
HRAS Mutations in Head and Neck Carcinomas in Japanese Patients: Clinical Significance, Prognosis, and Therapeutic Potential
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
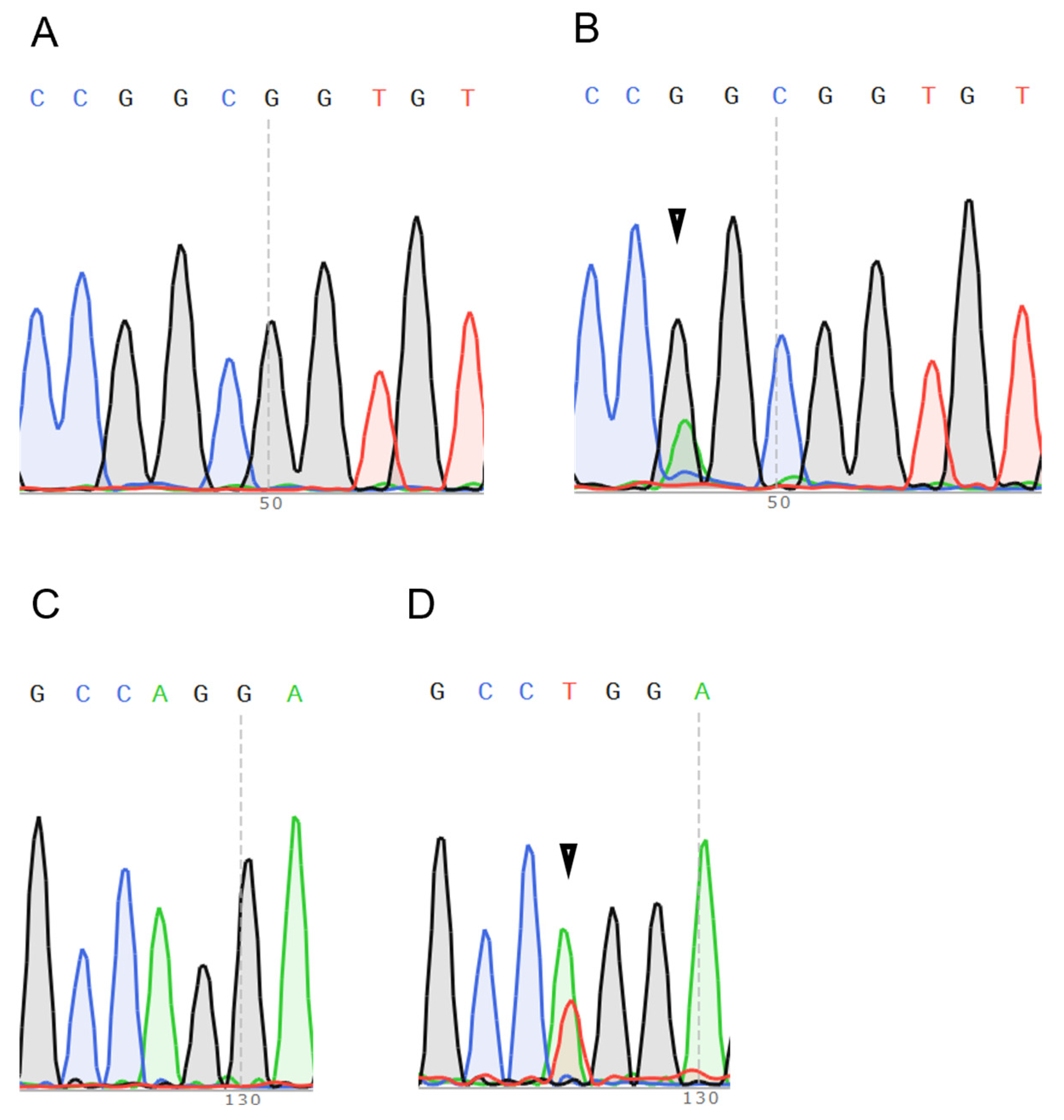
2.1. HRAS Mutations Are More Frequently Detected in Salivary Gland Carcinomas and Are Possibly Associated with Distant Metastasis
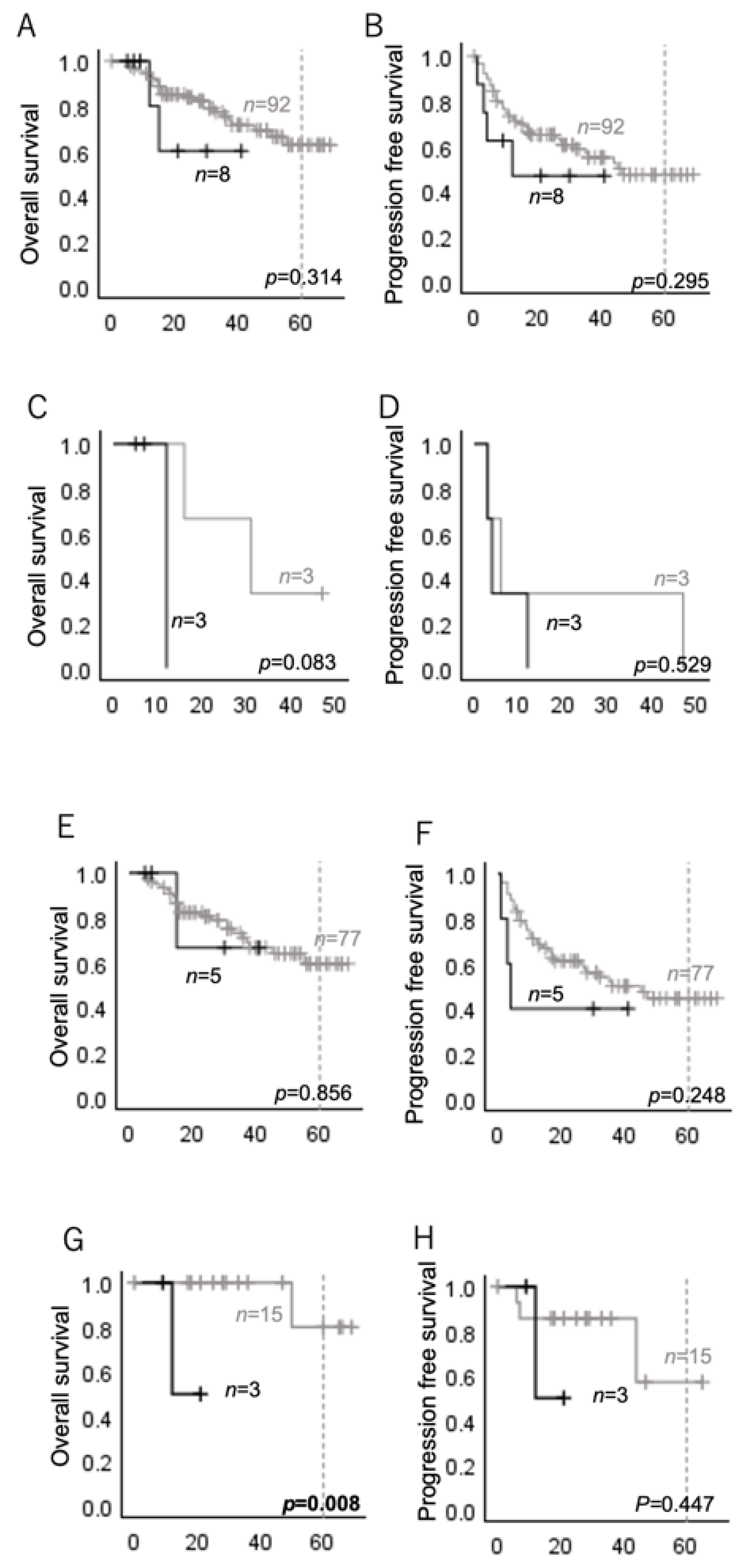
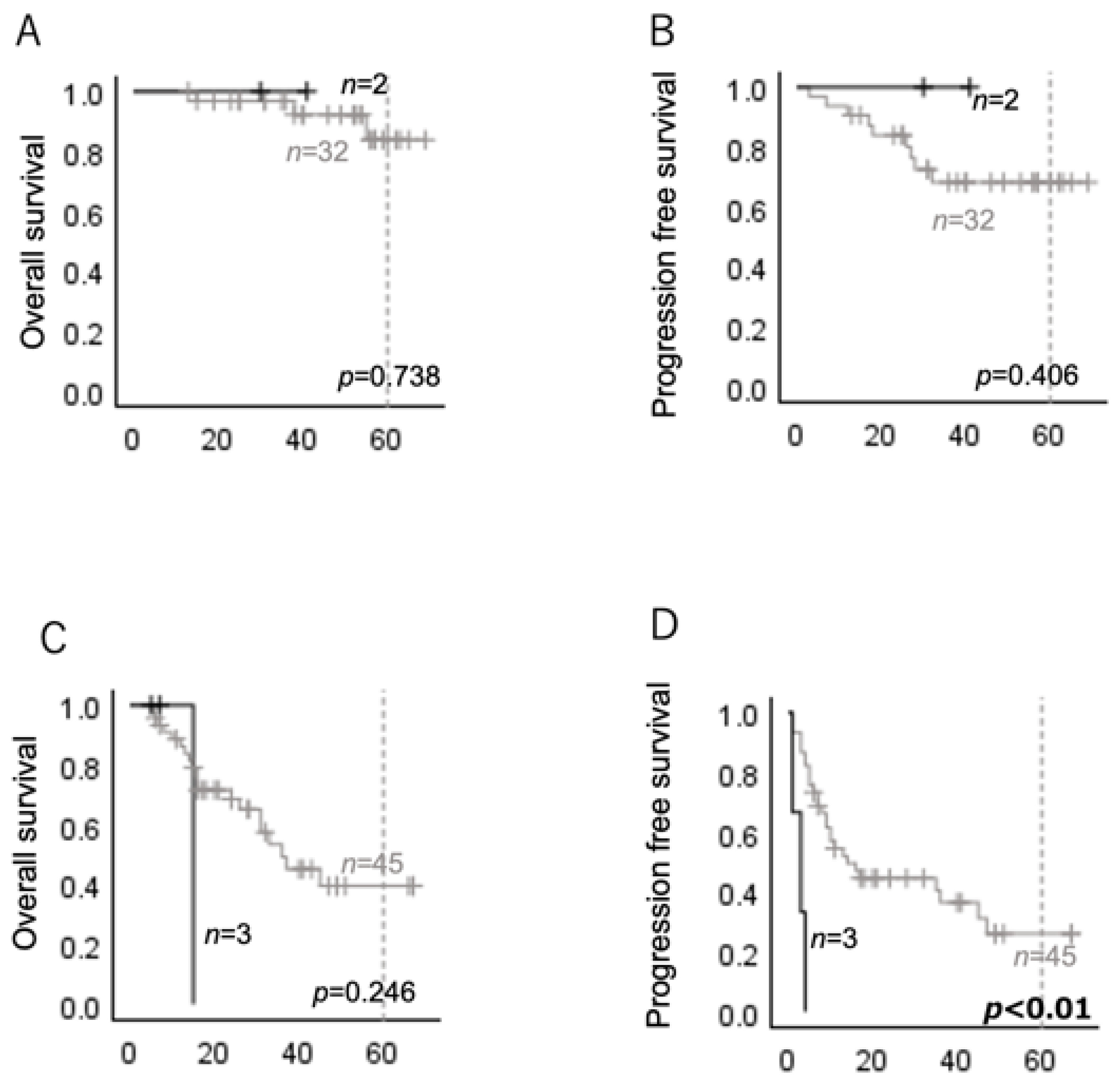
2.2. HRAS Mutations Are Associated with Poor Prognosis in Several Subgroups of Head and Neck Carcinomas
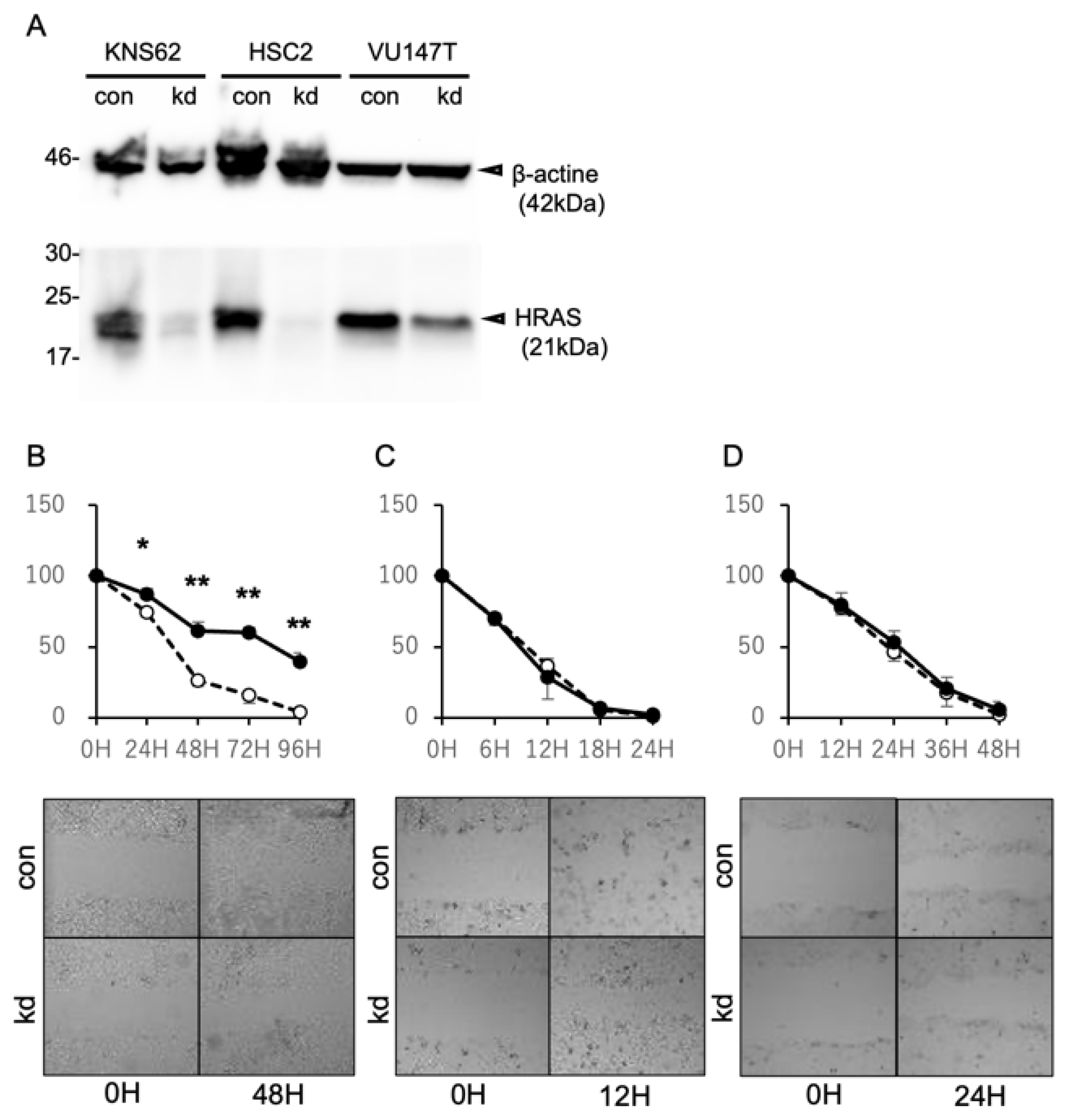
2.3. Suppression of HRAS Expression Inhibits the Cell Migration of HRAS Mutation-Positive Cells
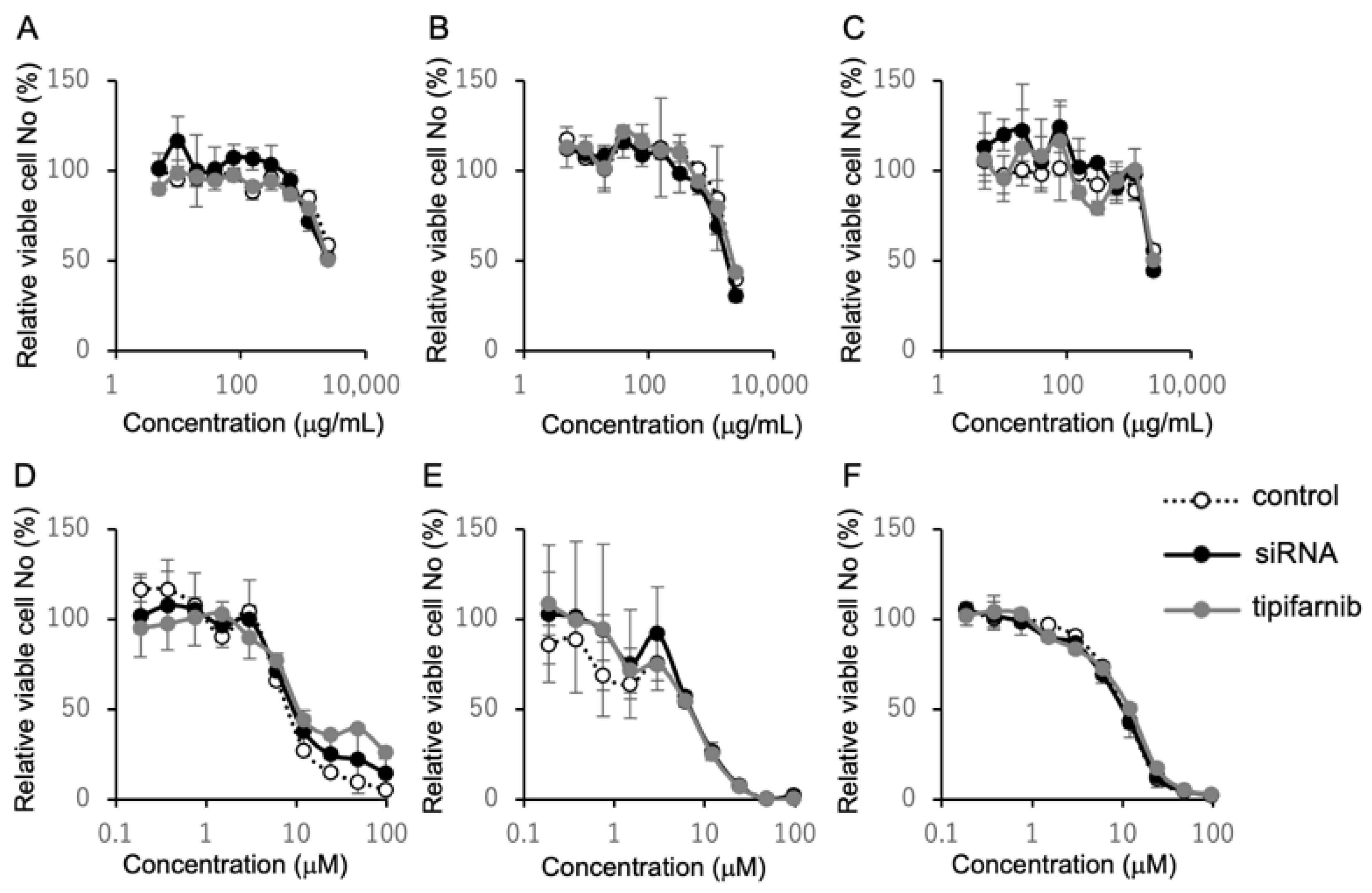
2.4. Inhibition of HRAS Does Not Affect Susceptibility to Cetuximab and Cisplatin
3. Discussion
4. Materials and Methods
4.1. Patients and Specimens
4.2. Cell Lines and Knockdown with siRNA
4.3. DNA Extraction and Detection of HRAS Mutations
4.4. Scratch Assay
4.5. MTS Assay
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, B.; Johnson, N.W.; Kumar, N. Global Epidemiology of Head and Neck Cancers: A Continuing Challenge. Oncology 2016, 91, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zolkind, P.; Lee, J.J.; Jackson, R.S.; Pipkorn, P.; Massa, S.T. Untreated head and neck cancer: Natural history and associated factors. Head Neck 2021, 43, 89–97. [Google Scholar] [CrossRef]
- Forastiere, A.A.; Zhang, Q.; Weber, R.S.; Maor, M.H.; Goepfert, H.; Pajak, T.F.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; et al. Long-term results of RTOG 91-11: A comparison of three nonsurgical treatment strategies to preserve the larynx in patients with locally advanced larynx cancer. J. Clin. Oncol. 2013, 31, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Forastiere, A.A.; Goepfert, H.; Maor, M.; Pajak, T.F.; Weber, R.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; Chao, C.; et al. Concurrent Chemotherapy and Radiotherapy for Organ Preservation in Advanced Laryngeal Cancer. New Engl. J. Med. 2003, 349, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef]
- Tibau, A.; Hwang, T.J.; Avorn, J.; Kesselheim, A.S. Clinical value of guideline recommended molecular targets and genome targeted cancer therapies: Cross sectional study. BMJ 2024, 386, e079126. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Lazar, V.; Leyland-Jones, B.; Schilsky, R.L.; Mendelsohn, J.; Kurzrock, R. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysis. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Yang, X.; Wu, H. RAS signaling in carcinogenesis, cancer therapy and resistance mechanisms. J. Hematol. Oncol. 2024, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Slebos, R.J.; Kibbelaar, R.E.; Dalesio, O.; Kooistra, A.; Stam, J.; Meijer, C.J.; Wagenaar, S.S.; Vanderschueren, R.G.; van Zandwijk, N.; Mooi, W.J.; et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N. Engl. J. Med. 1990, 323, 561–565. [Google Scholar] [CrossRef]
- Huang, S.; Ye, J.; Gao, X.; Huang, X.; Huang, J.; Lu, L.; Lu, C.; Li, Y.; Luo, M.; Xie, M.; et al. Progress of research on molecular targeted therapies for colorectal cancer. Front. Pharmacol. 2023, 14, 1160949. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Marcelo, K.L.; Hopkins, J.F.; Khan, N.I.; Du, R.; Hong, L.; Park, E.; Balsara, B.; Leoni, M.; Pickering, C.; et al. HRAS Mutations Define a Distinct Subgroup in Head and Neck Squamous Cell Carcinoma. JCO Precis. Oncol. 2023, 7, e2200211. [Google Scholar] [CrossRef]
- Stieglitz, E.; Ward, A.F.; Gerbing, R.B.; Alonzo, T.A.; Arceci, R.J.; Liu, Y.L.; Emanuel, P.D.; Widemann, B.C.; Cheng, J.W.; Jayaprakash, N.; et al. Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 629–636. [Google Scholar] [CrossRef]
- Macdonald, J.S.; McCoy, S.; Whitehead, R.P.; Iqbal, S.; Wade, J.L., 3rd; Giguere, J.K.; Abbruzzese, J.L. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: A Southwest oncology group (SWOG 9924) study. Investig. New Drugs 2005, 23, 485–487. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chilla, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma With HRAS Mutations. J. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Kareff, S.A.; Trabolsi, A.; Krause, H.B.; Samec, T.; Elliott, A.; Rodriguez, E.; Olazagasti, C.; Watson, D.C.; Bustos, M.A.; Hoon, D.S.B.; et al. The Genomic, Transcriptomic, and Immunologic Landscape of HRAS Mutations in Solid Tumors. Cancers 2024, 16, 1572. [Google Scholar] [CrossRef] [PubMed]
- Hideaki Takahashi, M.; Yuichiro Tada, M.; Takashi Saotome, M.; Kohei Akazawa, P.; Hiroya Ojiri, M.; Chihiro Fushimi, D.; Tatsuo Masubuchi, M. Phase II Trial of Trastuzumab and Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2–Positive Salivary Duct Carcinoma. J. Clin. Oncol. 2018, 37, 125–134. [Google Scholar] [CrossRef]
- Kato, S.; Elkin, S.K.; Schwaederle, M.; Tomson, B.N.; Helsten, T.; Carter, J.L.; Kurzrock, R. Genomic landscape of salivary gland tumors. Oncotarget 2015, 6, 25631. [Google Scholar] [CrossRef]
- Yoo, J.; Robinson, R.A. ras gene mutations in salivary gland tumors. Arch. Pathol. Lab. Med. 2000, 124, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Zaccarini, D.J.; Sivapiragasam, A.; Sokol, E.; Huang, R.S.P.; Pavlick, D.C.; Janovitz, T.; Nasr, M.R.; Ross, J.S. Comprehensive Molecular Profiling of Oncocytic Salivary Gland Malignancies. Appl. Immunohistochem. Mol. Morphol. 2022, 30, 609–613. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Kumar, P.A.; Zaccarini, D.; Ross, J.; Huang, R.; Sivapiragasam, A. Molecular Targets in Salivary Gland Cancers: A Comprehensive Genomic Analysis of 118 Mucoepidermoid Carcinoma Tumors. Biomedicines 2023, 11, 519. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Kobayashi, E.; Hwang, D.; Bheda-Malge, A.; Whitehurst, C.B.; Kabanov, A.V.; Kondo, S.; Aga, M.; Yoshizaki, T.; Pagano, J.S.; Sokolsky, M.; et al. Inhibition of UCH-L1 Deubiquitinating Activity with Two Forms of LDN-57444 Has Anti-Invasive Effects in Metastatic Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 3733. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, P.; Anderson, H.; Biorklund, A.; Moller, T.; Perfekt, R. Carcinoma of the parotid and submandibular glands—A study of survival in 2465 patients. Oral Oncol. 2002, 38, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Urano, M.; Nakaguro, M.; Yamamoto, Y.; Hirai, H.; Tanigawa, M.; Saigusa, N.; Shimizu, A.; Tsukahara, K.; Tada, Y.; Sakurai, K.; et al. Diagnostic Significance of HRAS Mutations in Epithelial-Myoepithelial Carcinomas Exhibiting a Broad Histopathologic Spectrum. Am. J. Surg. Pathol. 2019, 43, 984–994. [Google Scholar] [CrossRef]
- Yasuda, T.; Osaki, M.; Sugitani, M. HRAS mutation as a diagnostic molecular analysis for epithelial-myoepithelial carcinoma of the parotid gland: A case report. Clin. Case Rep. 2023, 11, e8099. [Google Scholar] [CrossRef]
- Saida, K.; Murase, T.; Ito, M.; Fujii, K.; Takino, H.; Masaki, A.; Kawakita, D.; Ijichi, K.; Tada, Y.; Kusafuka, K.; et al. Mutation analysis of the EGFR pathway genes, EGFR, RAS, PIK3CA, BRAF, and AKT1, in salivary gland adenoid cystic carcinoma. Oncotarget 2018, 9, 17043–17055. [Google Scholar] [CrossRef]
- Hanna, G.J.; Guenette, J.P.; Chau, N.G.; Sayehli, C.M.; Wilhelm, C.; Metcalf, R.; Wong, D.J.; Brose, M.; Razaq, M.; Perez-Ruiz, E.; et al. Tipifarnib in recurrent, metastatic HRAS-mutant salivary gland cancer. Cancer 2020, 126, 3972–3981. [Google Scholar] [CrossRef]
- Jagadeeshan, S.; Prasad, M.; Badarni, M.; Ben-Lulu, T.; Liju, V.B.; Mathukkada, S.; Saunders, C.; Shnerb, A.B.; Zorea, J.; Yegodayev, K.M.; et al. Mutated HRAS Activates YAP1-AXL Signaling to Drive Metastasis of Head and Neck Cancer. Cancer Res. 2023, 83, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef]
- Rieke, D.T.; Schroder, S.; Schafhausen, P.; Blanc, E.; Zuljan, E.; von der Emde, B.; Beule, D.; Keller, U.; Keilholz, U.; Klinghammer, K. Targeted treatment in a case series of AR+, HRAS/PIK3CA co-mutated salivary duct carcinoma. Front. Oncol. 2023, 13, 1107134. [Google Scholar] [CrossRef]
- Kobayashi, E.; Aga, M.; Kondo, S.; Whitehurst, C.; Yoshizaki, T.; Pagano, J.S.; Shackelford, J. C-Terminal Farnesylation of UCH-L1 Plays a Role in Transport of Epstein-Barr Virus Primary Oncoprotein LMP1 to Exosomes. mSphere 2018, 3, e00030-18. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | Mutation | p-Value | ||
|---|---|---|---|---|
| Positive | Negative | |||
| Numbers (%) | 100 | 8 (8.0) | 92 (92.0) | |
| Codon (%) | ||||
| Mutation at codon12 | 4 (50.0) | |||
| Mutation at codon13 | 0 (0.0) | |||
| Mutation at codon61 | 4 (50.0) | |||
| Male:Female | 73:27 | 3:5 | 70:22 | 0.032 * |
| Race Asian | 100 | 8 (100.0) | 92 (100.0) | |
| Age median (range) years | 70 (22–89) | 70.5 (47–82) | 70 (22–89) | 0.746 |
| Primary site (%) | 0.286 | |||
| Oral cavity | 26 (26.0) | 1 (12.5) | 25 (27.2) | |
| Oropharynx | 56 (56.0) | 4 (50.0) | 52 (56.5) | 0.726 |
| P16 positive | 34 | 2 | 32 | |
| P16 negative | 19 | 2 | 17 | |
| P16 unknown | 3 | 0 | 3 | |
| Salivary glands | 18 (18.0) | 3 (37.5) | 15 (16.3) | |
| Stage (%) 1 | 29 (29.0) | 3 (37.5) | 26 (28.3) | 0.431 |
| 2 | 24 (24.0) | 0 (0.0) | 24 (26.1) | |
| 3 | 10 (10.0) | 1 (12.5) | 9 (9.8) | |
| 4 | 37 (37.0) | 4 (50.0) | 33 (35.9) | |
| Tumor stage (%) 0 | 1 (1.0) | 1 (12.5) | 0 (0.0) | 0.008 * |
| 1 | 19 (19.0) | 0 (0.0) | 19 (20.7) | |
| 2 | 33 (33.0) | 2 (25.0) | 31 (33.7) | |
| 3 | 21 (21.0) | 2 (25.0) | 19 (25.0) | |
| 4 | 26 (26.0) | 3 (37.5) | 23 (31.5) | |
| Lymph node metastasis (%) | 60 (60.0) | 7 (87.5) | 53 (57.6) | 0.14 |
| Distant metastasis (%) | 6 (6.0) | 3 (37.5) | 3 (3.3) | 0.006 * |
| Initial treatment (%) | 64 (64.0) | 5 (62.5) | 59 (64.1) | 0.206 |
| Operation | 73 (73.0) | 4 (50.0) | 69 (75.0) | |
| Chemoradiation | 27 (27.0) | 4 (50.0) | 23 (25.0) | |
| Observation period median (range) months | 31 (0–69) | 13.5 (5–41) | 31.5 (0–69) | 0.011 * |
| Total | Mutation | p-Value | ||
|---|---|---|---|---|
| Positive | Negative | |||
| Numbers (%) | 82 | 5 (6.1) | 77 (93.9) | |
| Codon (%) | ||||
| Mutation at codon12 | 3 (60.0) | |||
| Mutation at codon13 | 0 (0.0) | |||
| Mutation at codon61 | 2 (40.0) | |||
| Male:Female | 64:18 | 2:3 | 62:15 | 0.068 |
| Race Asian | 82 | 5 (100.0) | 77 (100.0) | |
| Age median (range) years | 69.5 (23–86) | 71 (47–82) | 69 (23–86) | 0.572 |
| P16 positive | 34 (41.5) | 2 (40.0) | 32 (41.6) | 1.0 |
| Stage (%) 1 | 25 (30.5) | 2 (40.0) | 23 (29.9) | 0.379 |
| 2 | 21 (25.6) | 0 (0.0) | 21 (27.3) | |
| 3 | 8 (9.8) | 0 (0.0) | 8 (10.4) | |
| 4 | 28 (34.1) | 3 (60.0) | 25 (32.5) | |
| Tumor Stage (%) 0 | 1 (1.2) | 1 (20.0) | 0 (0.0) | 0.002 * |
| 1 | 16 (19.5) | 0 (0.0) | 16 (20.8) | |
| 2 | 29 (35.4) | 1 (20.0) | 28 (36.4) | |
| 3 | 16 (19.5) | 1 (20.0) | 15 (19.5) | |
| 4 | 20 (24.4) | 2 (40.0) | 18 (23.4) | |
| Lymph node metastasis (%) | 53 (64.6) | 5 (100) | 48 (62.3) | 0.156 |
| Distant metastasis (%) | 5 (6.1) | 2 (40.0) | 3 (3.9) | 0.028 * |
| Initial treatment (%) | 0.163 | |||
| Operation | 57 (69.5) | 2 (40.0) | 55 (71.4) | |
| Chemoradiation | 25 (30.5) | 3 (60.0) | 22 (28.6) | |
| Observation period median (range) months | 31 (5–69) | 15 (5–41) | 31 (5–69) | 0.089 |
| Total | Mutation | p-Value | ||
|---|---|---|---|---|
| Positive | Negative | |||
| Numbers (%) | 18 | 3 (16.7) | 15(83.3) | |
| Codon (%) | ||||
| Mutation at codon12 | 1 (33.3) | |||
| Mutation at codon13 | 0 (0.0) | |||
| Mutation at codon61 | 2 (66.7) | |||
| Male:Female | 9:9 | 1:2 | 8:7 | 1 |
| Race (%) Asian | 18 | 3 (100.0) | 15 (100.0) | |
| Age median (range) years | 71 (22–89) | 70 (60–72) | 72 (22–89) | 0.654 |
| Primary site (%) | 1 | |||
| Parotid gland | 15 (83.3) | 3 (20.0) | 12 (80.0) | |
| Submandibular gland | 3 (16.7) | 0 (0.0) | 3 (100.0) | |
| Sublingual gland | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Minor salivary gland | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Histopathology (%) | 0.65 | |||
| Squamous cell | 2 (11.1) | 0 (0.0) | 2 (13.3) | |
| Myoepithelial | 2 (11.1) | 1 (33.3) | 1 (6.7) | |
| Acinic cell | 2 (11.1) | 0 (0.0) | 2 (13.3) | |
| Adenoid cystic | 1 (5.6) | 0 (0.0) | 1 (6.7) | |
| Salivary duct | 3 (16.7) | 1 (33.3) | 2 (13.3) | |
| Mucoepidermoid | 4 (22.2) | 1 (33.3) | 3 (20.0) | |
| Secretory | 4 (22.2) | 0 (0.0) | 4 (26.7) | |
| Stage (%) 1 | 4 (22.2) | 1 (33.3) | 3 (20.0) | 0.457 |
| 2 | 3 (16.7) | 0 (0.0) | 3 (20.0) | |
| 3 | 2 (11.1) | 1 (33.3) | 1 (6.7) | |
| 4 | 9 (50.0) | 1 (33.3) | 8 (53.3) | |
| Tumor stage (%) 1 | 3 (16.7) | 0 (0.0) | 3 (20.0) | 0.84 |
| 2 | 4 (22.2) | 1 (33.3) | 3 (20.0) | |
| 3 | 5 (27.8) | 1 (33.3) | 4 (26.7) | |
| 4 | 6 (33.3) | 1 (33.3) | 5 (33.3) | |
| Lymph node metastasis (%) | 7 (38.9) | 2 (66.7) | 5 (33.3) | 0.528 |
| Distant metastasis (%) | 1 (5.6) | 1 (33.3) | 0 (0.0) | 0.167 |
| Initial treatment (%) | 0.314 | |||
| Operation | 16 (88.9) | 2 (66.7) | 14 (93.3) | |
| Chemoradiation | 2 (11.1) | 1 (33.3) | 1 (6.7) | |
| Observation period median (range) months | 28.5 (2–69) | 12 (9–21) | 33 (0–69) | 0.039 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohshima, H.; Kobayashi, E.; Inaba, M.; Nakazawa, R.; Hirai, N.; Ueno, T.; Nakanishi, Y.; Endo, K.; Kondo, S.; Moriyama-Kita, M.; et al. HRAS Mutations in Head and Neck Carcinomas in Japanese Patients: Clinical Significance, Prognosis, and Therapeutic Potential. Int. J. Mol. Sci. 2025, 26, 3093. https://doi.org/10.3390/ijms26073093
Ohshima H, Kobayashi E, Inaba M, Nakazawa R, Hirai N, Ueno T, Nakanishi Y, Endo K, Kondo S, Moriyama-Kita M, et al. HRAS Mutations in Head and Neck Carcinomas in Japanese Patients: Clinical Significance, Prognosis, and Therapeutic Potential. International Journal of Molecular Sciences. 2025; 26(7):3093. https://doi.org/10.3390/ijms26073093
Chicago/Turabian StyleOhshima, Hidemi, Eiji Kobayashi, Manabu Inaba, Ryotaro Nakazawa, Nobuyuki Hirai, Takayoshi Ueno, Yosuke Nakanishi, Kazuhira Endo, Satoru Kondo, Makiko Moriyama-Kita, and et al. 2025. "HRAS Mutations in Head and Neck Carcinomas in Japanese Patients: Clinical Significance, Prognosis, and Therapeutic Potential" International Journal of Molecular Sciences 26, no. 7: 3093. https://doi.org/10.3390/ijms26073093
APA StyleOhshima, H., Kobayashi, E., Inaba, M., Nakazawa, R., Hirai, N., Ueno, T., Nakanishi, Y., Endo, K., Kondo, S., Moriyama-Kita, M., Sugimoto, H., & Yoshizaki, T. (2025). HRAS Mutations in Head and Neck Carcinomas in Japanese Patients: Clinical Significance, Prognosis, and Therapeutic Potential. International Journal of Molecular Sciences, 26(7), 3093. https://doi.org/10.3390/ijms26073093