
Kumquat Fruit Administration Counteracts Dysmetabolism-Related Neurodegeneration and the Associated Brain Insulin Resistance in the High-Fat Diet-Fed Mice

 , ,
, ,  , ,
, ,  ,
, 
 ,
,  ,
, 
Abstract
1. Introduction
2. Results
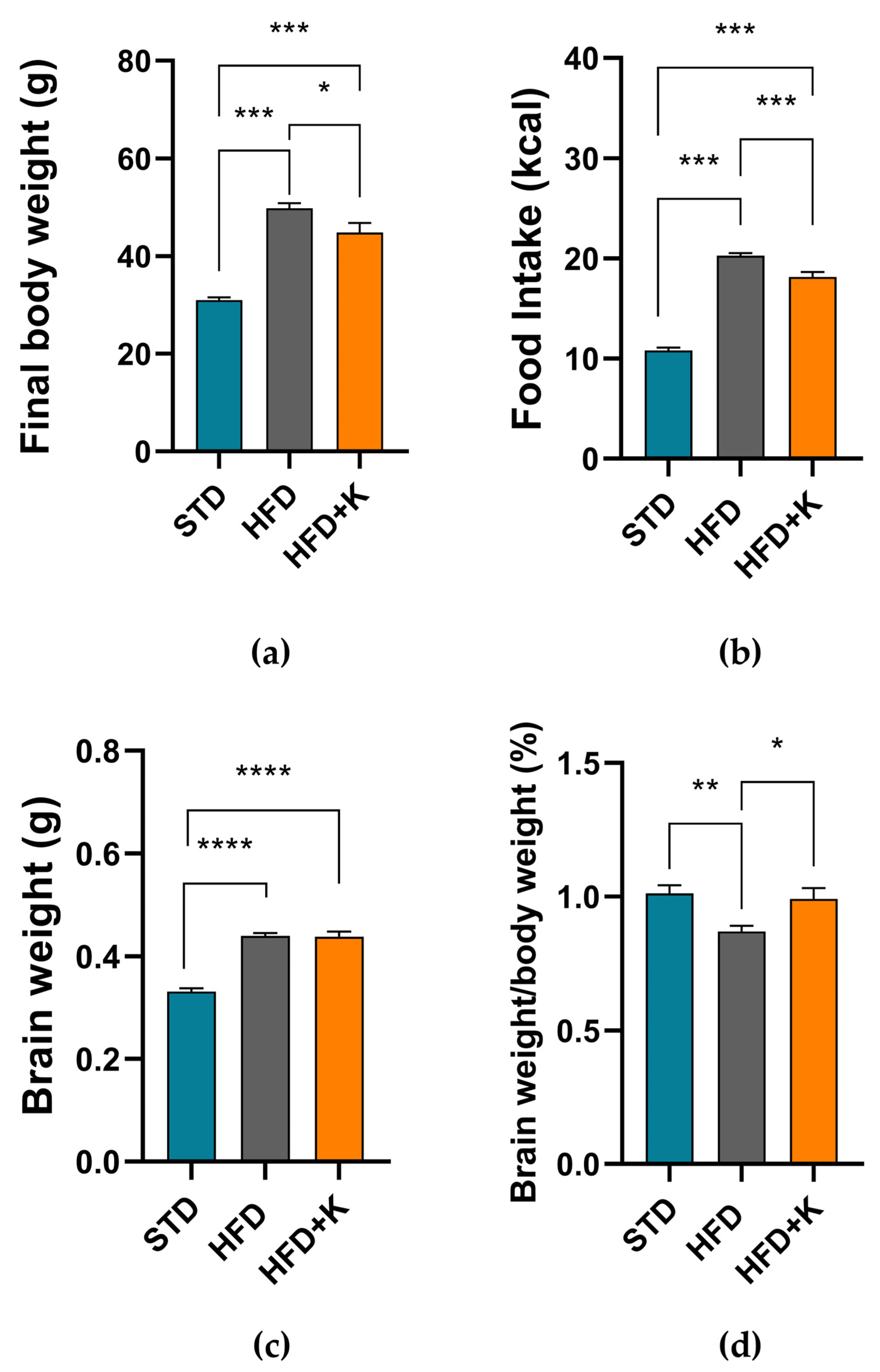
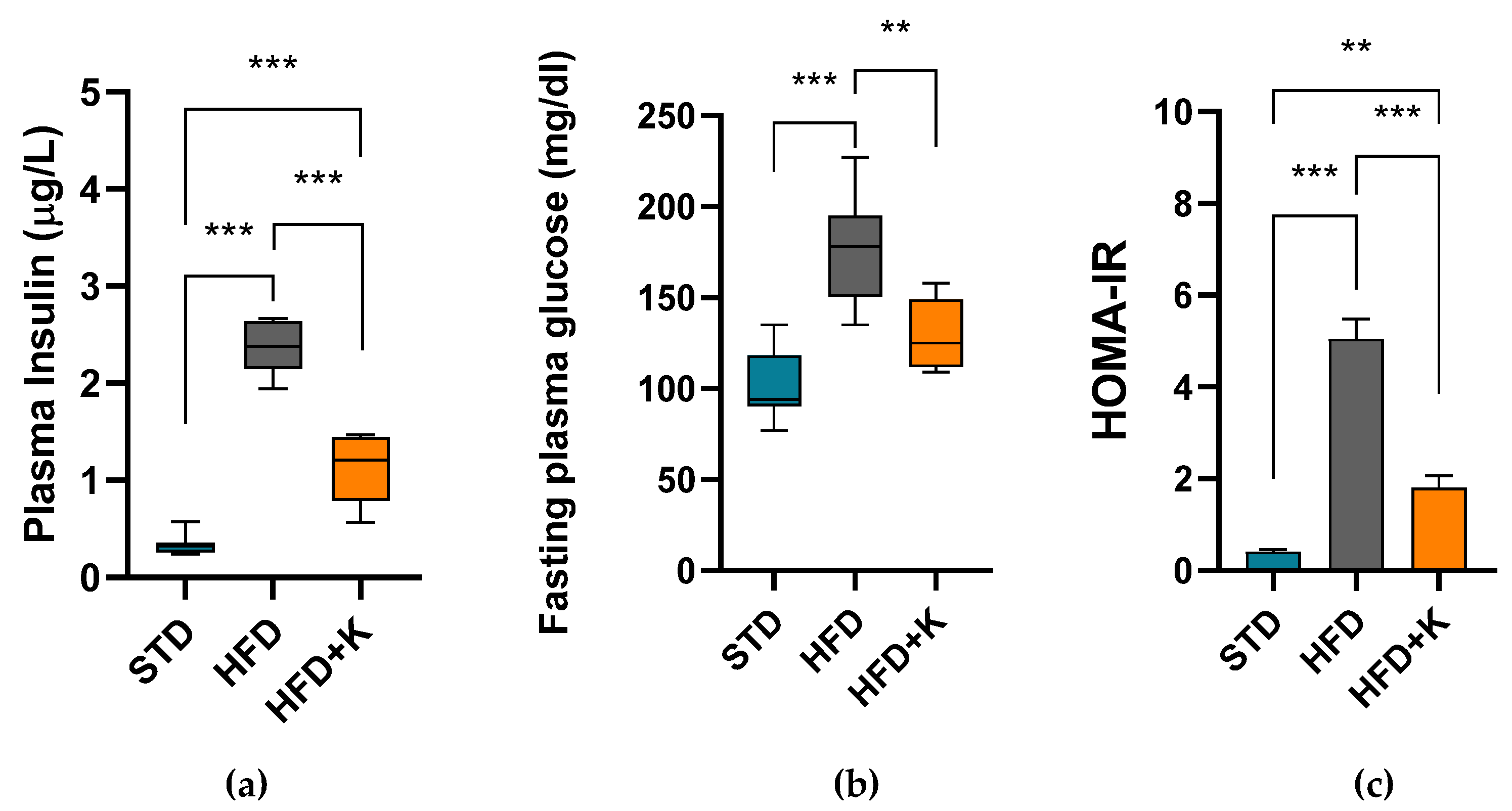
2.1. Metabolic Parameters
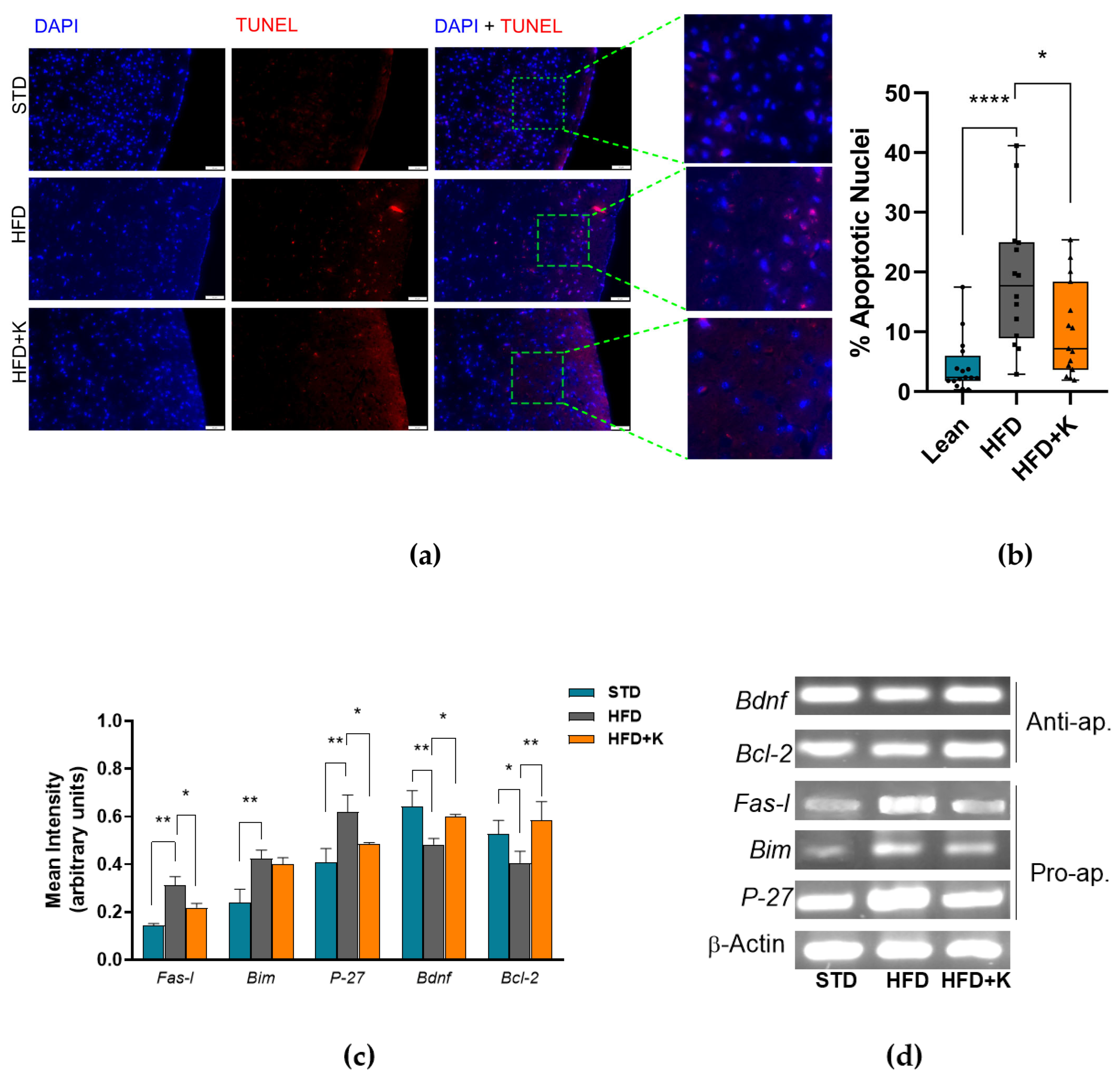
2.2. KF Supplementation Reduces HFD-Induced Apoptosis in the Brain Cortex
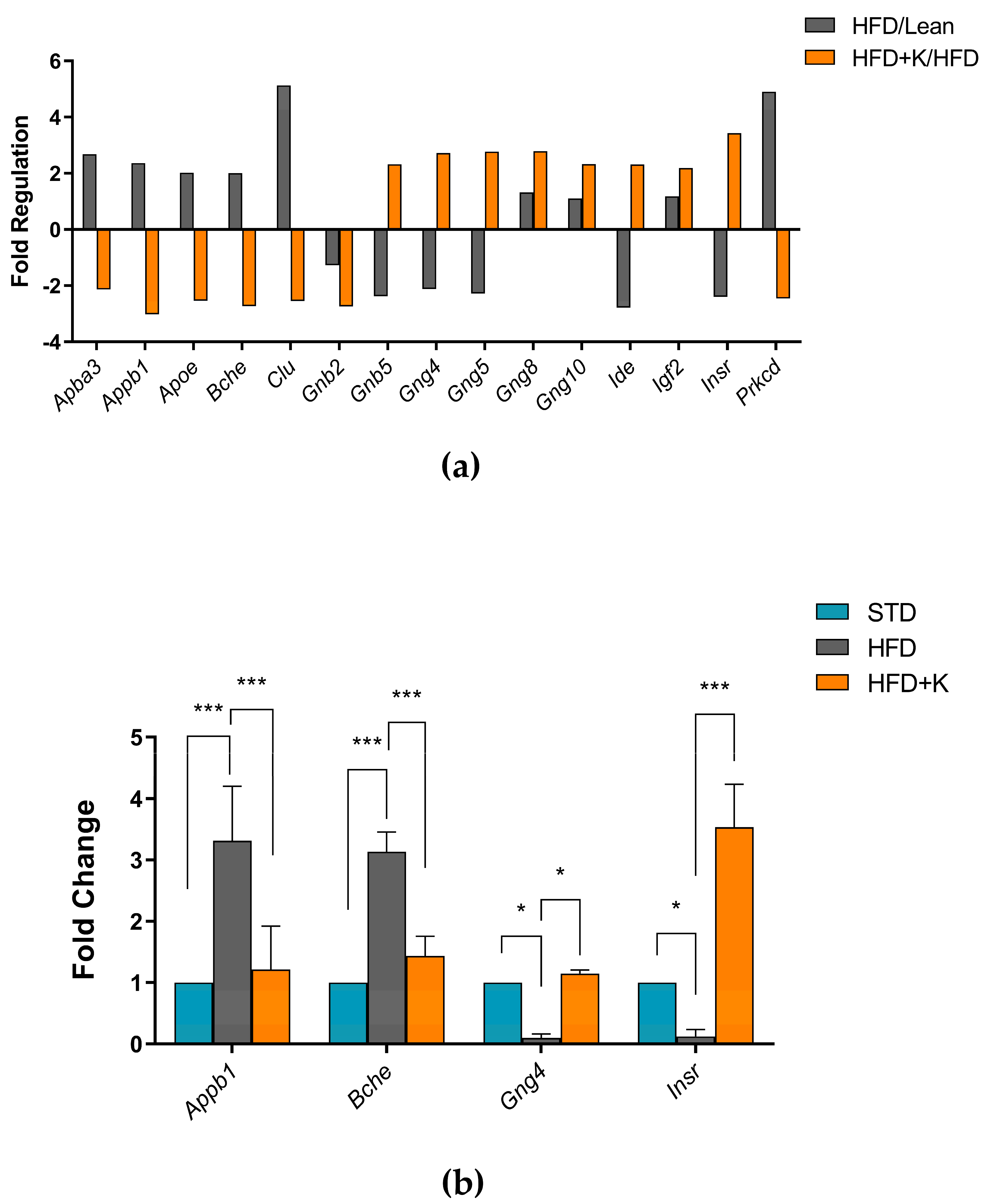
2.3. Exploration of KF Influence on Genes Related to Alzheimer’s Disease
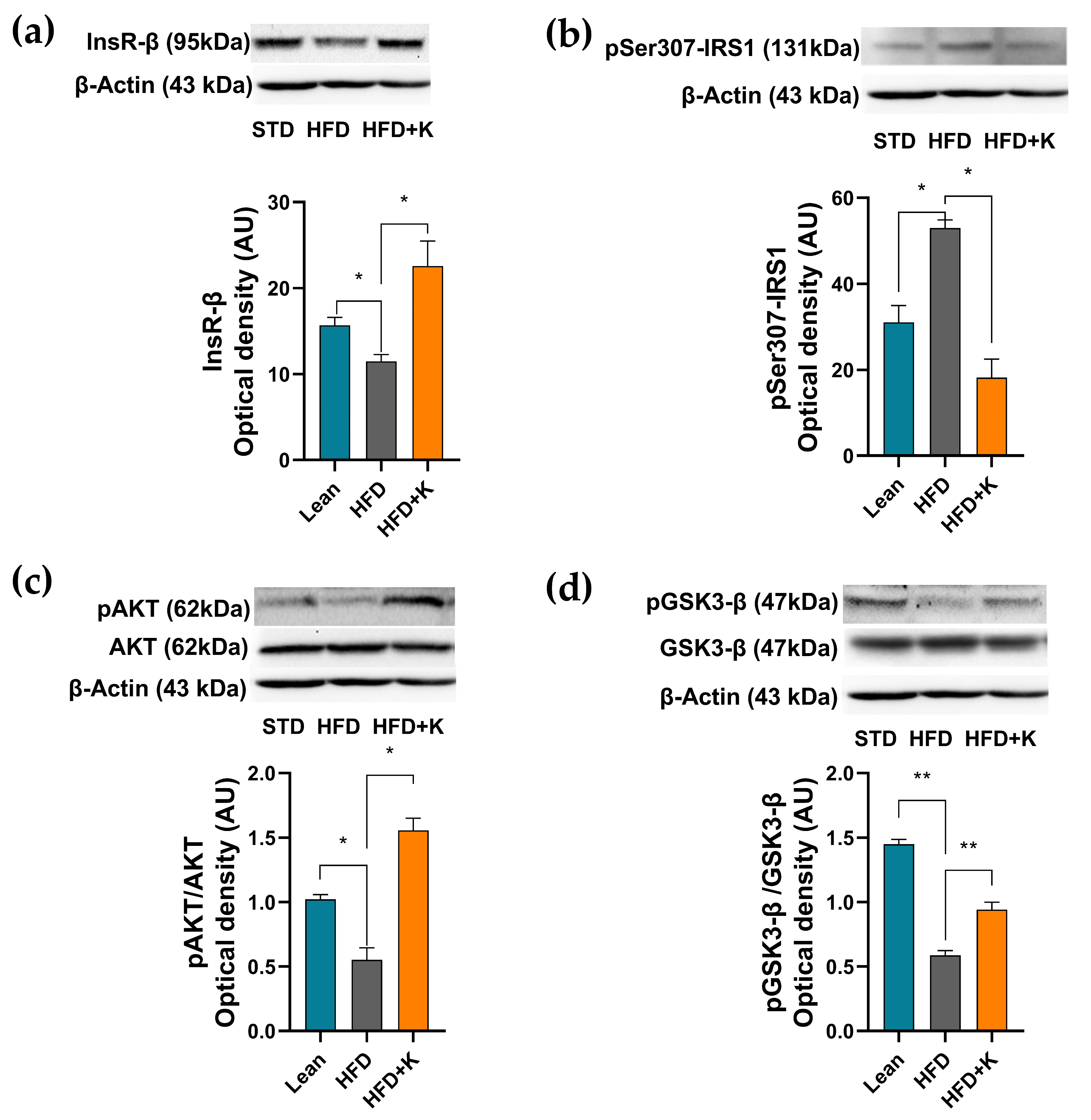
2.4. KF Supplementation Ameliorates the HFD-Induced Dysfunction of the Insulin Signalling Axis in Brain Cortex
2.5. KF Lowers HFD-Linked Neuroinflammation
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Animals and Diet
4.3. Metabolic Parameters
4.4. Brain Tissue Preparation
4.5. Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) Assay
4.6. Semiquantitative Polymerase Chain Reaction Experiments
4.7. RT2 Profiler PCR Array
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β |
| AD | Alzheimer’s Disease |
| HFD | High-Fat Diet |
| HFD+K | High Fat Diet supplemented with Kumquat fruit |
| HOMA-IR | Homeostatic Model Assessment for Insulin Resistance |
| IL1β | InterLeukin 1 β |
| Ins-Rβ | Insulin Receptor subunit β |
| IR | Insulin Resistance |
| IRS1 | Insulin Receptor Substrate 1 |
| KF | Kumquat Fruit |
| NFT | NeuroFibrillary Tangles |
| qPCR | quantitative real-time PCR |
| STD | Standard Diet |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction |
| T2DM | Type 2 Diabetes Mellitus |
| TNF-α | Tumor Necrosis Factor α |
| TUNEL | Terminal deoxynucleotidyl transferase biotin-dUTP Nick End Labeling |
References
- Barbaresko, J.; Koch, M.; Schulze, M.B.; Nöthlings, U. Dietary Pattern Analysis and Biomarkers of Low-Grade Inflammation: A Systematic Literature Review. Nutr. Rev. 2013, 71, 511–527. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diab. Rep. 2021, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cai, W.; Hoover, B.; Kahn, C.R. Insulin Action in the Brain: Cell Types, Circuits, and Diseases. Trends Neurosci. 2022, 45, 384–400. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L.; Rostagno, A.A.; Baranowska-Bik, A.; Orzechowski, A. Insulin and Insulin Resistance in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, C.; Hu, M.; Li, Q.; Li, J.; Quan, S.; Zhang, X.; Gu, L. Research Progress on the Association of Insulin Resistance with Type 2 Diabetes Mellitus and Alzheimer’s Disease. Metab. Brain Dis. 2024, 40, 35. [Google Scholar] [CrossRef]
- Zhao, W.Q.; Townsend, M. Insulin Resistance and Amyloidogenesis as Common Molecular Foundation for Type 2 Diabetes and Alzheimer’s Disease. Biochim. Biophys. Acta 2009, 1792, 482–496. [Google Scholar] [CrossRef]
- Woo, J.R.; Bae, S.H.; Wales, T.E.; Engen, J.R.; Lee, J.; Jang, H.; Park, S.Y. The Serine Phosphorylations in the IRS-1 PIR Domain Abrogate IRS-1 and IR Interaction. Proc. Natl. Acad. Sci. USA 2024, 121, e2401716121. [Google Scholar] [CrossRef]
- Ono, H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1317. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Feferman, L.; Tobacman, J.K. Carrageenan Inhibits Insulin Signaling through GRB10-Mediated Decrease in Tyr(P)-IRS1 and through Inflammation-Induced Increase in Ser(P)307-IRS1. J. Biol. Chem. 2015, 290, 10764–10774. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of Insulin Sensitivity by Serine/Threonine Phosphorylation of Insulin Receptor Substrate Proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Wang, Y.; Liu, N.; Shen, L.; Ma, Q.; Wang, M.; Han, B. Chronic Stress Induces Insulin Resistance and Enhances Cognitive Impairment in AD. Brain Res. Bull. 2024, 217, 111083. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Solch, R.J.; Aigbogun, J.O.; Voyiadjis, A.G.; Talkington, G.M.; Darensbourg, R.M.; O’Connell, S.; Pickett, K.M.; Perez, S.R.; Maraganore, D.M. Mediterranean Diet Adherence, Gut Microbiota, and Alzheimer’s or Parkinson’s Disease Risk: A Systematic Review. J. Neurol. Sci. 2022, 434, 120166. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Blake, S.M. Diet’s Role in Modifying Risk of Alzheimer’s Disease: History and Present Understanding. J. Alzheimer’s Dis. 2023, 96, 1353–1382. [Google Scholar] [CrossRef]
- Stefaniak, O.; Dobrzyńska, M.; Drzymała-Czyż, S.; Przysławski, J. Diet in the Prevention of Alzheimer’s Disease: Current Knowledge and Future Research Requirements. Nutrients 2022, 14, 4564. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Farokhi Larijani, S.; Hassanzadeh, G.; Zahmatkesh, M.; Radfar, F.; Farahmandfar, M. Intranasal Insulin Intake and Exercise Improve Memory Function in Amyloid-β Induced Alzheimer’s-like Disease in Rats: Involvement of Hippocampal BDNF-TrkB Receptor. Behav. Brain Res. 2024, 460, 114814. [Google Scholar] [CrossRef]
- AboEl-Azm, Y.H.; El-Samahy, M.; Hendi, N.I.; Arar, A.; Yasen, N.S.; Ramadan, S.; Zedan, E.M.; Al-dardery, N.M.; Khaity, A. Safety and Efficacy of Intranasal Insulin in Patients with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Clin. Transl. Res. 2023, 9, 222. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.J.; Chen, J.B.; Cao, J.P.; Li, X.; Sun, C. De Citrus Flavonoids and Their Antioxidant Evaluation. Crit. Rev. Food Sci. Nutr. 2022, 62, 3833–3854. [Google Scholar] [CrossRef]
- Singh, B.; Singh, J.P.; Kaur, A.; Singh, N. Phenolic Composition, Antioxidant Potential and Health Benefits of Citrus Peel. Food Res. Int. 2020, 132, 109114. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Ohizumi, Y. Beneficial Effects of Citrus-Derived Polymethoxylated Flavones for Central Nervous System Disorders. Nutrients 2021, 13, 145. [Google Scholar] [CrossRef] [PubMed]
- Azhar, S.; Sabahat, R.; Sajjad, R.; Nadeem, F.; Amjad, A.; Hafeez, N.; Nayab, T.; Wahid, S.; Tanweer, A. Effect of Citrus Flavanones on Diabetes: A Systematic Review. Curr. Diabetes Rev. 2023, 19, 94–102. [Google Scholar] [CrossRef]
- Hwang, S.L.; Shih, P.H.; Yen, G.C. Neuroprotective Effects of Citrus Flavonoids. J. Agric. Food Chem. 2012, 60, 877–885. [Google Scholar] [CrossRef]
- Gandhi, G.R.; Vasconcelos, A.B.S.; Wu, D.T.; Li, H.B.; Antony, P.J.; Li, H.; Geng, F.; Gurgel, R.Q.; Narain, N.; Gan, R.Y. Citrus Flavonoids as Promising Phytochemicals Targeting Diabetes and Related Complications: A Systematic Review of In Vitro and In Vivo Studies. Nutrients 2020, 12, 2907. [Google Scholar] [CrossRef]
- Pontifex, M.G.; Malik, M.M.A.H.; Connell, E.; Müller, M.; Vauzour, D. Citrus Polyphenols in Brain Health and Disease: Current Perspectives. Front. Neurosci. 2021, 15, 640648. [Google Scholar] [CrossRef]
- Miles, E.A.; Calder, P.C. Effects of Citrus Fruit Juices and Their Bioactive Components on Inflammation and Immunity: A Narrative Review. Front. Immunol. 2021, 12, 712608. [Google Scholar] [CrossRef]
- Tan, S.; Li, M.; Ding, X.; Fan, S.; Guo, L.; Gu, M.; Zhang, Y.; Feng, L.; Jiang, D.; Li, Y.; et al. Effects of Fortunella Margarita Fruit Extract on Metabolic Disorders in High-Fat Diet-Induced Obese C57BL/6 Mice. PLoS ONE 2014, 9, e93510. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, P.; Chang, Q.; Zheng, B.; Zhang, Y. Hypolipidemic Effect of Polysaccharides from Fortunella Margarita (Lour.) Swingle in Hyperlipidemic Rats. Food Chem. Toxicol. 2019, 132, 110663. [Google Scholar] [CrossRef]
- Lou, S.N.; Ho, C.T. Phenolic Compounds and Biological Activities of Small-Size Citrus: Kumquat and Calamondin. J. Food Drug Anal. 2017, 25, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Al-Sayed, H.M.A.; Abdelaleem, M.A.; Shawky, H.A. Physiochemical and Nutritional Evaluation of Whole Kumquat Fruits Powder and Its Protective Effect on Thyroid Hormones and Blood Sugar Levels in Diabetic Rats. Braz. J. Biol. 2021, 83, e247071. [Google Scholar] [CrossRef]
- Eddin, L.B.; Azimullah, S.; Jha, N.K.; Nagoor Meeran, M.F.; Beiram, R.; Ojha, S. Limonene, a Monoterpene, Mitigates Rotenone-Induced Dopaminergic Neurodegeneration by Modulating Neuroinflammation, Hippo Signaling and Apoptosis in Rats. Int. J. Mol. Sci. 2023, 24, 5222. [Google Scholar] [CrossRef]
- Zhang, J.; Pandey, M.; Awe, A.; Lue, N.; Kittock, C.; Fikse, E.; Degner, K.; Staples, J.; Mokhasi, N.; Chen, W.; et al. The Association of GNB5 with Alzheimer Disease Revealed by Genomic Analysis Restricted to Variants Impacting Gene Function. Am. J. Hum. Genet. 2024, 111, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Lauer, A.A.; Grösgen, S.; Thiel, A.; Lehmann, J.; Winkler, J.; Janitschke, D.; Herr, C.; Beisswenger, C.; Bals, R.; et al. Profiling of Alzheimer’s Disease Related Genes in Mild to Moderate Vitamin D Hypovitaminosis. J. Nutr. Biochem. 2019, 67, 123–137. [Google Scholar] [CrossRef]
- Bonham, L.W.; Evans, D.S.; Liu, Y.; Cummings, S.R.; Yaffe, K.; Yokoyama, J.S. Neurotransmitter Pathway Genes in Cognitive Decline During Aging: Evidence for GNG4 and KCNQ2 Genes. Am. J. Alzheimer’s Dis. Other Demen. 2018, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Choi, T.I.; Kim, Y.M.; Lee, S.; Han, B.; Bak, I.S.; Moon, S.A.; Yu, D.Y.; Shin, K.S.; Kwon, Y.K.; et al. Regulation of Habenular G-Protein Gamma 8 on Learning and Memory via Modulation of the Central Acetylcholine System. Mol. Psychiatry 2021, 26, 3737–3750. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, Y.H.; Park, S.J.; Huh, J.W.; Kim, S.H.; Kim, S.U.; Kim, J.S.; Jeong, K.J.; Lee, K.M.; Hong, Y.; et al. Insulin/IGF Signaling-Related Gene Expression in the Brain of a Sporadic Alzheimer’s Disease Monkey Model Induced by Intracerebroventricular Injection of Streptozotocin. J. Alzheimer’s Dis. 2014, 38, 251–267. [Google Scholar] [CrossRef]
- Vepsäläinen, S.; Helisalmi, S.; Mannermaa, A.; Pirttilä, T.; Soininen, H.; Hiltunen, M. Combined Risk Effects of IDE and NEP Gene Variants on Alzheimer Disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1268–1270. [Google Scholar] [CrossRef]
- Du, Y.; Guo, T.; Hao, Y.; Li, C.; Tang, L.; Li, X.; Zhang, X.; Li, L.; Yao, D.; Xu, X.; et al. PKCδ Serves as a Potential Biomarker and Therapeutic Target for Microglia-Mediated Neuroinflammation in Alzheimer’s Disease. Alzheimer’s Dement. 2024, 20, 5511–5527. [Google Scholar] [CrossRef]
- Kitanaka, J.; Wang, X.B.; Kitanaka, N.; Hembree, C.M.; Uhl, G.R. Genomic Organization of the Murine G Protein Beta Subunit Genes and Related Processed Pseudogenes. DNA Seq. 2001, 12, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Housny, M.; Gulkarov, S.; Hossain, T.; Locke, B.; Srivastava, A.; Pinkhasov, A.; Gomolin, I.H.; Wisniewski, T.; De Leon, J. Role of Apolipoprotein E in Alzheimer’s Disease Pathogenesis, Prognosis and Treatment. Discov. Med. 2024, 36, 1917. [Google Scholar] [CrossRef] [PubMed]
- Bonifácio, M.J.; Sousa, F.; Aires, C.; Loureiro, A.I.; Fernandes-Lopes, C.; Pires, N.M.; Palma, P.N.; Moser, P.; Soares-da-Silva, P. Preclinical Pharmacological Evaluation of the Fatty Acid Amide Hydrolase Inhibitor BIA 10-2474. Br. J. Pharmacol. 2020, 177, 2123–2142. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Stopa, E.G.; Johanson, C.E.; Baird, A.; Silverberg, G.D. Choroid Plexus Genes for CSF Production and Brain Homeostasis Are Altered in Alzheimer’s Disease. Fluids Barriers CNS 2018, 15, 34. [Google Scholar] [CrossRef]
- Ghosh, S.; Das, B.; Jana, S.; Singh, K.O.; Sharma, N.; Mukherjee, P.K.; Haldar, P.K. Mechanistic Insight into Neuroprotective Effect of Standardized Ginger Chemo Varieties from Manipur, India in Scopolamine Induced Learning and Memory Impaired Mice. Metab. Brain Dis. 2025, 40, 101. [Google Scholar] [CrossRef]
- Laslo, A.; Laslo, L.; Arbănași, E.M.; Ujlaki-Nagi, A.A.; Chinezu, L.; Ivănescu, A.D.; Arbănași, E.M.; Cărare, R.O.; Cordoș, B.A.; Popa, I.A.; et al. Pathways to Alzheimer’s Disease: The Intersecting Roles of Clusterin and Apolipoprotein E in Amyloid-β Regulation and Neuronal Health. Pathophysiology 2024, 31, 545–558. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Gammazza, A.M.; Cappello, F.; Mulè, F.; Carlo, M. Di Insulin Resistance as Common Molecular Denominator Linking Obesity to Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef]
- Vinuesa, A.; Pomilio, C.; Gregosa, A.; Bentivegna, M.; Presa, J.; Bellotto, M.; Saravia, F.; Beauquis, J. Inflammation and Insulin Resistance as Risk Factors and Potential Therapeutic Targets for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 653651. [Google Scholar] [CrossRef]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible Implications of Insulin Resistance and Glucose Metabolism in Alzheimer’s Disease Pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef]
- Petrov, D.; Pedrós, I.; Artiach, G.; Sureda, F.X.; Barroso, E.; Pallàs, M.; Casadesús, G.; Beas-Zarate, C.; Carro, E.; Ferrer, I.; et al. High-Fat Diet-Induced Deregulation of Hippocampal Insulin Signaling and Mitochondrial Homeostasis Deficiences Contribute to Alzheimer Disease Pathology in Rodents. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1687–1699. [Google Scholar] [CrossRef]
- Turdi, S.; Ge, W.; Hu, N.; Bradley, K.M.; Wang, X.; Ren, J. Interaction between Maternal and Postnatal High Fat Diet Leads to a Greater Risk of Myocardial Dysfunction in Offspring via Enhanced Lipotoxicity, IRS-1 Serine Phosphorylation and Mitochondrial Defects. J. Mol. Cell. Cardiol. 2013, 55, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, Q.; Wang, X.; Tang, B.; Zhang, X.; Yu, H.; Li, Z. Time-Dependent Effects of High-Fat Diet on Cognition and Cerebral Insulin Signaling: Window for Recovery and Potential Therapeutic Target. Mech. Ageing Dev. 2024, 220, 111955. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, P. Role of Insulin Receptor Substance-1 Modulating PI3K/Akt Insulin Signaling Pathway in Alzheimer’s Disease. 3 Biotech 2021, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Duronio, V. The Life of a Cell: Apoptosis Regulation by the PI3K/PKB Pathway. Biochem. J. 2008, 415, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Shareena, G.; Kumar, D.; Thorat, N. Exploring the Diverse Roles of GSK-3β Kinase in Alzheimer’s Disease. In Deciphering Drug Targets for Alzheimer’s Disease; Springer Nature: Singapore, 2023; pp. 219–244. [Google Scholar] [CrossRef]
- Katashima, C.K.; Silva, V.R.R.; Lenhare, L.; Marin, R.M.; Carvalheira, J.B.C. INOS Promotes Hypothalamic Insulin Resistance Associated with Deregulation of Energy Balance and Obesity in Rodents. Sci. Rep. 2017, 7, 9265. [Google Scholar] [CrossRef]
- Patel, B.; New, L.E.; Griffiths, J.C.; Deuchars, J.; Filippi, B.M. Inhibition of Mitochondrial Fission and INOS in the Dorsal Vagal Complex Protects from Overeating and Weight Gain. Mol. Metab. 2021, 43, 101123. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alexiou, A.; Papadakis, M.; Bahaa, M.M.; Alibrahim, F.; Batiha, G.E.S. New Insight on the Potential Detrimental Effect of Metabolic Syndrome on the Alzheimer Disease Neuropathology: Mechanistic Role. J. Cell. Mol. Med. 2024, 28, e70118. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Kim, J.B.; Han, A.R.; Park, E.Y.; Kim, J.Y.; Cho, W.; Lee, J.; Seo, E.K.; Lee, K.T. Inhibition of LPS-Induced INOS, COX-2 and Cytokines Expression by Poncirin through the NF-ΚB Inactivation in RAW 264.7 Macrophage Cells. Biol. Pharm. Bull. 2007, 30, 2345–2351. [Google Scholar] [CrossRef]
- Alkanat, M.; Alkanat, H.Ö. D-Limonene Reduces Depression-like Behaviour and Enhances Learning and Memory through an Anti-Neuroinflammatory Mechanism in Male Rats Subjected to Chronic Restraint Stress. Eur. J. Neurosci. 2024, 60, 4491–4502. [Google Scholar] [CrossRef]
- Lorigooini, Z.; Boroujeni, S.N.; Sayyadi-Shahraki, M.; Rahimi-Madiseh, M.; Bijad, E.; Amini-Khoei, H. Limonene through Attenuation of Neuroinflammation and Nitrite Level Exerts Antidepressant-Like Effect on Mouse Model of Maternal Separation Stress. Behav. Neurol. 2021, 2021, 8817309. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.C.; De Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J.A. S-Nitrosation of the Insulin Receptor, Insulin Receptor Substrate 1, and Protein Kinase B/Akt: A Novel Mechanism of Insulin Resistance. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, H.J.; Lee, Y.S.; Kang, G.M.; Lim, H.S.; Lee, S.H.; Song, D.K.; Kwon, O.; Hwang, I.; Son, M.; et al. Hypothalamic Macrophage Inducible Nitric Oxide Synthase Mediates Obesity-Associated Hypothalamic Inflammation. Cell Rep. 2018, 25, 934–946.e5. [Google Scholar] [CrossRef] [PubMed]
- Hewett, S.J.; Uliasz, T.F.; Vidwans, A.S.; Hewett, J.A. Cyclooxygenase-2 Contributes ToN-Methyl-d-Aspartate-Mediated Neuronal Cell Death in Primary Cortical Cell Culture. J. Pharmacol. Exp. Ther. 2000, 293, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Luo, Y.; Kuang, S.; Yang, Y.; Tian, X.; Ma, J.; Mai, S.; Xue, L.; Yang, J. Cyclooxygenase-2 Signalling Pathway in the Cortex Is Involved in the Pathophysiological Mechanisms in the Rat Model of Depression. Sci. Rep. 2017, 7, 488. [Google Scholar] [CrossRef]
- Terzo, S.; Calvi, P.; Nuzzo, D.; Picone, P.; Allegra, M.; Mulè, F.; Amato, A. Long-Term Ingestion of Sicilian Black Bee Chestnut Honey and/or D-Limonene Counteracts Brain Damage Induced by High Fat-Diet in Obese Mice. Int. J. Mol. Sci. 2023, 24, 3467. [Google Scholar] [CrossRef]
- Terzo, S.; Amato, A.; Calvi, P.; Giardina, M.; Nuzzo, D.; Picone, P.; Palumbo-Piccionello, A.; Amata, S.; Giardina, I.C.; Massaro, A.; et al. Positive Impact of Indicaxanthin from Opuntia Ficus-Indica Fruit on High-Fat Diet–Induced Neuronal Damage and Gut Microbiota Dysbiosis. Neural Regen. Res. 2025. [Google Scholar] [CrossRef]
- Restivo, I.; Basilicata, M.G.; Giardina, I.C.; Massaro, A.; Pepe, G.; Salviati, E.; Pecoraro, C.; Carbone, D.; Cascioferro, S.; Parrino, B.; et al. A Combination of Polymethoxyflavones from Citrus Sinensis and Prenylflavonoids from Humulus Lupulus Counteracts IL-1β-Induced Differentiated Caco-2 Cells Dysfunction via a Modulation of NF-ΚB/Nrf2 Activation. Antioxidants 2023, 12, 1621. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | T° Annealing |
|---|---|---|---|
| Fas-l | 5′-CAAGTCCAACTCAAGGTCCATGCC-3′ | 5′-AGAGAGGCTCAGATACGTTTGAC-3′ | 58 °C |
| Bim | 5′-GGAGGAGGCGGAGGATGAT-3′ | 5′-TCCTGTCTTGCGGTTCTGTC-3′ | 58 °C |
| P 27 | 5′-TGCGAGTGTCTAACGGGAG-3′ | 5′-GTTTGACGTCTTCTGAGGCC-3′ | 59 °C |
| Bcl-2 | 5′-ATGTGTGTGGAGAGCGTCAA-3′ | 5′-AGAGACAGCCAGGAGAAATCA-3′ | 47 °C |
| bdnf | 5′-GGCTGACACTTTTGAGCACGTC-3′ | 5′-CTCCAAAGGCACTTGACTGCTG-3′ | 52 °C |
| β-actin | 5′-CGGGATCCCCGCCCTAGGCACCAGGGT-3′ | 5′-GGAAATTCGGCTGGGGTGTTGAAGGTCTCAAA-3′ | 60 °C |
| Protein | Manufacturer | Cat # | Host Organism | Molecular Weight |
|---|---|---|---|---|
| Primary antibodies | ||||
| AKT 1 | Santa Cruz Biotechnologies (Milan, Italy) | SC-5298 | Mouse | 62 kDa |
| p-AKT 1 | Sigma-Aldritch (Milan, Italy) | SAB4504331 | Rabbit | 62KDa |
| β-Actin | Santa Cruz Biotechnologies (Milan, Italy) | SC 47778 | Mouse | 43 kDa |
| COX-2 2 | Invitrogen (Milan, Italy) | 35-8200 | Mouse | 72 kDa |
| GSK3 β 3 | Santa Cruz Biotechnologies (Milan, Italy) | SC-377213 | Mouse | 47 kDa |
| p-GSK3 β 3 | Santa Cruz Biotechnologies (Milan, Italy) | SC-373800 | Mouse | 47 kDa |
| iNOS 4 | Invitrogen (Milan, Italy) | PA1-036 | Rabbit | 130 kDa |
| InsR-β 5 | Santa Cruz Biotechnologies (Milan, Italy) | SC-57342 | Mouse | 95 kDa |
| pSer307-IRS1 6 | Merck (Milan, Italy) | SAB4504442 | Rabbit | 131 kDa |
| NF-κB 7 | Santa Cruz Biotechnologies (Milan, Italy) | SC-8008 | Mouse | 65 kDa |
| Secondary antibodies | ||||
| IgG-HRP-Conjugated Anti-Mouse | Sigma-Aldrich (Milan, Italy) | A9044 | Rabbit | / |
| IgG-HRP-Conjugated Anti-Rabbit | Sigma-Aldrich (Milan, Italy) | A0545 | Goat | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massaro, A.; Calvi, P.; Restivo, I.; Giardina, M.; Mulè, F.; Tesoriere, L.; Amato, A.; Nuzzo, D.; Picone, P.; Terzo, S.; et al. Kumquat Fruit Administration Counteracts Dysmetabolism-Related Neurodegeneration and the Associated Brain Insulin Resistance in the High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2025, 26, 3077. https://doi.org/10.3390/ijms26073077
Massaro A, Calvi P, Restivo I, Giardina M, Mulè F, Tesoriere L, Amato A, Nuzzo D, Picone P, Terzo S, et al. Kumquat Fruit Administration Counteracts Dysmetabolism-Related Neurodegeneration and the Associated Brain Insulin Resistance in the High-Fat Diet-Fed Mice. International Journal of Molecular Sciences. 2025; 26(7):3077. https://doi.org/10.3390/ijms26073077
Chicago/Turabian StyleMassaro, Alessandro, Pasquale Calvi, Ignazio Restivo, Marta Giardina, Flavia Mulè, Luisa Tesoriere, Antonella Amato, Domenico Nuzzo, Pasquale Picone, Simona Terzo, and et al. 2025. "Kumquat Fruit Administration Counteracts Dysmetabolism-Related Neurodegeneration and the Associated Brain Insulin Resistance in the High-Fat Diet-Fed Mice" International Journal of Molecular Sciences 26, no. 7: 3077. https://doi.org/10.3390/ijms26073077
APA StyleMassaro, A., Calvi, P., Restivo, I., Giardina, M., Mulè, F., Tesoriere, L., Amato, A., Nuzzo, D., Picone, P., Terzo, S., & Allegra, M. (2025). Kumquat Fruit Administration Counteracts Dysmetabolism-Related Neurodegeneration and the Associated Brain Insulin Resistance in the High-Fat Diet-Fed Mice. International Journal of Molecular Sciences, 26(7), 3077. https://doi.org/10.3390/ijms26073077