
Multifaceted Cardioprotective Potential of Reduced Glutathione Against Doxorubicin-Induced Cardiotoxicity via Modulating Inflammation–Oxidative Stress Axis

 , , ,
, , ,
Abstract
1. Introduction
2. Results
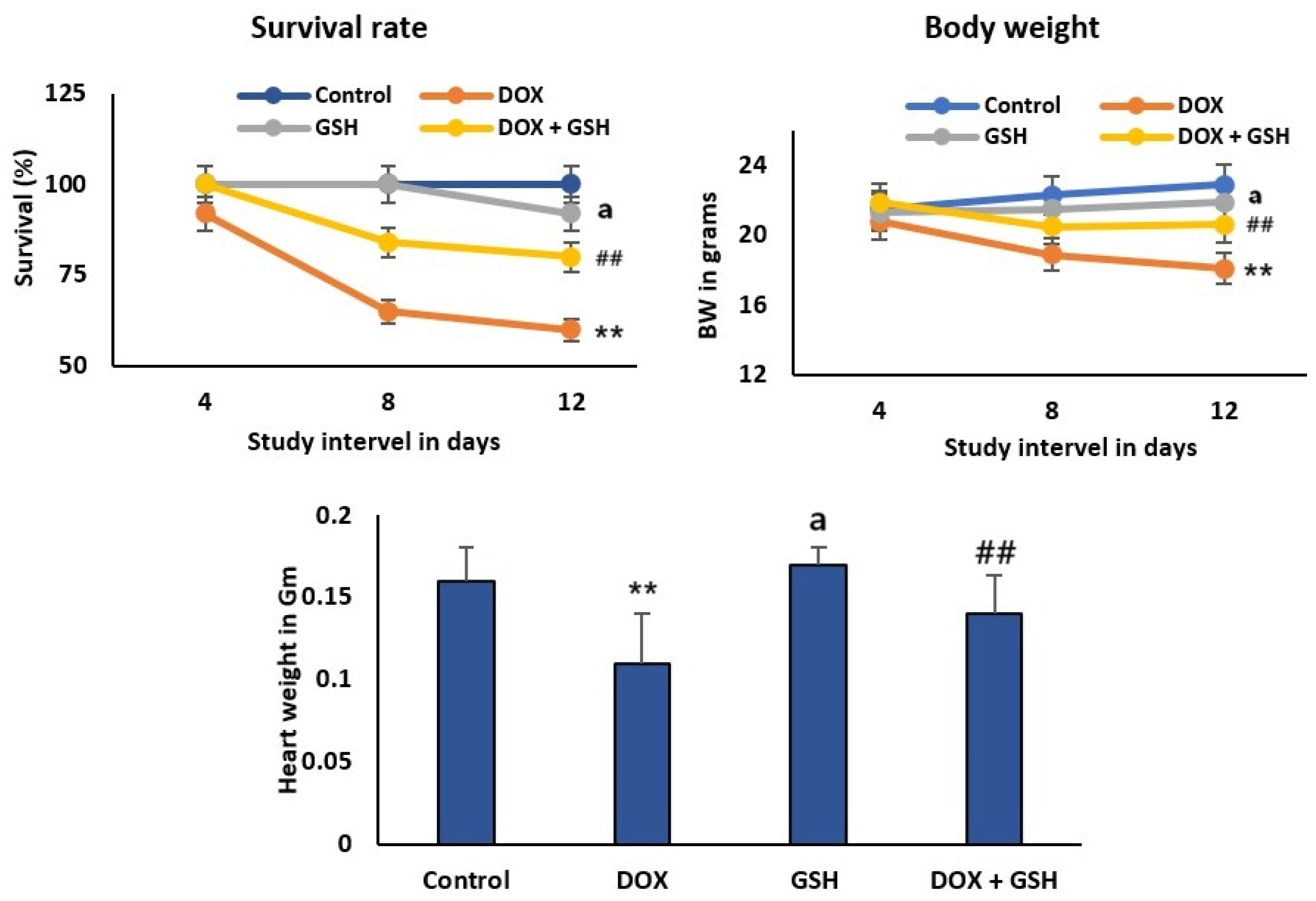
2.1. Effect of Dox and GSH on Survival Rate, Body Weight, and Heart Weight for Different Treatment Groups
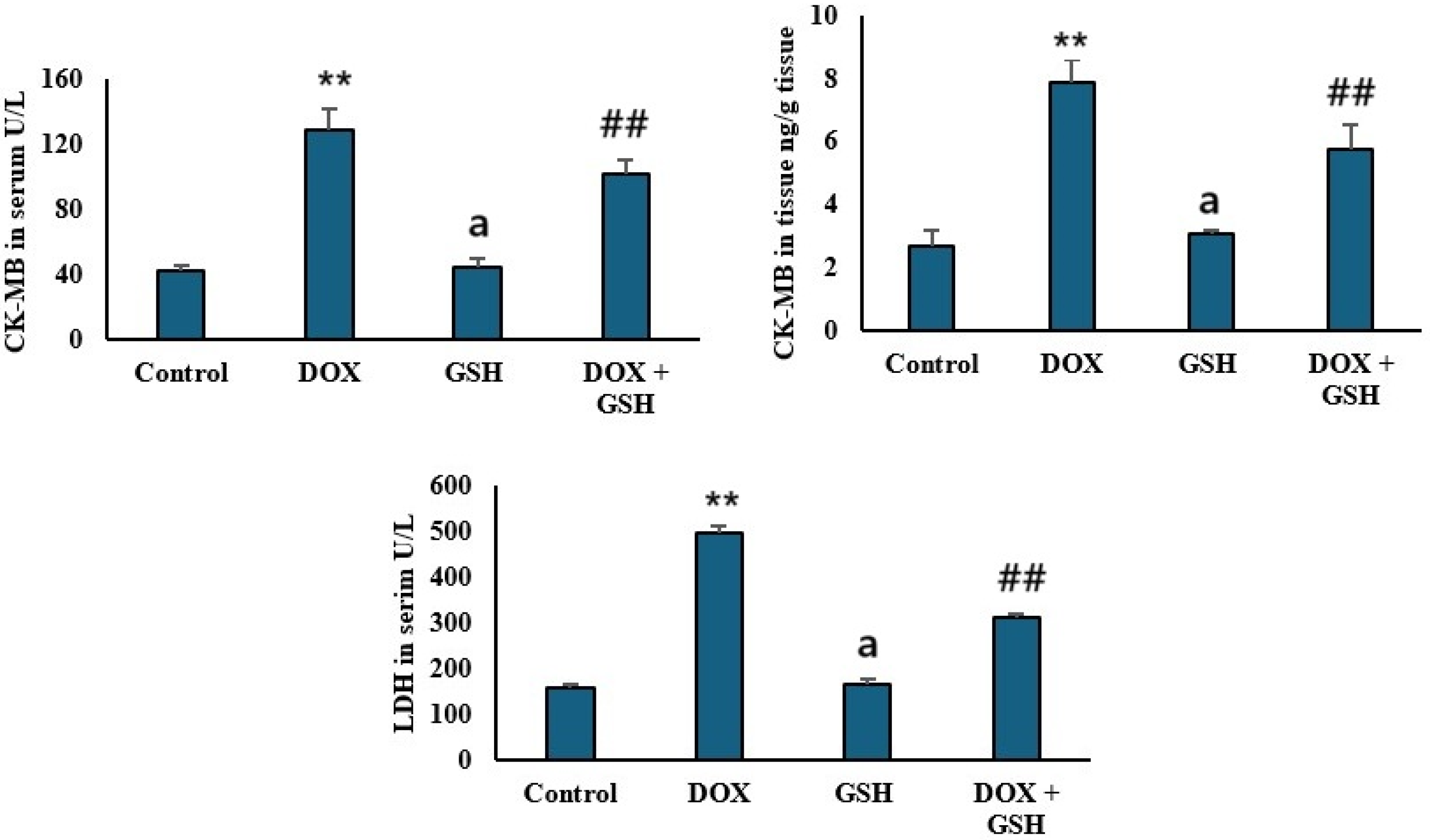
2.2. Effect of Dox and GSH on Serum and Tissue Creatine Kinase-MB (CK-MB) and Serum Lactate Dehydrogenase (LDH)
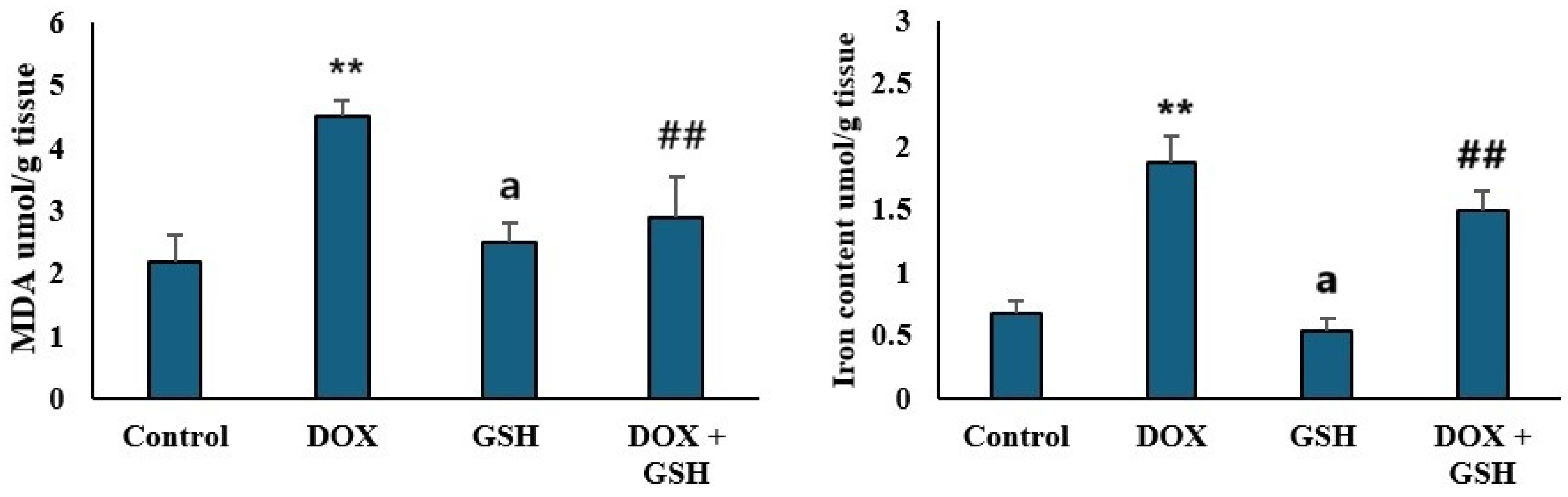
2.3. Malondialdehyde (MDA) Levels and Iron Content in Tissue
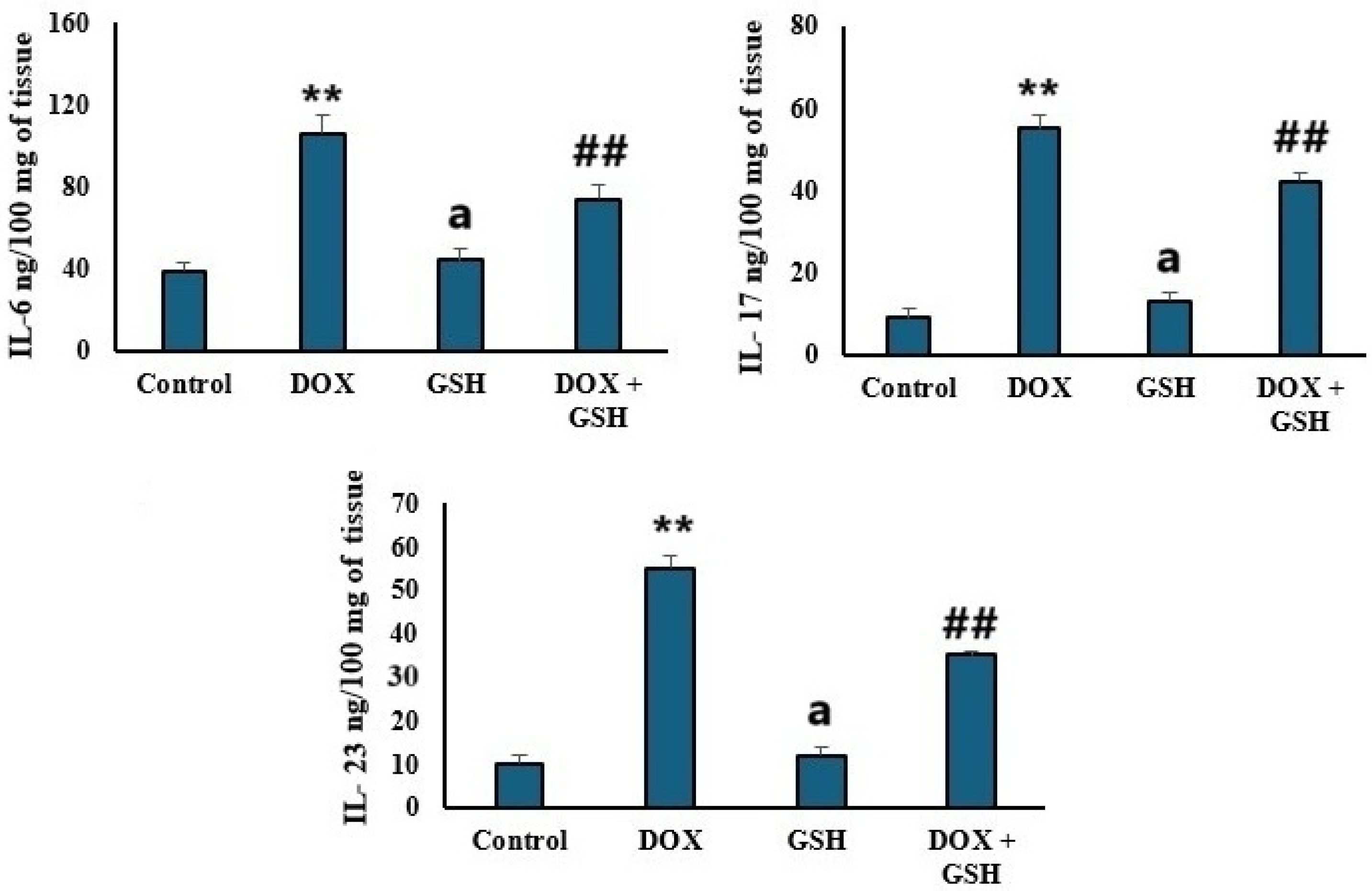
2.4. Pro-Inflammatory Cytokines Levels (IL-6, IL-17, and IL-23)
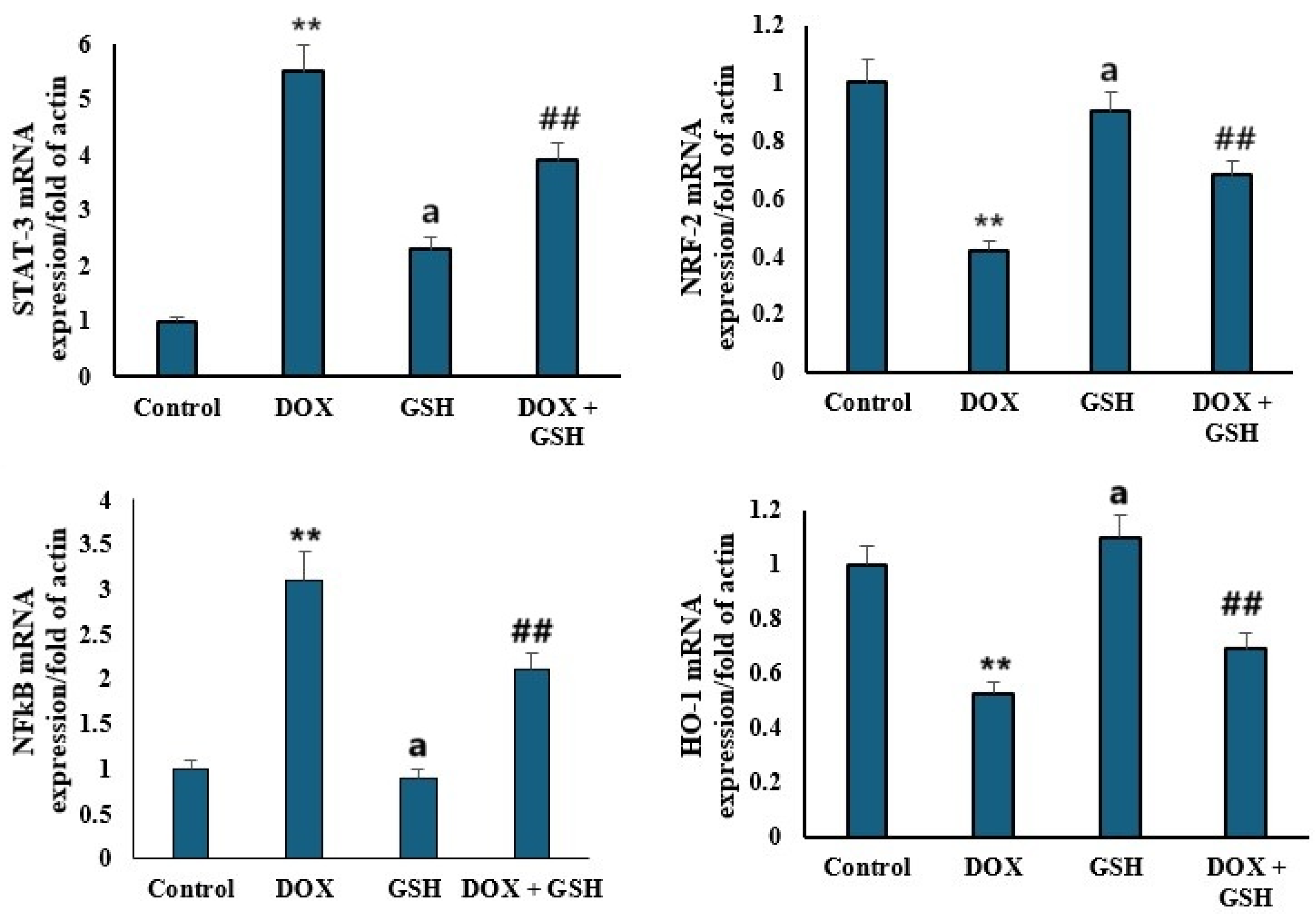
2.5. mRNA Expression Levels of STAT-3, NRF-2, NFκB, and HO-1
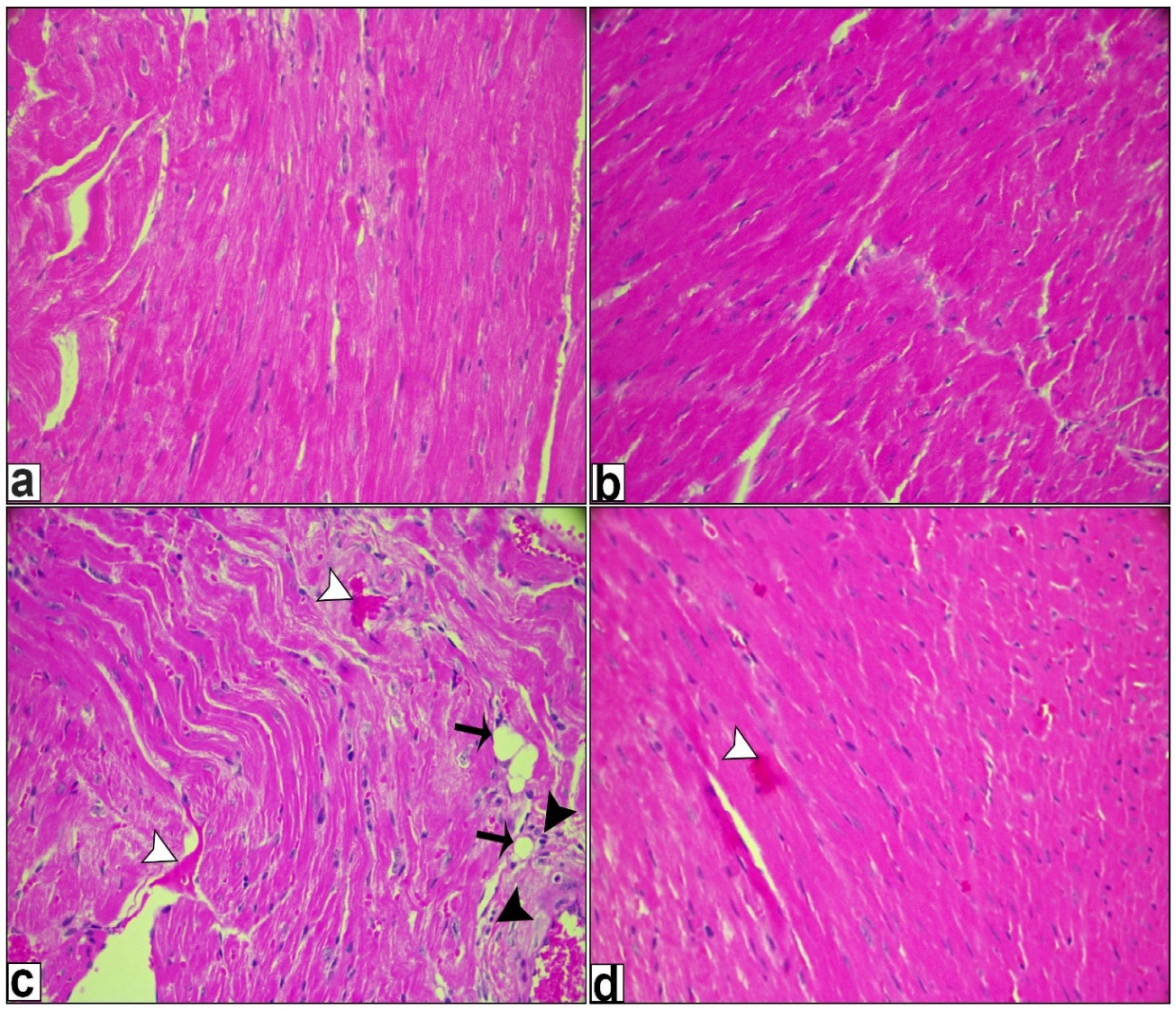
2.6. Histological Examination
3. Discussion
4. Materials and Methods
4.1. Animal Study and Biochemical Analysis
4.2. CK-MB and LDH Assay
4.3. Iron Content in Tissue
4.4. Cytokine Estimation
4.5. Quantitative Real-Time PCR
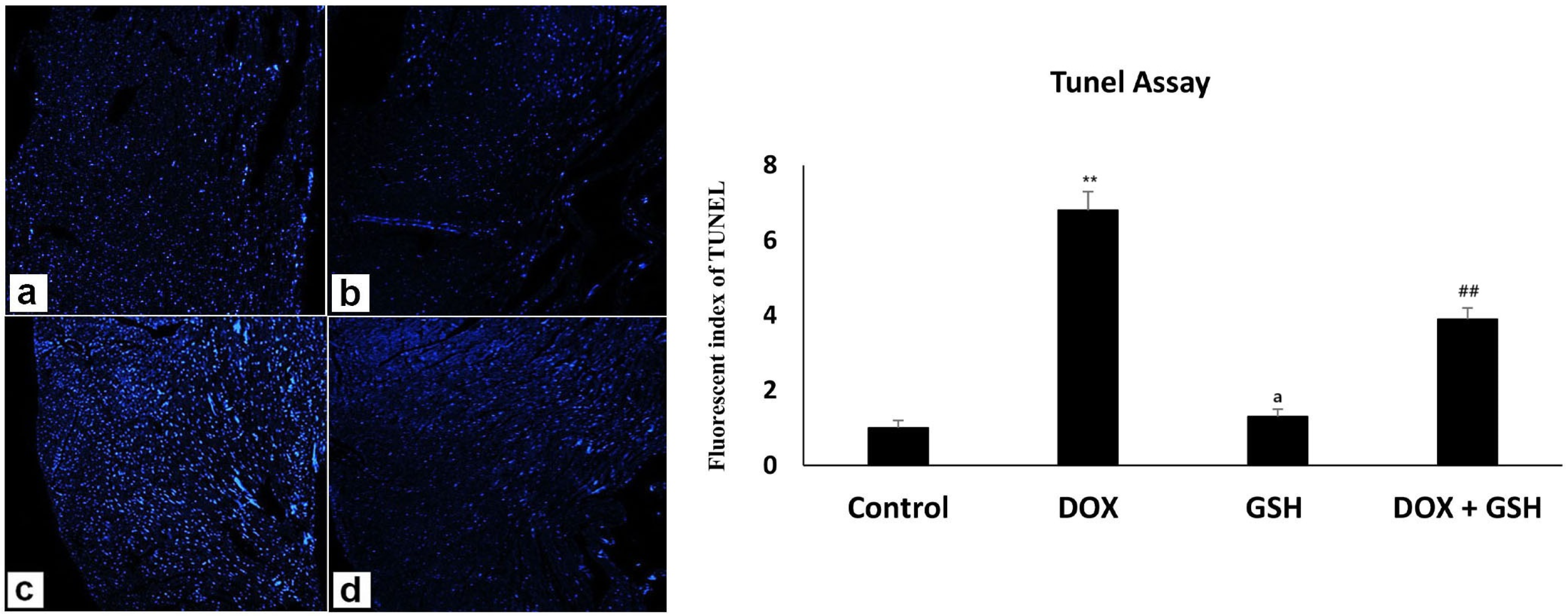
4.6. Microscopic Examination (H and E Staining) and TUNEL Assay
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenetics Genom. 2011, 21, 440–446. [Google Scholar]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [PubMed]
- Mobaraki, M.; Faraji, A.; Zare, M.; Dolati, P.; Ataei, M.; Manshadi, H.D. Molecular mechanisms of cardiotoxicity: A review on major side-effect of doxorubicin. Indian J Pharm Sci 2017, 79, 335–344. [Google Scholar]
- Benjanuwattra, J.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Doxorubicin and its proarrhythmic effects: A comprehensive review of the evidence from experimental and clinical studies. Pharmacol. Res. 2020, 151, 104542. [Google Scholar] [PubMed]
- Sangweni, N.F.; van Vuuren, D.; Mabasa, L.; Gabuza, K.; Huisamen, B.; Naidoo, S.; Barry, R.; Johnson, R. Prevention of anthracycline-induced cardiotoxicity: The good and bad of current and alternative therapies. Front. Cardiovasc. Med. 2022, 9, 907266. [Google Scholar]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. IJC Heart Vasc. 2016, 10, 17–24. [Google Scholar]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun. Signal. 2023, 21, 61. [Google Scholar]
- Kalyanaraman, B.; Joseph, J.; Kalivendi, S.; Wang, S.; Konorev, E.; Kotamraju, S. Doxorubicin-induced apoptosis: Implications in cardiotoxicity. Mol. Cell. Biochem. 2002, 234, 119–124. [Google Scholar]
- Guven, C.; Sevgiler, Y.; Taskin, E. Mitochondrial dysfunction associated with doxorubicin. In Mitochondrial Diseases; IntechOpen: London, UK, 2018; pp. 323–360. [Google Scholar]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin induces endotheliotoxicity and mitochondrial dysfunction via ROS/eNOS/NO pathway. Front. Pharmacol. 2020, 10, 1531. [Google Scholar]
- Elblehi, S.S.; El-Sayed, Y.S.; Soliman, M.M.; Shukry, M. Date palm pollen extract avert doxorubicin-induced cardiomyopathy fibrosis and associated oxidative/nitrosative stress, inflammatory cascade, and apoptosis-targeting Bax/Bcl-2 and Caspase-3 signaling pathways. Animals 2021, 11, 886. [Google Scholar] [CrossRef]
- Jones, I.C.; Dass, C.R. Doxorubicin-induced cardiotoxicity: Causative fators and possible interventions. J. Pharm. Pharmacol. 2022, 74, 1677–1688. [Google Scholar]
- Yu, J.; Wang, C.; Kong, Q.; Wu, X.; Lu, J.-J.; Chen, X. Recent progress in doxorubicin-induced cardiotoxicity and protective potential of natural products. Phytomedicine 2018, 40, 125–139. [Google Scholar]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar]
- Koss-Mikołajczyk, I.; Todorovic, V.; Sobajic, S.; Mahajna, J.; Gerić, M.; Tur, J.A.; Bartoszek, A. Natural products counteracting cardiotoxicity during cancer chemotherapy: The special case of doxorubicin, a comprehensive review. Int. J. Mol. Sci. 2021, 22, 10037. [Google Scholar] [CrossRef] [PubMed]
- Cadeddu Dessalvi, C.; Deidda, M.; Noto, A.; Madeddu, C.; Cugusi, L.; Santoro, C.; López-Fernández, T.; Galderisi, M.; Mercuro, G. Antioxidant approach as a cardioprotective strategy in chemotherapy-induced cardiotoxicity. Antioxid. Redox Signal. 2021, 34, 572–588. [Google Scholar] [PubMed]
- Yousefian, M.; Hosseinzadeh, H.; Hayes, A.W.; Hadizadeh, F.; Karimi, G. The protective effect of natural compounds on doxorubicin-induced cardiotoxicity via nicotinamide adenine dinucleotide phosphate oxidase inhibition. J. Pharm. Pharmacol. 2022, 74, 351–359. [Google Scholar]
- Yarmohammadi, F.; Hesari, M.; Shackebaei, D. The role of mTOR in doxorubicin-altered cardiac metabolism: A promising therapeutic target of natural compounds. Cardiovasc. Toxicol. 2024, 24, 146–157. [Google Scholar]
- Szponar, J.; Niziński, P.; Dudka, J.; Kasprzak-Drozd, K.; Oniszczuk, A. Natural products for preventing and managing anthracycline-induced cardiotoxicity: A comprehensive review. Cells 2024, 13, 1151. [Google Scholar] [CrossRef]
- Mohamad, E.A.; Ahmed, S.M.; Masoud, M.A.; Mohamed, F.A.; Mohammed, H.S. Cardioprotective potential of moringa oleifera leaf extract loaded niosomes nanoparticles-against doxorubicin toxicity in rats. Curr. Pharm. Biotechnol. 2025, 26, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Luo, H.; Li, W.; Ling, G.; Wei, Y.; Wang, W.; Wang, Y. The Dual Function of Autophagy in Doxorubicin-induced Cardiotoxicity: Mechanism and Natural products. Semin. Cancer Biol. 2025, 109, 83–90. [Google Scholar]
- Averill-Bates, D.A. The antioxidant glutathione. In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2023; Volume 121, pp. 109–141. [Google Scholar]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxidative Med. Cell. Longev. 2019, 2019, 5028181. [Google Scholar]
- Chen, T.-H.; Wang, H.-C.; Chang, C.-J.; Lee, S.-Y. Mitochondrial glutathione in cellular redox homeostasis and disease manifestation. Int. J. Mol. Sci. 2024, 25, 1314. [Google Scholar] [CrossRef] [PubMed]
- Corso, C.R.; Acco, A. Glutathione system in animal model of solid tumors: From regulation to therapeutic target. Crit. Rev. Oncol. /Hematol. 2018, 128, 43–57. [Google Scholar]
- Tan, M.; Yin, Y.; Ma, X.; Zhang, J.; Pan, W.; Tan, M.; Zhao, Y.; Yang, T.; Jiang, T.; Li, H. Glutathione system enhancement for cardiac protection: Pharmacological options against oxidative stress and ferroptosis. Cell Death Dis. 2023, 14, 131. [Google Scholar]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar]
- Gould, R.L.; Pazdro, R. Impact of supplementary amino acids, micronutrients, and overall diet on glutathione homeostasis. Nutrients 2019, 11, 1056. [Google Scholar] [CrossRef]
- Atmaca, G. Antioxidant effects of sulfur-containing amino acids. Yonsei Med. J. 2004, 45, 776–788. [Google Scholar]
- Wierzbicka, G.T.; Hagen, T.M.; Tones, D.P. Glutathione in food. J. Food Compos. Anal. 1989, 2, 327–337. [Google Scholar]
- Wu, G.; Lupton, J.R.; Turner, N.D.; Fang, Y.-Z.; Yang, S. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in glutathione content in liver diseases: An update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef]
- Alhowail, A.; Almogbel, Y. Metformin administration increases the survival rate of doxorubicin-treated mice. Die Pharm. Int. J. Pharm. Sci. 2019, 74, 737–739. [Google Scholar]
- To, H.; Ohdo, S.; Shin, M.; Uchimaru, H.; Yukawa, E.; Higuchi, S.; Fujimura, A.; Kobayashi, E. Dosing time dependency of doxorubicin-induced cardiotoxicity and bone marrow toxicity in rats. J. Pharm. Pharmacol. 2003, 55, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Biondo, L.A.; Lima Junior, E.A.; Souza, C.O.; Cruz, M.M.; Cunha, R.D.; Alonso-Vale, M.I.; Oyama, L.M.; Nascimento, C.M.O.; Pimentel, G.D.; Dos Santos, R.V. Impact of doxorubicin treatment on the physiological functions of white adipose tissue. PLoS ONE 2016, 11, e0151548. [Google Scholar] [CrossRef]
- Kenar, Z.Ç.; Ertan, O. Effects of Doxorubicin Administration at Different Doses and Durations on the Body Weight of Rats. Res. Pract. Vet. Anim. Sci. 2025, 2, 49–54. [Google Scholar]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Tangudu, N.K.; Alan, B.; Vinchi, F.; Wörle, K.; Lai, D.; Vettorazzi, S.; Leopold, K.; Vujić Spasić, M. Scavenging reactive oxygen species production normalizes ferroportin expression and ameliorates cellular and systemic iron disbalances in hemolytic mouse model. Antioxid. Redox Signal. 2018, 29, 484–499. [Google Scholar] [CrossRef]
- Mischley, L.K.; Vespignani, M.F.; Finnell, J.S. Safety survey of intranasal glutathione. J. Altern. Complement. Med. 2013, 19, 459–463. [Google Scholar] [CrossRef]
- Mouli, S.; Nanayakkara, G.; AlAlasmari, A.; Eldoumani, H.; Fu, X.; Berlin, A.; Lohani, M.; Nie, B.; Arnold, R.D.; Kavazis, A. The role of frataxin in doxorubicin-mediated cardiac hypertrophy. Am. J. Physiol. -Heart Circ. Physiol. 2015, 309, H844–H859. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Bai, N.; Zheng, M.; Wang, Y.; Wang, Y.; Zhang, L.; Li, J.; Li, G.; Zhao, H.; Liu, G. Tranilast prevents doxorubicin-induced myocardial hypertrophy and angiotensin II synthesis in rats. Life Sci. 2021, 267, 118984. [Google Scholar] [CrossRef]
- Adwas, A.A.; Azab, A.E.; Adous, A.A.; Adous, M.A. Targeting Oxidative Stress: A Comparative Study on the Effects of Doxorubicin on Antioxidant Enzymes Activities in Heart and Tumor Tissues in Mice. Int. J. Community Health Med. Res. 2024, 8, 70–81. [Google Scholar]
- Ferreira, A.d.A.; Matsubara, L.S.; Matsubara, B.B. Anthracycline—induced cardiotoxicity. Cardiovasc. Hematol. Agents Med. Chem. 2008, 6, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Levis, B.E.; Binkley, P.F.; Shapiro, C.L. Cardiotoxic effects of anthracycline-based therapy: What is the evidence and what are the potential harms? Lancet Oncol. 2017, 18, e445–e456. [Google Scholar] [CrossRef]
- Arola, O.J.; Saraste, A.; Pulkki, K.; Kallajoki, M.; Parvinen, M.; Voipio-Pulkki, L.-M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000, 60, 1789–1792. [Google Scholar]
- Xu, M.F.; Tang, P.L.; Qian, Z.M.; Ashraf, M. Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sci. 2001, 68, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Guo, T.; Wang, Z.; Zhao, Y. The role of iron in doxorubicin-induced cardiotoxicity: Recent advances and implication for drug delivery. J. Mater. Chem. B 2021, 9, 4793–4803. [Google Scholar]
- Han, H.; Li, J.; Santos, H.A. Recent advances in Fenton and Fenton-like reaction mediated nanoparticle in cancer therapy. Biomed. Technol. 2023, 3, 40–51. [Google Scholar] [CrossRef]
- Gumilar, K.E.; Priangga, B.; Lu, C.H.; Dachlan, E.G.; Tan, M. Iron metabolism and ferroptosis: A pathway for understanding preeclampsia. Biomed Pharmacother. 2023, 167, 115565. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Ikura, H.; Moriyama, H.; Shirakawa, K.; Katsumata, Y.; Sano, M. Therapeutic targets for DOX-induced cardiomyopathy: Role of apoptosis vs. ferroptosis. Int. J. Mol. Sci. 2022, 23, 1414. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Zhang, S.; Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 2021, 11, 3052–3059. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [PubMed]
- Chatterjee, S. Oxidative stress, inflammation, and disease. In Oxidative Stress and Biomaterials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 35–58. [Google Scholar]
- Yang, Y.; Pan, Y.; Liu, B.; Zhang, Y.; Yin, C.; Wang, J.; Nie, H.; Xu, R.; Tai, Y.; He, X. Neutrophil-derived oxidative stress contributes to skin inflammation and scratching in a mouse model of allergic contact dermatitis via triggering pro-inflammatory cytokine and pruritogen production in skin. Biochem. Pharmacol. 2024, 223, 116163. [Google Scholar]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Q.; Huang, Y.; Yang, Z.; Li, G.; Sun, X.; Gu, X.; Qiao, Y.; Wu, Q.; Xie, T. Natural products for enhancing the sensitivity or decreasing the adverse effects of anticancer drugs through regulating the redox balance. Chin. Med. 2024, 19, 110. [Google Scholar] [PubMed]
- Reis-Mendes, A.; Ferreira, M.; Padrão, A.I.; Duarte, J.A.; Duarte-Araújo, M.; Remião, F.; Carvalho, F.; Sousa, E.; Bastos, M.L.; Costa, V.M. The Role of Nrf2 and Inflammation on the Dissimilar Cardiotoxicity of Doxorubicin in Two-Time Points: A Cardio-Oncology In Vivo Study Through Time. Inflammation 2024, 47, 264–284. [Google Scholar]
- Lu, C.; Wei, J.; Gao, C.; Sun, M.; Dong, D.; Mu, Z. Molecular signaling pathways in doxorubicin-induced nephrotoxicity and potential therapeutic agents. Int. Immunopharmacol. 2025, 144, 113373. [Google Scholar]
- Kim, D.-S.; Chae, S.-W.; Kim, H.-R.; Chae, H.-J. CO and bilirubin inhibit doxorubicin-induced cardiac cell death. Immunopharmacol. Immunotoxicol. 2009, 31, 64–70. [Google Scholar]
- Lee, E.J.; Jang, W.B.; Choi, J.; Lim, H.J.; Park, S.; Rethineswaran, V.K.; Ha, J.S.; Yun, J.; Hong, Y.J.; Choi, Y.J.; et al. The Protective Role of Glutathione against Doxorubicin-Induced Cardiotoxicity in Human Cardiac Progenitor Cells. Int. J. Mol. Sci. 2023, 24, 12070. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Esworthy, R.S.; Chu, F.F. Control of doxorubicin-induced, reactive oxygen-related apoptosis by glutathione peroxidase 1 in cardiac fibroblasts. Biochem. Biophys. Rep. 2020, 21, 100709. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Pan, J.-A.; Zhang, H.; Lin, H.; Gao, L.; Zhang, H.-L.; Zhang, J.-F.; Wang, C.-Q.; Gu, J. Irisin ameliorates doxorubicin-induced cardiac perivascular fibrosis through inhibiting endothelial-to-mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021, 46, 102120. [Google Scholar]
- AlQahtani, A.A.; Osman, A.-M.M.; Damanhouri, Z.A.; Al-Kreathy, H.M.; Al-Malky, H.S.; Ramadan, W.S.; Alharthi, S.E.; Kamel, F.O. Cardioprotective effect of marine Astaxanthin on doxorubicin-induced Cardiotoxicity in Normal rats. J. Pharm. Res. Int. 2019, 27, 1–11. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, F.; Huang, L.; Ling, Y.; Xing, R.; Lu, J.; Nie, H. ITGB4/BNIP3 Activates Autophagy and Reduces MHC-I Expression to Mediate Tumour Immune Escape in Pancreatic Cancer Cell Lines. Immunology 2025, 174, 264–277. [Google Scholar] [PubMed]
- He, F. Bradford protein assay. Bio-Protoc. 2011, 101, e45. [Google Scholar]
- Flores, S.E.; Day, A.S.; Keenan, J.I. Measurement of total iron in Helicobacter pylori-infected gastric epithelial cells. Biometals 2015, 28, 143–150. [Google Scholar] [PubMed]
- Shukla, P.; Mansoori, M.N.; Singh, D. Efficacy of anti-IL-23 monotherapy versus combination therapy with anti-IL-17 in estrogen deficiency induced bone loss conditions. Bone 2018, 110, 84–95. [Google Scholar]
- Hu, Z.; Luo, D.; Wang, D.; Ma, L.; Zhao, Y.; Li, L. IL-17 activates the IL-6/STAT3 signal pathway in the proliferation of hepatitis B virus-related hepatocellular carcinoma. Cell. Physiol. Biochem. 2017, 43, 2379–2390. [Google Scholar]
- Sun, G.; Chan, S.Y.; Yuan, Y.; Chan, K.W.; Qiu, G.; Sun, K.; Leung, M.P. Isolation of differentially expressed genes in human heart tissues. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2002, 1588, 241–246. [Google Scholar]
- Bai, Y.; Chen, Q.; Sun, Y.P.; Wang, X.; Lv, L.; Zhang, L.P.; Liu, J.S.; Zhao, S.; Wang, X.L. Sulforaphane protection against the development of doxorubicin-induced chronic heart failure is associated with Nrf2 Upregulation. Cardiovasc. Ther. 2017, 35, e12277. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Batkai, S.; Kashiwaya, Y.; Hasko, G.; Liaudet, L.; Szabo, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| STAT-3 | AGGAGTCTAACAACGGCAGCCT | GTGGTACACCTCAGTCTCGAAG |
| NRF-2 | GGCAACAGTAGCCACATTGGCT | GTCTGGATGGTCATTTCACCGC |
| NFκB | GCTGCCAAAGAAGGACACGACA | GGCAGGCTATTGCTCATCACAG |
| HO-1 | CCAGGCAGAGAATGCTGAGTTC | AAGACTGGGCTCTCCTTGTTGC |
| GAPDH | CATCACTGCCACCCAGAAGACTG | ATGCCAGTGAGCTTCCCGTTCAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negm, A.; Mersal, E.A.; Dawood, A.F.; Abd El-Azim, A.O.; Hasan, O.; Alaqidi, R.; Alotaibi, A.; Alshahrani, M.; Alheraiz, A.; Shawky, T.M. Multifaceted Cardioprotective Potential of Reduced Glutathione Against Doxorubicin-Induced Cardiotoxicity via Modulating Inflammation–Oxidative Stress Axis. Int. J. Mol. Sci. 2025, 26, 3201. https://doi.org/10.3390/ijms26073201
Negm A, Mersal EA, Dawood AF, Abd El-Azim AO, Hasan O, Alaqidi R, Alotaibi A, Alshahrani M, Alheraiz A, Shawky TM. Multifaceted Cardioprotective Potential of Reduced Glutathione Against Doxorubicin-Induced Cardiotoxicity via Modulating Inflammation–Oxidative Stress Axis. International Journal of Molecular Sciences. 2025; 26(7):3201. https://doi.org/10.3390/ijms26073201
Chicago/Turabian StyleNegm, Amr, Ezat A. Mersal, Amal F. Dawood, Amira O. Abd El-Azim, Omar Hasan, Rayan Alaqidi, Ahmed Alotaibi, Mohammed Alshahrani, Abdullah Alheraiz, and Tamer M. Shawky. 2025. "Multifaceted Cardioprotective Potential of Reduced Glutathione Against Doxorubicin-Induced Cardiotoxicity via Modulating Inflammation–Oxidative Stress Axis" International Journal of Molecular Sciences 26, no. 7: 3201. https://doi.org/10.3390/ijms26073201
APA StyleNegm, A., Mersal, E. A., Dawood, A. F., Abd El-Azim, A. O., Hasan, O., Alaqidi, R., Alotaibi, A., Alshahrani, M., Alheraiz, A., & Shawky, T. M. (2025). Multifaceted Cardioprotective Potential of Reduced Glutathione Against Doxorubicin-Induced Cardiotoxicity via Modulating Inflammation–Oxidative Stress Axis. International Journal of Molecular Sciences, 26(7), 3201. https://doi.org/10.3390/ijms26073201