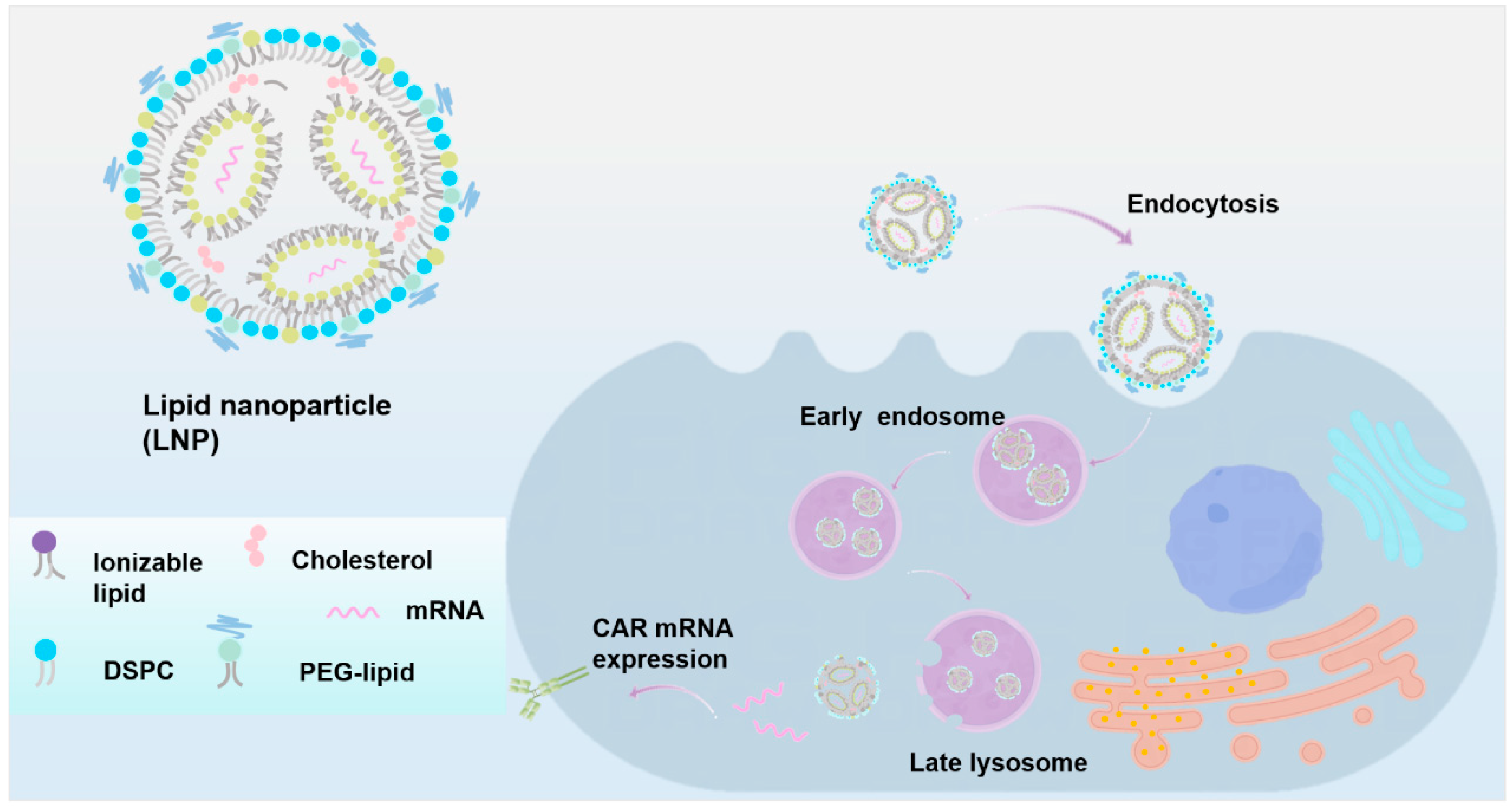
Lipid Nanoparticles
Delivering CAR Plasmid
Delivering CAR mRNA
2.3. Synergistic Treatment
2.4. Challenges and Solutions
2.4.1. Cytotoxicity
Cytokine Release Syndrome (CRS)
Off-Target Effects
Neurotoxicity
2.4.2. Delivery of Materials
2.4.3. Technical Means
3. Summary and Outlook
Funding
Conflicts of Interest
References
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef] [PubMed]
- King, A.C.; Orozco, J.S. Axicabtagene Ciloleucel: The First FDA-Approved CAR T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma. J. Adv. Pract. Oncol. 2019, 10, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Maus, M.V.; Mooney, D.J.; Wong, W.W. The future of engineered immune cell therapies. Science 2022, 378, 853–858. [Google Scholar] [CrossRef]
- Ye, B.; Stary, C.M.; Li, X.; Gao, Q.; Kang, C.; Xiong, X. Engineering chimeric antigen receptor-T cells for cancer treatment. Mol. Cancer 2018, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef]
- Huang, H.; Wu, H.-W.; Hu, Y.-X. Current advances in chimeric antigen receptor T-cell therapy for refractory/relapsed multiple myeloma. J. Zhejiang Univ. B 2019, 21, 29–41. [Google Scholar] [CrossRef]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, M.; Chen, H.; He, C.; Lin, Z.; Yang, X.; Li, H.; Shen, W.; Lu, D.; Xu, X. Double-negative T cells: A promising avenue of adoptive cell therapy in transplant oncology. J. Zhejiang Univ. B 2023, 24, 387–396. [Google Scholar] [CrossRef]
- Niu, Z.; Wu, J.; Zhao, Q.; Zhang, J.; Zhang, P.; Yang, Y. CAR-based immunotherapy for breast cancer: Peculiarities, ongoing investigations, and future strategies. Front. Immunol. 2024, 15, 1385571. [Google Scholar] [CrossRef]
- Lungova, K.; Putman, M. Barriers to CAR T-cell therapy in rheumatology. Lancet Rheumatol. 2024, 7, e212–e216. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- D’aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Short, L.; Holt, R.A.; Cullis, P.R.; Evgin, L. Direct in vivo CAR T cell engineering. Trends Pharmacol. Sci. 2024, 45, 406–418. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef]
- Wang, L.; Yao, R.; Zhang, L.; Fan, C.; Ma, L.; Liu, J. Chimeric antigen receptor T cell therapy and other therapeutics for malignancies: Combination and opportunity. Int. Immunopharmacol. 2019, 70, 498–503. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Roddie, C.; O’Reilly, M.; Pinto, J.D.A.; Vispute, K.; Lowdell, M. Manufacturing chimeric antigen receptor T cells: Issues and challenges. Cytotherapy 2019, 21, 327–340. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Hughes-Parry, H.E.; Cross, R.S.; Jenkins, M.R. The Evolving Protein Engineering in the Design of Chimeric Antigen Receptor T Cells. Int. J. Mol. Sci. 2019, 21, 204. [Google Scholar] [CrossRef]
- Bao, C.; Gao, Q.; Li, L.-L.; Han, L.; Zhang, B.; Ding, Y.; Song, Z.; Zhang, R.; Zhang, J.; Wu, X.-H. The Application of Nanobody in CAR-T Therapy. Biomolecules 2021, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237.e8. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef]
- Yang, Y.; Gu, X.; He, J.; Hu, Y.; Cai, Z. Waldenström macroglobulinemia: A challenging case treated with anti-CD19 CAR-T cell therapy. J. Zhejiang Univ. B 2024, 25, 719–722. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Leick, M.B.; Maus, M.V. CAR-T cells beyond CD19, UnCAR-Ted territory. Am. J. Hematol. 2019, 94, S34–S41. [Google Scholar] [CrossRef]
- Aghajanian, H.; Rurik, J.G.; Epstein, J.A. CAR-based therapies: Opportunities for immuno-medicine beyond cancer. Nat. Metab. 2022, 4, 163–169. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2018, 120, 26–37. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Rota, G.; Kosti, P.; Ronet, C.; Spill, A.; Seijo, B.; Romero, P.; Dangaj, D.; Coukos, G.; Irving, M. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J. Exp. Med. 2020, 218, e20192203. [Google Scholar] [CrossRef]
- Sun, W.; Jiang, Z.; Jiang, W.; Yang, R. Universal chimeric antigen receptor T cell therapy—The future of cell therapy: A review providing clinical evidence. Cancer Treat. Res. Commun. 2022, 33, 100638. [Google Scholar] [CrossRef]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next generations of CAR-T cells–new therapeutic opportunities in hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef] [PubMed]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef]
- Duan, D.; Wang, K.; Wei, C.; Feng, D.; Liu, Y.; He, Q.; Xu, X.; Wang, C.; Zhao, S.; Lv, L.; et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front. Immunol. 2021, 12, 609421. [Google Scholar] [CrossRef] [PubMed]
- Grover, N.S.; Ivanova, A.; Moore, D.T.; Cheng, C.J.A.; Babinec, C.; West, J.; Cavallo, T.; Morrison, J.K.; Buchanan, F.B.; Bowers, E.; et al. CD30-Directed CAR-T Cells Co-Expressing CCR4 in Relapsed/Refractory Hodgkin Lymphoma and CD30+ Cutaneous T Cell Lymphoma. Blood 2021, 138, 742. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Caruso, H.G.; Tanaka, R.; Liang, J.; Ling, X.; Sabbagh, A.; Henry, V.K.; Collier, T.L.; Heimberger, A.B. Shortened ex vivo manufacturing time of EGFRvIII-specific chimeric antigen receptor (CAR) T cells reduces immune exhaustion and enhances antiglioma therapeutic function. J. Neuro-Oncol. 2019, 145, 429–439. [Google Scholar] [CrossRef]
- Tanaka, K.; Kato, I.; Tanaka, M.; Morita, D.; Matsuda, K.; Takahashi, Y.; Nakahata, T.; Umeda, K.; Hiramatsu, H.; Adachi, S.; et al. Direct Delivery of piggyBac CD19 CAR T Cells Has Potent Anti-tumor Activity against ALL Cells in CNS in a Xenograft Mouse Model. Mol. Ther. Oncolytics 2020, 18, 37–46. [Google Scholar] [CrossRef]
- Lai, P.; Chen, X.; Wang, Y.; Wang, J.; Zhang, Y.; Geng, S.; Li, P.; Du, X.; Weng, J.; Pei, D. C3aR costimulation enhances the antitumor efficacy of CAR-T cell therapy through Th17 expansion and memory T cell induction. J. Hematol. Oncol. 2022, 15, 68. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19, an Anti-CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy, in Patients (Pts) with Relapsed/Refractory Mantle Cell Lymphoma (R/R MCL): Results of the Phase 2 ZUMA-2 Study. Biol. Blood Marrow Transplant. 2020, 26, S1. [Google Scholar] [CrossRef]
- Rive, C.M.; Yung, E.; Dreolini, L.; Brown, S.D.; May, C.G.; Woodsworth, D.J.; Holt, R.A. Selective B cell depletion upon intravenous infusion of replication-incompetent anti-CD19 CAR lentivirus. Mol. Ther. Methods Clin. Dev. 2022, 26, 4–14. [Google Scholar] [CrossRef]
- Shen, L.; Li, H.; Bin, S.; Li, P.; Chen, J.; Gu, H.; Yuan, W. The efficacy of third generation anti-HER2 chimeric antigen receptor T cells in combination with PD1 blockade against malignant glioblastoma cells. Oncol. Rep. 2019, 42, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef]
- Klampatsa, A.; Haas, A.R.; Moon, E.K.; Albelda, S.M. Chimeric Antigen Receptor (CAR) T Cell Therapy for Malignant Pleural Mesothelioma (MPM). Cancers 2017, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Thayaparan, T.; Petrovic, R.M.; Achkova, D.Y.; Zabinski, T.; Davies, D.M.; Klampatsa, A.; Parente-Pereira, A.C.; Whilding, L.M.; van der Stegen, S.J.; Woodman, N.; et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology 2017, 6, e1363137. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Mulazzani, M.; Fräßle, S.P.; von Mücke-Heim, I.; Langer, S.; Zhou, X.; Ishikawa-Ankerhold, H.; Leube, J.; Zhang, W.; Dötsch, S.; Svec, M.; et al. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 24275–24284. [Google Scholar] [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef]
- Alghamdi, M.; Gumbleton, M.; Newland, B. Local delivery to malignant brain tumors: Potential biomaterial-based therapeutic/adjuvant strategies. Biomater. Sci. 2021, 9, 6037–6051. [Google Scholar] [CrossRef]
- Sun, Z.; Song, C.; Wang, C.; Hu, Y.; Wu, J. Hydrogel-Based Controlled Drug Delivery for Cancer Treatment: A Review. Mol. Pharm. 2019, 17, 373–391. [Google Scholar] [CrossRef]
- Ogunnaike, E.A.; Valdivia, A.; Yazdimamaghani, M.; Leon, E.; Nandi, S.; Hudson, H.; Du, H.; Khagi, S.; Gu, Z.; Savoldo, B.; et al. Fibrin gel enhances the antitumor effects of chimeric antigen receptor T cells in glioblastoma. Sci. Adv. 2021, 7, eabg5841. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Archibong, E.; Chen, Q.; Ruan, H.; Ahn, S.; Dukhovlinova, E.; Kang, Y.; Wen, D.; Dotti, G.; et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat. Biomed. Eng. 2021, 5, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Bujoli, B.; Scimeca, J.-C.; Verron, E. Fibrin as a Multipurpose Physiological Platform for Bone Tissue Engineering and Targeted Delivery of Bioactive Compounds. Pharmaceutics 2019, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Heher, P.; Mühleder, S.; Mittermayr, R.; Redl, H.; Slezak, P. Fibrin-based delivery strategies for acute and chronic wound healing. Adv. Drug Deliv. Rev. 2018, 129, 134–147. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, R.; Nakazawa, Y.; Sakashita, K.; Saito, S.; Tanaka, M.; Shiohara, M.; Shimodaira, S.; Koike, K. Intrathecal donor lymphocyte infusion for isolated leukemia relapse in the central nervous system following allogeneic stem cell transplantation: A case report and literature review. Int. J. Hematol. 2015, 103, 107–111. [Google Scholar] [CrossRef]
- Agarwal, S.; Weidner, T.; Thalheimer, F.B.; Buchholz, C.J. In Vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology 2019, 8, e1671761. [Google Scholar] [CrossRef]
- Fidler, B. ‘In Vivo’ Cell Therapy: Expanding Beyond CAR-T. 2022. Available online: https://www.biopharmadive.com/news/in-vivo-cell-therapy-car-t-biotech-startups/634287/ (accessed on 15 January 2024).
- Bender, R.R.; Muth, A.; Schneider, I.C.; Friedel, T.; Hartmann, J.; Plückthun, A.; Maisner, A.; Buchholz, C.J. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 2016, 12, e1005641. [Google Scholar] [CrossRef]
- Mhaidly, R.; Verhoeyen, E. The Future: In Vivo CAR T Cell Gene Therapy. Mol. Ther. 2019, 27, 707–709. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Thalheimer, F.B.; Hartmann, S.; Frank, A.M.; Bender, R.R.; Danisch, S.; Costa, C.; Wels, W.S.; Modlich, U.; Stripecke, R.; et al. In Vivo generation of human CD 19- CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018, 10, e9158. [Google Scholar] [CrossRef]
- Xin, T.; Cheng, L.; Zhou, C.; Zhao, Y.; Hu, Z.; Wu, X. In-Vivo Induced CAR-T Cell for the Potential Breakthrough to Overcome the Barriers of Current CAR-T Cell Therapy. Front. Oncol. 2022, 12, 809754. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.; Ho, N.; Buchholz, C.J. Precision medicine: In vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol. Ther. 2022, 30, 2401–2415. [Google Scholar] [CrossRef]
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef]
- FDA Approved Drug Products: Kymriah (Tisagenlecleucel) Suspension for Intravenous Infusion. Available online: https://www.fda.gov/media/107296/download (accessed on 15 January 2024).
- Wei, G.; Ding, L.; Wang, J.; Hu, Y.; Huang, H. Advances of CD19-directed chimeric antigen receptor-modified T cells in refractory/relapsed acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- FDA Approved Drug Products: YESCARTA (Axicabtagene Ciloleucel) Suspension for Intravenous Infusion. Available online: https://www.fda.gov/media/108377/download (accessed on 15 January 2024).
- FDA Approved Drug Products: Tecartus (Brexucabtagene Autoleucel) Suspension. Available online: https://www.fda.gov/media/140409/download (accessed on 15 January 2024).
- FDA Approved Drug Products: Breyanzi (Lisocabtagene Maraleucel) Intravenous Infusion. Available online: https://fda.report/media/145711/Package-Insert-BREYANZI_0.pdf (accessed on 15 January 2024).
- FDA Approved Drug Products: Abecma (Idecabtagene Vicleucel) Intravenous Infusion. Available online: https://www.fda.gov/media/147055/download?attachment (accessed on 15 January 2024).
- FDA Approved Drug Products: Carvykti (Ciltacabtagene Autoleucel) Suspension for Intravenous Infusion. Available online: https://www.fda.gov/media/156560/download?attachment (accessed on 15 January 2024).
- Poorebrahim, M.; Sadeghi, S.; Fakhr, E.; Abazari, M.F.; Poortahmasebi, V.; Kheirollahi, A.; Askari, H.; Rajabzadeh, A.; Rastegarpanah, M.; Linē, A.; et al. Production of CAR T-cells by GMP-grade lentiviral vectors: Latest advances and future prospects. Crit. Rev. Clin. Lab. Sci. 2019, 56, 393–419. [Google Scholar] [CrossRef]
- Huckaby, J.T.; Landoni, E.; Jacobs, T.M.; Savoldo, B.; Dotti, G.; Lai, S.K. Bispecific binder redirected lentiviral vector enables in vivo engineering of CAR-T cells. J. Immunother. Cancer 2021, 9, e002737. [Google Scholar] [CrossRef] [PubMed]
- Liu, A. AACR: Exuma Aims to be First to the Clinic with Under-the-Skin Cancer CAR Cell Therapy Without Complex Manufacturing. 2022. Available online: https://www.fiercebiotech.com/research/aacr-exuma-eyes-first-clinic-under-skin-cancer-car-cell-therapy-without-complex (accessed on 15 January 2024).
- Umoja’s Solution: “Off the Shelf” Cell Therapies. Available online: https://www.umoja-biopharma.com/our-science (accessed on 20 January 2024).
- Michels, A.; Frank, A.M.; Günther, D.M.; Mataei, M.; Börner, K.; Grimm, D.; Hartmann, J.; Buchholz, C.J. Lentiviral and adeno-associated vectors efficiently transduce mouse T lymphocytes when targeted to murine CD8. Mol. Ther. Methods Clin. Dev. 2021, 23, 334–347. [Google Scholar] [CrossRef]
- Mustang Bio Collaborates with Mayo Clinic on Novel CAR T Technology. 2021. Available online: https://ir.mustangbio.com/news-events/press-releases/detail/13/mustang-bio-collaborates-with-mayo-clinic-on-novel-car-t (accessed on 1 March 2024).
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef]
- McCarron, A.; Donnelley, M.; McIntyre, C.; Parsons, D. Challenges of up-scaling lentivirus production and processing. J. Biotechnol. 2016, 240, 23–30. [Google Scholar] [CrossRef]
- Piscopo, N.J.; Mueller, K.P.; Das, A.; Hematti, P.; Murphy, W.L.; Palecek, S.P.; Capitini, C.M.; Saha, K. Bioengineering Solutions for Manufacturing Challenges in CAR T Cells. Biotechnol. J. 2017, 13, 1700095. [Google Scholar] [CrossRef]
- Picanço-Castro, V.; Pereira, C.G.; Covas, D.T.; Porto, G.S.; Athanassiadou, A.; Figueiredo, M.L. Emerging patent landscape for non-viral vectors used for gene therapy. Nat. Biotechnol. 2020, 38, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2016, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Morneau-Brosnan, D. Non-viral vectors for chimeric antigen receptor immunotherapy. Nat. Rev. Methods Prim. 2024, 4, 75. [Google Scholar] [CrossRef]
- Tretbar, U.S.; Rurik, J.G.; Rustad, E.H.; Sürün, D.; Köhl, U.; Olweus, J.; Buchholz, F.; Ivics, Z.; Fricke, S.; Blache, U. Non-viral vectors for chimeric antigen receptor immunotherapy. Nat. Rev. Methods Prim. 2024, 4, 74. [Google Scholar] [CrossRef]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Fernández, P.O.M.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Zhou, J.-E.; Sun, L.; Jia, Y.; Wang, Z.; Luo, T.; Tan, J.; Fang, X.; Zhu, H.; Wang, J.; Yu, L.; et al. Lipid nanoparticles produce chimeric antigen receptor T cells with interleukin-6 knockdown in vivo. J. Control. Release 2022, 350, 298–307. [Google Scholar] [CrossRef]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef]
- Moffett, H.F.; Coon, M.E.; Radtke, S.; Stephan, S.B.; McKnight, L.; Lambert, A.; Stoddard, B.L.; Kiem, H.P.; Stephan, M.T. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017, 8, 389. [Google Scholar] [CrossRef]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef]
- Olden, B.R.; Cheng, Y.; Yu, J.L.; Pun, S.H. Cationic polymers for non-viral gene delivery to human T cells. J. Control. Release 2018, 282, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going? Cells 2020, 9, 1337. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cells 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Precigen Receives FDA Orphan Drug Designation for PRGN-3006 UltraCAR-T™ in Patients with Acute Myeloid Leukemia (AML). 2020. Available online: https://www.biospace.com/article/releases/precigen-receives-fda-orphan-drug-designation-for-prgn-3006-ultracar-t-in-patients-with-acute-myeloid-leukemia-aml (accessed on 1 March 2024).
- Shy, B.R.; Vykunta, V.S.; Ha, A.; Talbot, A.; Roth, T.L.; Nguyen, D.N.; Pfeifer, W.G.; Chen, Y.Y.; Blaeschke, F.; Shifrut, E.; et al. High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 2022, 41, 521–531. [Google Scholar] [CrossRef]
- Yagyu, S.; Nakazawa, Y. piggyBac-transposon-mediated CAR-T cells for the treatment of hematological and solid malignancies. Int. J. Clin. Oncol. 2023, 28, 736–747. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.M.; Hamilton, A.G.; Mai, D.; Patel, S.K.; Swingle, K.L.; Sheppard, N.C.; June, C.H.; Mitchell, M.J. Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for mRNA Engineering of CAR T Cells. Nano Lett. 2021, 22, 533–542. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef]
- Ye, B.; Hu, Y.; Zhang, M.; Huang, H. Research advance in lipid nanoparticle-mRNA delivery system and its application in CAR-T cell therapy. Zhejiang Da Xue Xue Bao Yi Xue Ban 2022, 51, 185–191. [Google Scholar] [CrossRef]
- Vaccine-Like mRNA Injection Can Be Used to Make CAR T Cells. 2022. Available online: https://www.pennmedicine.org/news/news-releases/2022/january/vaccine-like-mrna-injection-can-be-used-to-make-car-t-cells-in-the-body (accessed on 1 April 2024).
- Chen, C.; Jing, W.; Chen, Y.; Wang, G.; Abdalla, M.; Gao, L.; Han, M.; Shi, C.; Li, A.; Sun, P.; et al. Intracavity generation of glioma stem cell–specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 2022, 14, eabn1128. [Google Scholar] [CrossRef] [PubMed]
- Olweus, J. Manufacture of CAR-T cells in the body. Nat. Biotechnol. 2017, 35, 520–521. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Simon, B.; Fujii, S.-I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Interius BioTherapeutics Doses First Patient with in vivo Chimeric Antigen Receptor (CAR) Gene Therapy for B-cell Malignancies. Available online: https://interiusbio.com/press-release/interius-biotherapeutics-doses-first-patient-with-in-vivo-chimeric-antigen-receptor-car-gene-therapy-for-b-cell-malignancies/ (accessed on 1 April 2024).
- Garcia, J.; Dehner, C.; Teoh, J.; Wallis, W. A Phase 1, Multicenter, Open-Label Study of UB-VV111 in Combination with Rapamycin in Relapsed/Refractory CD19+ B-Cell Malignancies. Blood 2024, 144, 1750.1751. [Google Scholar] [CrossRef]
- Kong, Y.; Tang, L.; You, Y.; Li, Q.; Zhu, X. Analysis of causes for poor persistence of CAR-T cell therapy in vivo. Front. Immunol. 2023, 14, 1063454. [Google Scholar] [CrossRef]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, Z.; Sun, M.; Li, B.; Pan, F.; Ma, A.; Liao, J.; Yin, T.; Tang, X.; Huang, G.; et al. IL-12 nanochaperone-engineered CAR T cell for robust tumor-immunotherapy. Biomaterials 2021, 281, 121341. [Google Scholar] [CrossRef]
- Glienke, W.; Dragon, A.C.; Zimmermann, K.; Martyniszyn-Eiben, A.; Mertens, M.; Abken, H.; Rossig, C.; Altvater, B.; Aleksandrova, K.; Arseniev, L.; et al. GMP-Compliant Manufacturing of TRUCKs: CAR T Cells targeting GD2 and Releasing Inducible IL-18. Front. Immunol. 2022, 13, 839783. [Google Scholar] [CrossRef]
- Allen, G.M.; Frankel, N.W.; Reddy, N.R.; Bhargava, H.K.; Yoshida, M.A.; Stark, S.R.; Purl, M.; Lee, J.; Yee, J.L.; Yu, W.; et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science 2022, 378, eaba1624. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Nai, Y.; Shen, M.; Li, T.; Huang, J.; Han, X.; Wang, W.; Pang, D.; Jin, A. IL-21 Optimizes the CAR-T Cell Preparation Through Improving Lentivirus Mediated Transfection Efficiency of T Cells and Enhancing CAR-T Cell Cytotoxic Activities. Front. Mol. Biosci. 2021, 8, 675179. [Google Scholar] [CrossRef]
- Zhang, Z.; Miao, L.; Ren, Z.; Tang, F.; Li, Y. Gene-Edited Interleukin CAR-T Cells Therapy in the Treatment of Malignancies: Present and Future. Front. Immunol. 2021, 12, 718686. [Google Scholar] [CrossRef]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [PubMed]
- Oladapo, O.; Yeku, T.P.; David, R.; Spriggs, R.J. Brentjens Chimeric antigen receptor (CAR) T cells genetically engineered to deliver IL-12 to the tumor microenvironment in ovarian cancer. J. Clin. Oncol. 2017, 35, 3050. [Google Scholar]
- Avanzi, M.P.; van Leeuwen, D.G.; Li, X.; Cheung, K.; Park, H.; Purdon, T.J.; Brentjens, R.J. IL-18 Secreting CAR T Cells Enhance Cell Persistence, Induce Prolonged B Cell Aplasia and Eradicate CD19+ Tumor Cells without Need for Prior Conditioning. Blood 2016, 128, 816. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef]
- Schubert, M.-L.; Schmitt, M.; Wang, L.; Ramos, C.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Laomeephol, C.; Areecheewakul, S.; Tawinwung, S.; Suppipat, K.; Chunhacha, P.; Neves, N.M.; Luckanagul, J.A. Potential roles of hyaluronic acid in in vivo CAR T cell reprogramming for cancer immunotherapy. Nanoscale 2022, 14, 17821–17840. [Google Scholar] [CrossRef]
- Engineering CAR T Cells with Biomaterials. Cancer Discov. 2017, 7, 656–657. [CrossRef] [PubMed]
- Schroeder, T.; Martens, T.; Fransecky, L.; Valerius, T.; Schub, N.; Pott, C.; Baldus, C.; Stölzel, F. Management of chimeric antigen receptor T (CAR-T) cell-associated toxicities. Intensiv. Care Med. 2024, 50, 1459–1469. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2017, 15, 47–62. [Google Scholar] [CrossRef]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 897. [Google Scholar] [CrossRef]
- Rakhshandehroo, T.; Mantri, S.R.; Moravej, H.; Louis, B.B.V.; Farid, A.S.; Munaretto, L.; Regan, K.; Khan, R.M.M.; Wolff, A.; Farkash, Z.; et al. A CAR enhancer increases the activity and persistence of CAR T cells. Nat. Biotechnol. 2024, 1–12. [Google Scholar] [CrossRef]
- Liu, S.; Deng, B.; Yin, Z.; Pan, J.; Lin, Y.; Ling, Z.; Wu, T.; Chen, D.; Chang, A.H.; Gao, Z.; et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.-C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective Targeting of TAG72+ Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front. Immunol. 2018, 9, 2268. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Böll, B.; Schellongowski, P.; Valade, S.; Metaxa, V.; Azoulay, E.; von Bergwelt-Baildon, M. Critical care management of chimeric antigen receptor T-cell therapy recipients. CA A Cancer J. Clin. 2021, 72, 78–93. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e117. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qi, Y.; Li, H.; Liu, F.; Cao, J.; Chen, W.; Wang, Y.; Qi, K.; Yan, Z.; Zhu, F.; et al. Impact of glucocorticoids on short-term and long-term outcomes in patients with relapsed/refractory multiple myeloma treated with CAR-T therapy. Front. Immunol. 2022, 13, 943004. [Google Scholar] [CrossRef] [PubMed]
- Borrega, J.G.; Gödel, P.; Rüger, M.A.; Onur, Ö.A.; Shimabukuro-Vornhagen, A.; Kochanek, M.; Böll, B. In the Eye of the Storm: Immune-mediated Toxicities Associated With CAR-T Cell Therapy. HemaSphere 2019, 3, e191. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar]
- Gehrke, L.; Gonçalves, V.D.R.; Andrae, D.; Rasko, T.; Ho, P.; Einsele, H.; Hudecek, M.; Friedel, S.R. Current Non-Viral-Based Strategies to Manufacture CAR-T Cells. Int. J. Mol. Sci. 2024, 25, 13685. [Google Scholar] [CrossRef]
- Mei, H.; Chen, Z.; Hu, Y. Bio-Orthogonally Redirected Bispecific Lentiviral Vectors for In Vivo CAR-T Cell Generation. Blood 2023, 142, 2062. [Google Scholar] [CrossRef]
- Watanabe, N.; McKenna, M.K. Methods in Cell Biology; Spada, S., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 167, pp. 171–183. [Google Scholar]
- Kaštánková, I.; Štach, M.; Žižková, H.; Ptáčková, P.; Šmilauerová, K.; Mucha, M.; Šroller, V.; Otáhal, P. Enzymatically produced piggyBac transposon vectors for efficient non-viral manufacturing of CD19-specific CAR T cells. Mol. Ther. Methods Clin. Dev. 2021, 23, 119–127. [Google Scholar] [CrossRef]
- Huang, X.; Guo, H.; Tammana, S.; Jung, Y.-C.; Mellgren, E.; Bassi, P.; Cao, Q.; Tu, Z.J.; Kim, Y.C.; Ekker, S.C.; et al. Gene Transfer Efficiency and Genome-Wide Integration Profiling of Sleeping Beauty, Tol2, and PiggyBac Transposons in Human Primary T Cells. Mol. Ther. 2010, 18, 1803–1813. [Google Scholar] [CrossRef]
- Kon, E.; Ad-El, N.; Hazan-Halevy, I.; Stotsky-Oterin, L.; Peer, D. Targeting cancer with mRNA–lipid nanoparticles: Key considerations and future prospects. Nat. Rev. Clin. Oncol. 2023, 20, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.D.; Davis, K.L. Correlative studies reveal factors contributing to successful CAR-T cell therapies in cancer. Cancer Metastasis Rev. 2024, 44, 15. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Zhang, X.; Wang, X.; Zhao, X.; Li, S.; Guan, Z.; Cao, J.; Liu, D.; Zheng, J.; Shi, M. In vivo manufacture and manipulation of CAR-T cells for better druggability. Cancer Metastasis Rev. 2024, 43, 1075–1093. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Generation Mode | Company | Name | Indication | Antigen | Mode of Administration | Delivery Vector | Dose Range | Reference |
|---|---|---|---|---|---|---|---|---|
| In vitro | Novartis | Kymriah | RRFL | CD19 | Intravenous | lentivirus vector | For patients below 50 kg: 0.2~5.0 per kg body weight × 106 CAR-positive survival T cells For patients above 50 kg: 0.1 to 2.5 × 108 CAR-positive survival T cells | [75,76] |
| Gilead | Yescarta | R/R LBCL | CD19 | Intravenous | retroviral vector | 2 × 106 CAR-positive living cells/kg body weight | [77] | |
| Gilead | Tecartus | R/R B-ALL | CD19 | Intravenous | retroviral vector | 2 × 106 CAR-positive living cells/kg body weight | [78] | |
| Bristol Myers Squibb | Breyanzi | R/R LBCL | CD19 | Intravenous | lentivirus vector | 1.5 × 106 to 70 × 106 CAR-positive live T cells/mL | [79] | |
| Bristol Myers Squibb | Abecma | R/R MM | BCMA | Intravenous | lentivirus vector | 300 to 460 × 106 CAR-positive T cells | [80] | |
| Legend Biotech | Carvykti | R/R MM | BCMA | Intravenous | lentivirus vector | 1.0 × 106 CAR positive active T cells/kg body weight | [81] |
| Name/Identifier | Delivery Vector | Delivery Content | Target Head | Delivery Method | Size | Zeta Potential | Disease | Reference |
|---|---|---|---|---|---|---|---|---|
| CD5/LNP FAPCAR | Lipid nanoparticle | FAP-CAR mRNA | CD5 | Intravenousinjection | 80 nm | —— | Cardiac fibrosis | [95] |
| AntiCD3-LNP/CAR19 + shIL6 | Lipid nanoparticle | shRNA and the CAR gene | CD3 | Intravenous injection | 200 ± 16.5 nm | 1.6 ± 0.2 mV | acute lymphoblastic leukemia | [96] |
| Ionizable Lipid Nanoparticle C14−4LNPs | C14−4LNPs | CD19 CAR mRNA | CD19 | —— | 70.17 ± 0.41 nm | —— | —— | [97] |
| Foxo13A-NP | Engineered NPs | Foxo13A19–41BB mRNA | CD19 | Intravenous injection | 109.6 ± 26.6 nm | 1.1 ± 5.3 mV | lymphadenoma | [98] |
| Poly(B-amino) ester polymer (PBAE) NP | Engineered NPs | 194-1BBz CAR plasmid DNA | CD19 | Intravenous injection | 155 ± 40 nm | −7.8 ± 2.1 mV | leukemia | [99] |
| IVT mRNA nanoparticles | PBAE-447 polymer nanocarrier | CAR mRNA | CD8 | Intravenous injection | 106.9 ± 7.2 nm. | 4 ± 2 mV | leukemia | [100] |
| comb-shaped pHEMA-g-pDMAEMA polymer | polymers nanocarrier | plasmidDNA/mRNA | —— | —— | 100–150 nm | 25–30 mV | —— | [101] |
| Advantages | Disadvantages | Reference | |
|---|---|---|---|
| Viral vector | Rapid preparation | High cost | [150,151,152] |
| Stable integration | Low security | ||
| Mature Technology Pathways | Immunogenicity | ||
| Transposon carrier | Large gene fragments | Low transfection efficiency | [92,153,154] |
| High biosafety | Complex plasmid design | ||
| LNP | High RNA stability | Transient expression | [150,155,156] |
| High delivery efficiency | Low transfection efficiency |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Liu, Y.; Lin, G. Strategies for Altering Delivery Technologies to Optimize CAR Therapy. Int. J. Mol. Sci. 2025, 26, 3206. https://doi.org/10.3390/ijms26073206
Cao L, Liu Y, Lin G. Strategies for Altering Delivery Technologies to Optimize CAR Therapy. International Journal of Molecular Sciences. 2025; 26(7):3206. https://doi.org/10.3390/ijms26073206
Chicago/Turabian StyleCao, Lili, Yingying Liu, and Guimei Lin. 2025. "Strategies for Altering Delivery Technologies to Optimize CAR Therapy" International Journal of Molecular Sciences 26, no. 7: 3206. https://doi.org/10.3390/ijms26073206
APA StyleCao, L., Liu, Y., & Lin, G. (2025). Strategies for Altering Delivery Technologies to Optimize CAR Therapy. International Journal of Molecular Sciences, 26(7), 3206. https://doi.org/10.3390/ijms26073206