
RBM17 Promotes the Chemoresistance of Oral Squamous Cancer Cells Through Checkpoint Kinase 1

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
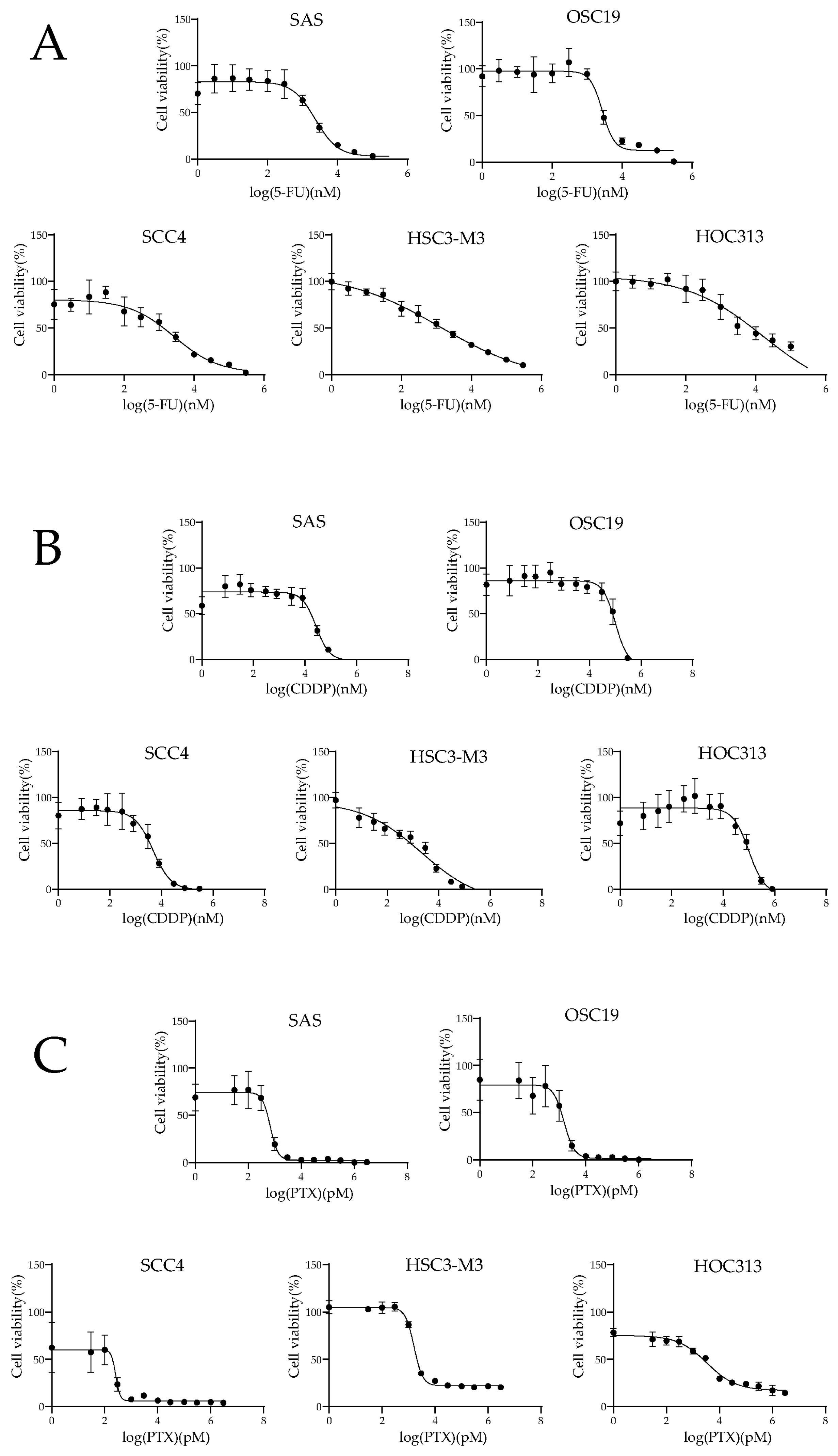
2.1. Inhibitory Effects of OSCC Cell Lines for 5-FU, CDDP, and PTX
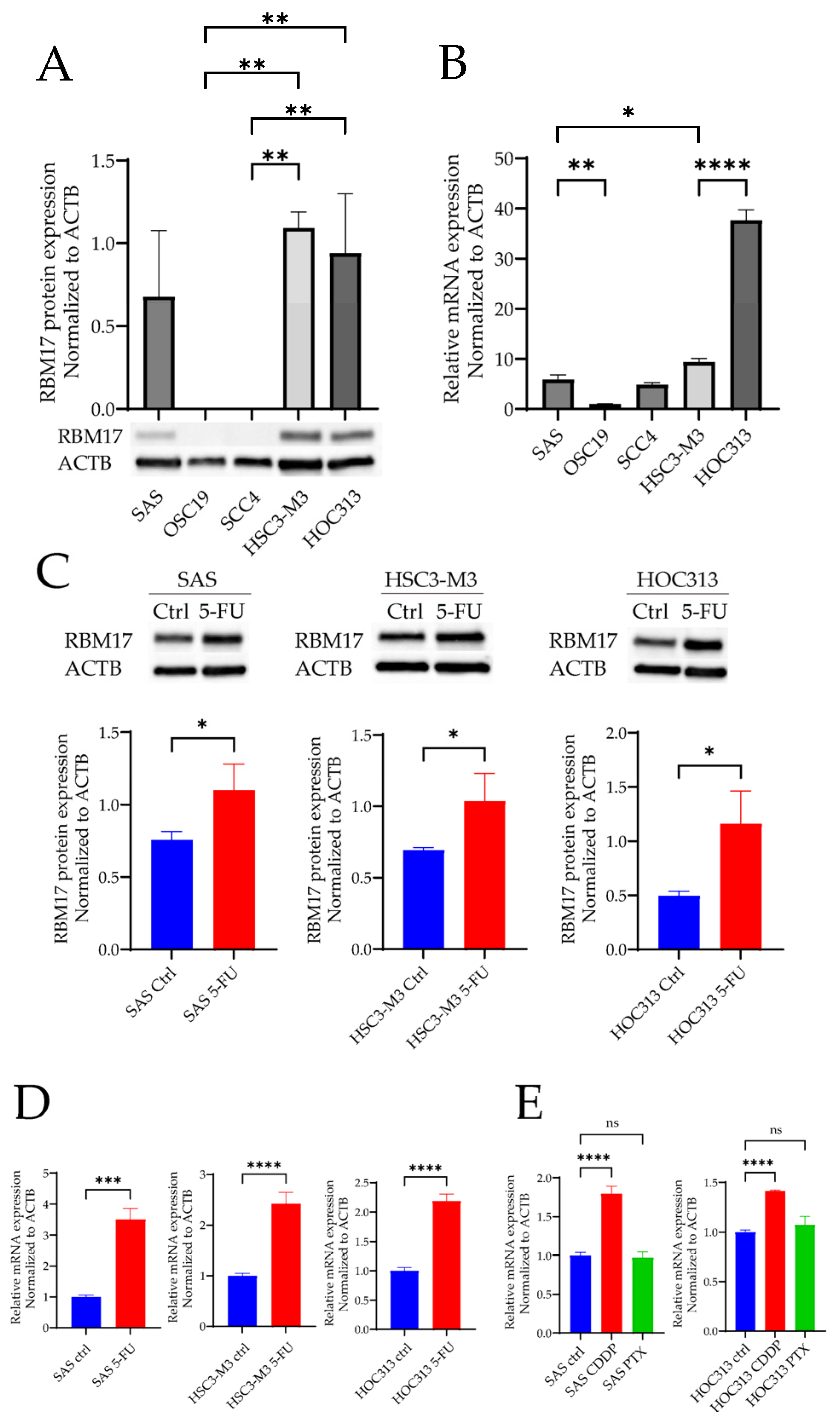
2.2. RBM17 mRNA and Protein Expressions After Cytotoxic Anticancer Drug Administration
2.3. Changes in 5-FU Drug Sensitivity After RBM17 Gene Knockdown
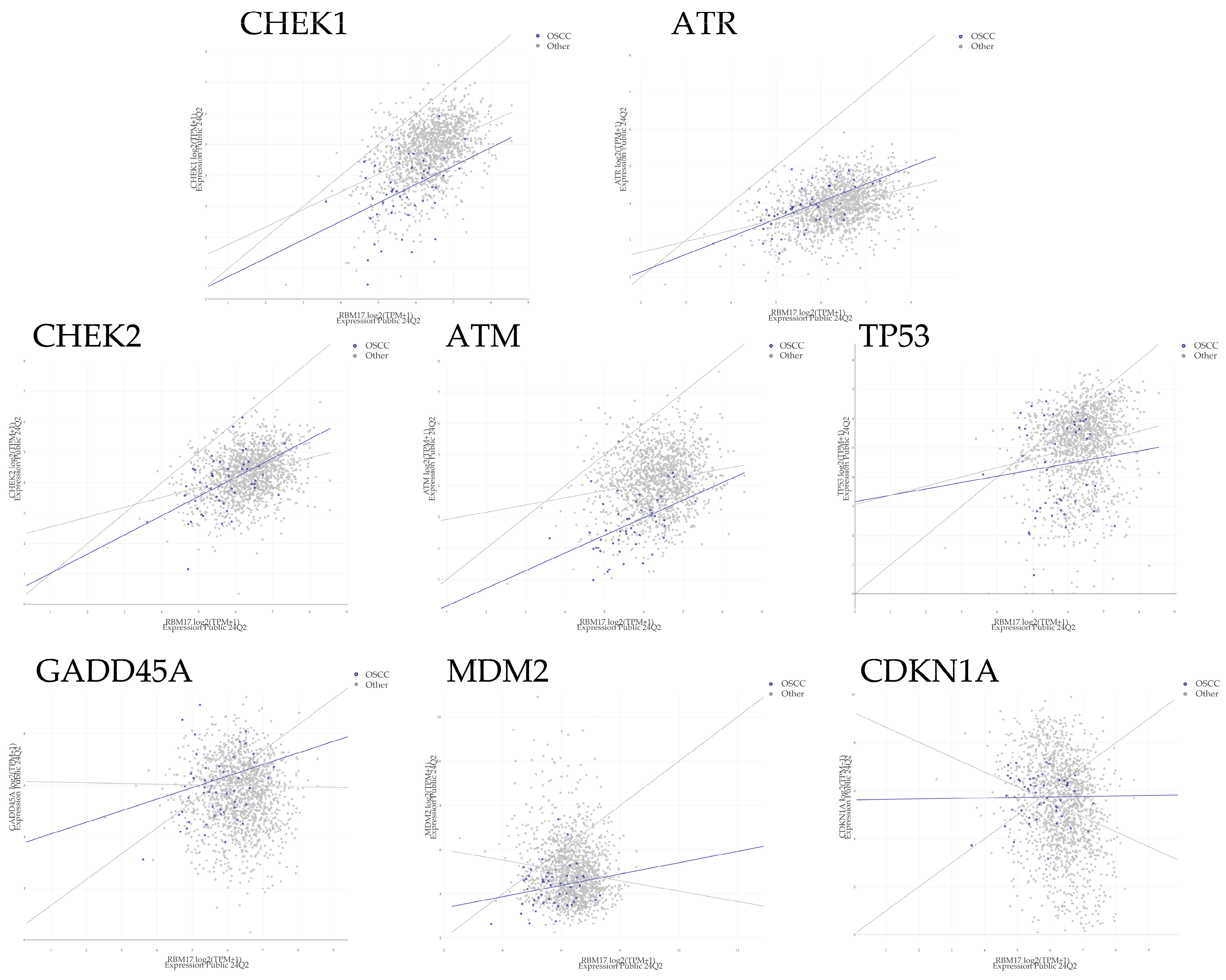
2.4. The Relationship Between RBM17 and CHEK1 Protein Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Assay for Measuring IC50 Values
4.3. RNA Extraction and RT-PCR
4.4. Protein Extraction and Western Blotting Analysis
4.5. siRNAs and Transfection
4.6. Proliferation Assay
4.7. MTT Assay for Chemosensitivity
4.8. Bioinformatics
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badwelan, M.; Muaddi, H.; Ahmed, A.; Lee, K.T.; Tran, S.D. Oral Squamous Cell Carcinoma and Concomitant Primary Tumors, What Do We Know? A Review of the Literature. Curr. Oncol. 2023, 30, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- The Global Cancer Observatory; IARC; WHO. Lip, Oral Cavity Cancer. Available online: https://gco.iarc.who.int/media/globocan/factsheets/cancers/1-lip-oral-cavity-fact-sheet.pdf (accessed on 2 August 2024).
- Dhar, H.; Vaish, R.; D’Cruz, A.K. Management of locally advanced oral cancers. Oral Oncol. 2020, 105, 104662. [Google Scholar] [CrossRef] [PubMed]
- Marta, G.N.; Riera, R.; Bossi, P.; Zhong, L.P.; Licitra, L.; Macedo, C.R.; de Castro Junior, G.; Carvalho, A.L.; William, W.N., Jr.; Kowalski, L.P. Induction chemotherapy prior to surgery with or without postoperative radiotherapy for oral cavity cancer patients: Systematic review and meta-analysis. Eur. J. Cancer 2015, 51, 2596–2603. [Google Scholar] [CrossRef]
- Bernier, J.; Cooper, J.S.; Pajak, T.F.; van Glabbeke, M.; Bourhis, J.; Forastiere, A.; Ozsahin, E.M.; Jacobs, J.R.; Jassem, J.; Ang, K.K.; et al. Defining risk levels in locally advanced head and neck cancers: A comparative analysis of concurrent postoperative radiation plus chemotherapy trials of the EORTC (#22931) and RTOG (#9501). Head Neck 2005, 27, 843–850. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Neubauer, G.; King, A.; Rappsilber, J.; Calvio, C.; Watson, M.; Ajuh, P.; Sleeman, J.; Lamond, A.; Mann, M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet. 1998, 20, 46–50. [Google Scholar] [CrossRef]
- Corsini, L.; Bonnal, S.; Basquin, J.; Hothorn, M.; Scheffzek, K.; Valcarcel, J.; Sattler, M. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat. Struct. Mol. Biol. 2007, 14, 620–629. [Google Scholar] [CrossRef]
- Fukumura, K.; Yoshimoto, R.; Sperotto, L.; Kang, H.S.; Hirose, T.; Inoue, K.; Sattler, M.; Mayeda, A. SPF45/RBM17-dependent, but not U2AF-dependent, splicing in a distinct subset of human short introns. Nat. Commun. 2021, 12, 4910. [Google Scholar] [CrossRef]
- Fukumura, K.; Sperotto, L.; Seuss, S.; Kang, H.S.; Yoshimoto, R.; Sattler, M.; Mayeda, A. SAP30BP interacts with RBM17/SPF45 to promote splicing in a subset of human short introns. Cell Rep. 2023, 42, 113534. [Google Scholar] [CrossRef]
- Sampath, J.; Long, P.R.; Shepard, R.L.; Xia, X.; Devanarayan, V.; Sandusky, G.E.; Perry, W.L., 3rd; Dantzig, A.H.; Williamson, M.; Rolfe, M.; et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am. J. Pathol. 2003, 163, 1781–1790. [Google Scholar] [CrossRef]
- Wang, X.; Chen, D.; Han, G.; Wang, X.; Liu, X.; Xu, B.; Liu, W.; Li, H.; Zhang, M.; Ma, S.; et al. Downregulation of RBM17 enhances cisplatin sensitivity and inhibits cell invasion in human hypopharyngeal cancer cells. Open Med. 2023, 18, 20230669. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, K.; Venables, J.P.; Mayeda, A. SPF45/RBM17-dependent splicing and multidrug resistance to cancer chemotherapy. Mol. Cell Oncol. 2021, 8, 1996318. [Google Scholar] [CrossRef] [PubMed]
- Perry, W.L., 3rd; Shepard, R.L.; Sampath, J.; Yaden, B.; Chin, W.W.; Iversen, P.W.; Jin, S.; Lesoon, A.; O’Brien, K.A.; Peek, V.L.; et al. Human splicing factor SPF45 (RBM17) confers broad multidrug resistance to anticancer drugs when overexpressed--a phenotype partially reversed by selective estrogen receptor modulators. Cancer Res. 2005, 65, 6593–6600. [Google Scholar] [CrossRef]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Kannampuzha, S.; Gopalakrishnan, A.V. Cancer chemoresistance and its mechanisms: Associated molecular factors and its regulatory role. Med. Oncol. 2023, 40, 264. [Google Scholar] [CrossRef]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef]
- Gadhikar, M.A.; Sciuto, M.R.; Alves, M.V.; Pickering, C.R.; Osman, A.A.; Neskey, D.M.; Zhao, M.; Fitzgerald, A.L.; Myers, J.N.; Frederick, M.J. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013, 12, 1860–1873. [Google Scholar] [CrossRef]
- Itamochi, H.; Nishimura, M.; Oumi, N.; Kato, M.; Oishi, T.; Shimada, M.; Sato, S.; Naniwa, J.; Sato, S.; Kudoh, A.; et al. Checkpoint kinase inhibitor AZD7762 overcomes cisplatin resistance in clear cell carcinoma of the ovary. Int. J. Gynecol. Cancer 2014, 24, 61–69. [Google Scholar] [CrossRef]
- Lodovichi, S.; Cervelli, T.; Pellicioli, A.; Galli, A. Inhibition of DNA Repair in Cancer Therapy: Toward a Multi-Target Approach. Int. J. Mol. Sci. 2020, 21, 6684. [Google Scholar] [CrossRef]
- Kaneda, Y.; Shimamoto, H.; Matsumura, K.; Arvind, R.; Zhang, S.; Sakai, E.; Omura, K.; Tsuchida, N. Role of caspase 8 as a determinant in chemosensitivity of p53-mutated head and neck squamous cell carcinoma cell lines. J. Med. Dent. Sci. 2006, 53, 57–66. [Google Scholar] [PubMed]
- McNeely, S.; Beckmann, R.; Bence Lin, A.K. CHEK again: Revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol. Ther. 2014, 142, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Nakamura, Y.; Nagase, H.; Nakagawara, A.; Koshinaga, T.; Wada, S.; Makishima, M. Co-Inhibition of the DNA Damage Response and CHK1 Enhances Apoptosis of Neuroblastoma Cells. Int. J. Mol. Sci. 2019, 20, 3700. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Broad DepMap. Figshare+; Dataset; DepMap 24Q2 Public: Cambridge, MA, USA, 2024. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | 5-FU (µM) | CDDP (µM) | PTX (nM) |
|---|---|---|---|
| SAS | 2.29 (1.67–3.23) | 26.79 (20.28–35.48) | 0.67 (2.53–0.86) |
| OSC19 | 2.73 (2.24–3.56) | 100.00 (77.45–148.25) | 1.51 (1.13–2.03) |
| SCC4 | 2.65 (1.40–6.41) | 4.73 (3.45–6.49) | 0.26 (0.18–0.30) |
| HSC3-M3 | 1.29 (0.62–5.09) | 2.10 (0.91–5.20) | 1.61 (1.50–1.74) |
| HOC313 | 16.29 (1.86–142.89) | 98.86 (62.09–232.81) | 3.36 (2.43–4.59) |
| Gene | Group | Points | Pearson | Spearman | Slope | Intercept | p-value |
|---|---|---|---|---|---|---|---|
| CHEK1 | All | 1517 | 0.468 | 0.445 | 0.601 | 0.950 | <0.001 |
| OSCC | 49 | 0.402 | 0.396 | 0.596 | 0.133 | 0.004 | |
| Other | 1468 | 0.451 | 0.429 | 0.567 | 1.20 | <0.001 | |
| ATR | All | 1517 | 0.387 | 0.367 | 0.295 | 2.08 | <0.001 |
| OSCC | 49 | 0.631 | 0.591 | 0.475 | 1.19 | <0.001 | |
| Other | 1468 | 0.380 | 0.360 | 0.294 | 2.08 | <0.001 | |
| CHEK2 | All | 1517 | 0.328 | 0.309 | 0.337 | 2.14 | <0.001 |
| OSCC | 49 | 0.512 | 0.496 | 0.630 | 0.395 | <0.001 | |
| Other | 1468 | 0.314 | 0.299 | 0.324 | 2.22 | <0.001 | |
| ATM | All | 1517 | 0.221 | 0.182 | 0.287 | 2.30 | <0.001 |
| OSCC | 49 | 0.453 | 0.493 | 0.566 | −0.419 | 0.001 | |
| Other | 1468 | 0.181 | 0.150 | 0.231 | 2.69 | <0.001 | |
| TP53 | All | 1517 | 0.185 | 0.189 | 0.328 | 2.96 | <0.001 |
| OSCC | 49 | 0.105 | 0.104 | 0.217 | 3.16 | 0.473 | |
| Other | 1468 | 0.177 | 0.182 | 0.314 | 3.06 | <0.001 | |
| GADD45A | All | 1517 | −0.011 | −0.021 | −0.019 | 5.76 | 0.670 |
| OSCC | 49 | 0.263 | 0.244 | 0.448 | 3.32 | 0.068 | |
| Other | 1468 | −0.015 | −0.027 | −0.026 | 5.80 | 0.572 | |
| MDM2 | All | 1517 | −0.104 | −0.073 | −0.188 | 6.14 | <0.001 |
| OSCC | 49 | 0.208 | 0.157 | 0.256 | 2.85 | 0.152 | |
| Other | 1468 | −0.128 | −0.092 | −0.234 | 6.46 | <0.001 | |
| CDKN1A | All | 1517 | −0.232 | −0.230 | −0.602 | 9.13 | <0.001 |
| OSCC | 49 | 0.017 | −0.055 | 0.022 | 5.61 | 0.907 | |
| Other | 1468 | −0.234 | −0.231 | −0.622 | 9.26 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakahara, M.; Arai, R.; Tokuoka, I.; Fukumura, K.; Mayeda, A.; Yashiro, M.; Nakahara, H. RBM17 Promotes the Chemoresistance of Oral Squamous Cancer Cells Through Checkpoint Kinase 1. Int. J. Mol. Sci. 2025, 26, 3127. https://doi.org/10.3390/ijms26073127
Nakahara M, Arai R, Tokuoka I, Fukumura K, Mayeda A, Yashiro M, Nakahara H. RBM17 Promotes the Chemoresistance of Oral Squamous Cancer Cells Through Checkpoint Kinase 1. International Journal of Molecular Sciences. 2025; 26(7):3127. https://doi.org/10.3390/ijms26073127
Chicago/Turabian StyleNakahara, Miyuka, Ryosuke Arai, Isao Tokuoka, Kazuhiro Fukumura, Akila Mayeda, Masakazu Yashiro, and Hirokazu Nakahara. 2025. "RBM17 Promotes the Chemoresistance of Oral Squamous Cancer Cells Through Checkpoint Kinase 1" International Journal of Molecular Sciences 26, no. 7: 3127. https://doi.org/10.3390/ijms26073127
APA StyleNakahara, M., Arai, R., Tokuoka, I., Fukumura, K., Mayeda, A., Yashiro, M., & Nakahara, H. (2025). RBM17 Promotes the Chemoresistance of Oral Squamous Cancer Cells Through Checkpoint Kinase 1. International Journal of Molecular Sciences, 26(7), 3127. https://doi.org/10.3390/ijms26073127