
Machine Learning-Guided Screening and Molecular Docking for Proposing Naturally Derived Drug Candidates Against MERS-CoV 3CL Protease

 , ,
, , 
 and
and
Abstract

1. Introduction
2. Results
2.1. Model Performance
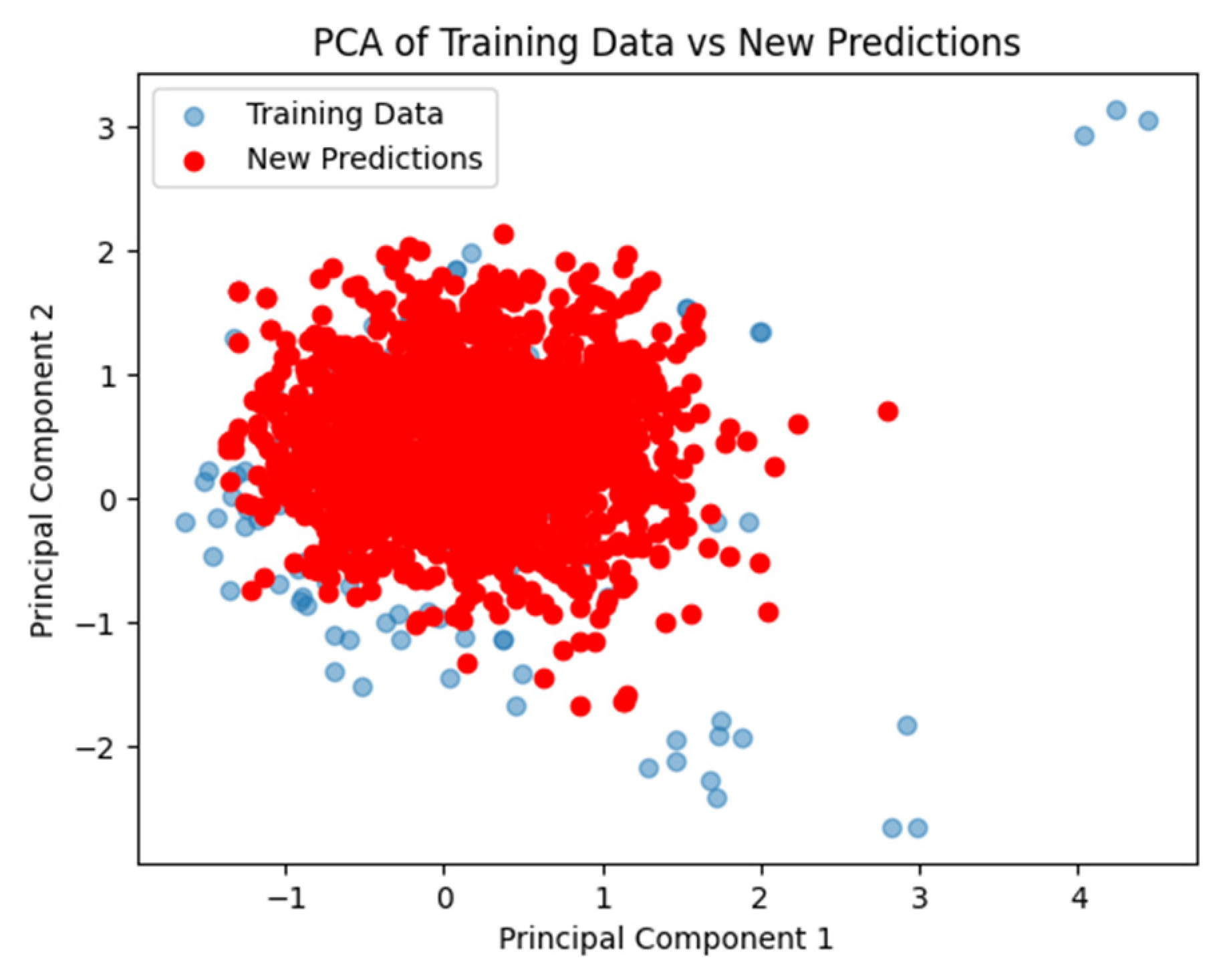
2.2. Validation and Visualization of Model Performance
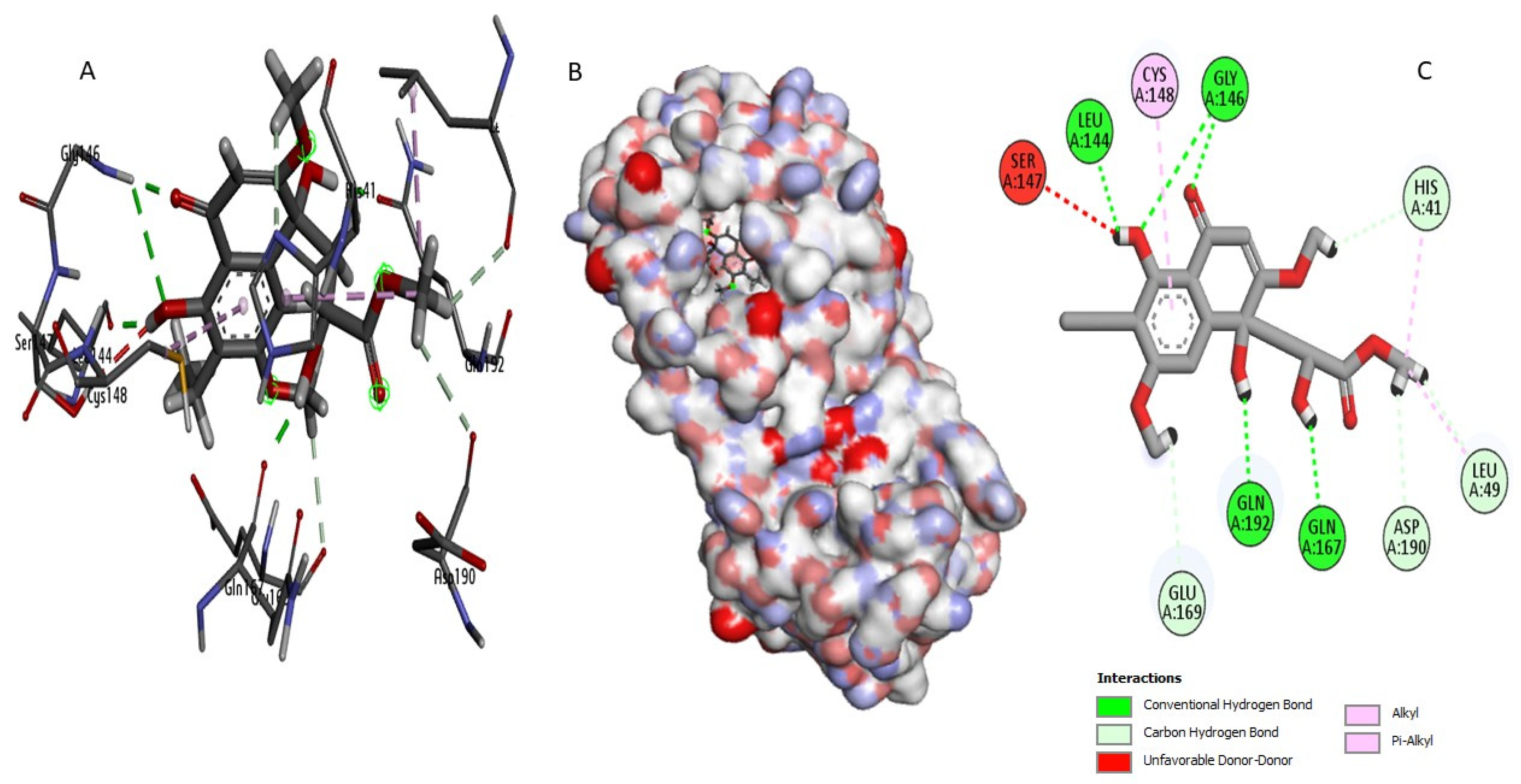
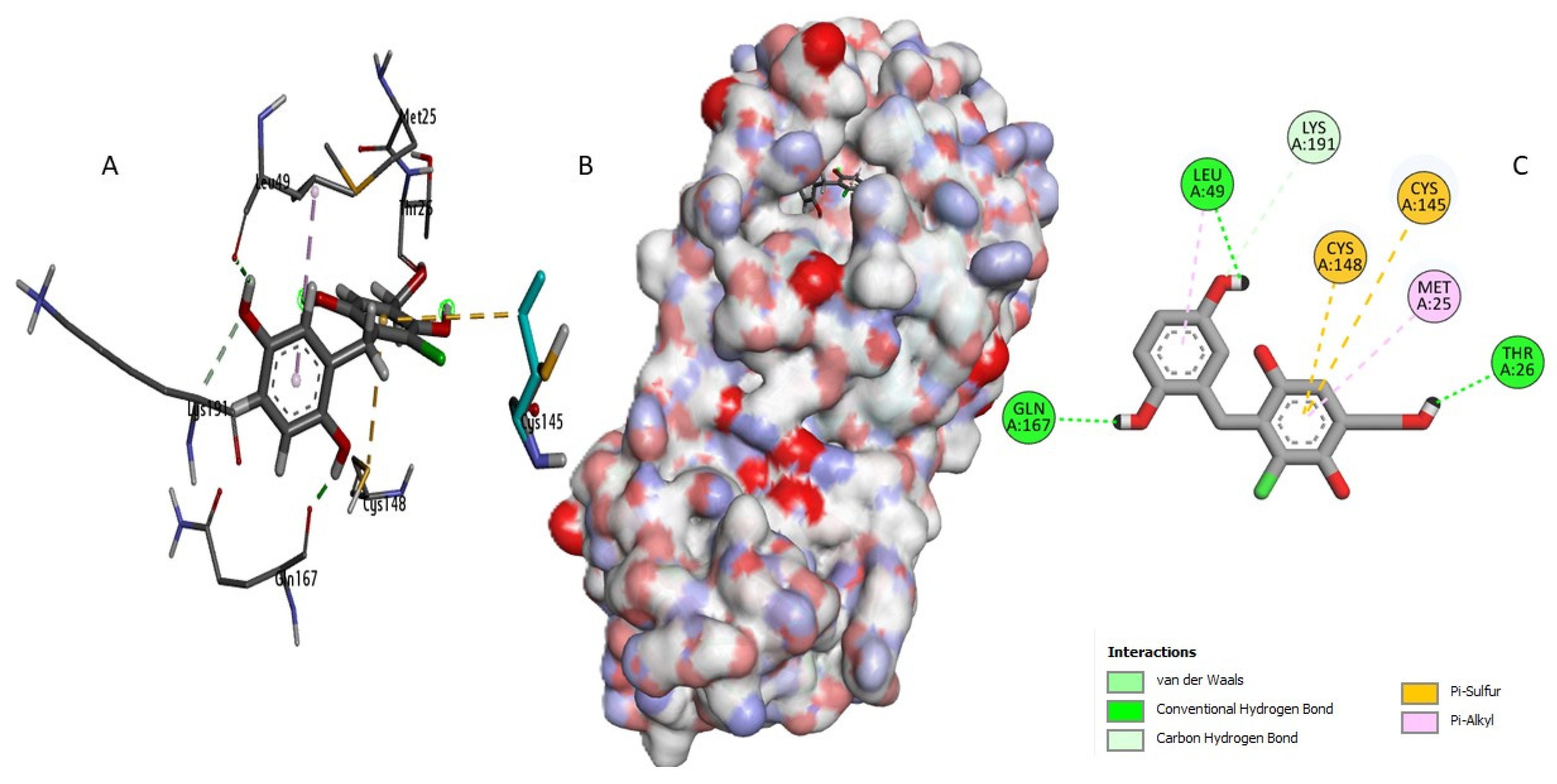
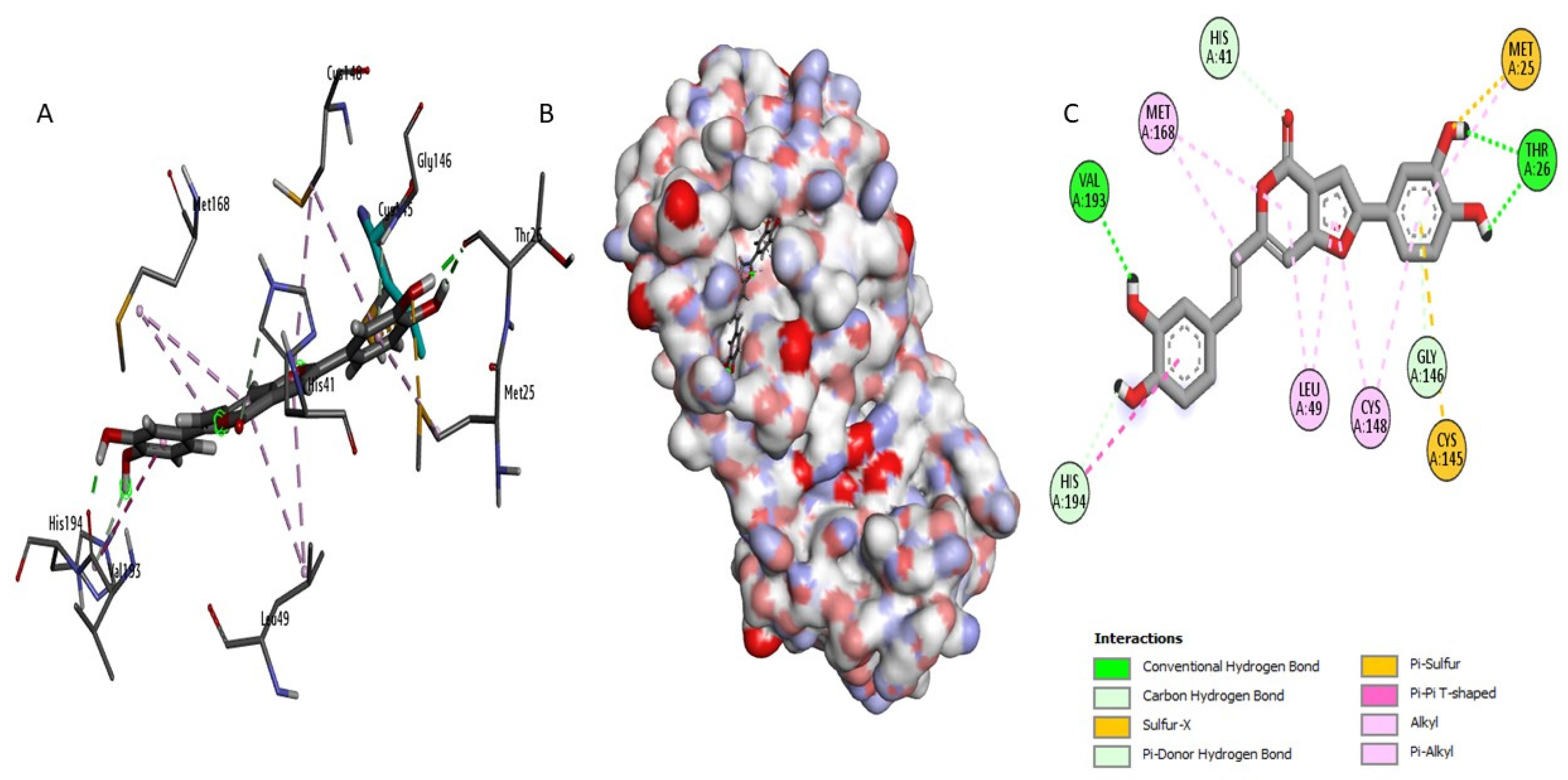
2.3. Docking Study and Binding Interaction Analysis of Compounds with MERS-CoV 3CLpro
2.4. Evaluation of Pharmacological Profiles and ADMET Characteristics
2.4.1. Prediction of Compound Physicochemical Properties
2.4.2. Evaluation of Medicinal Chemistry and Drug-Likeness Properties
2.4.3. Toxicity Profile and Pharmacological Implications
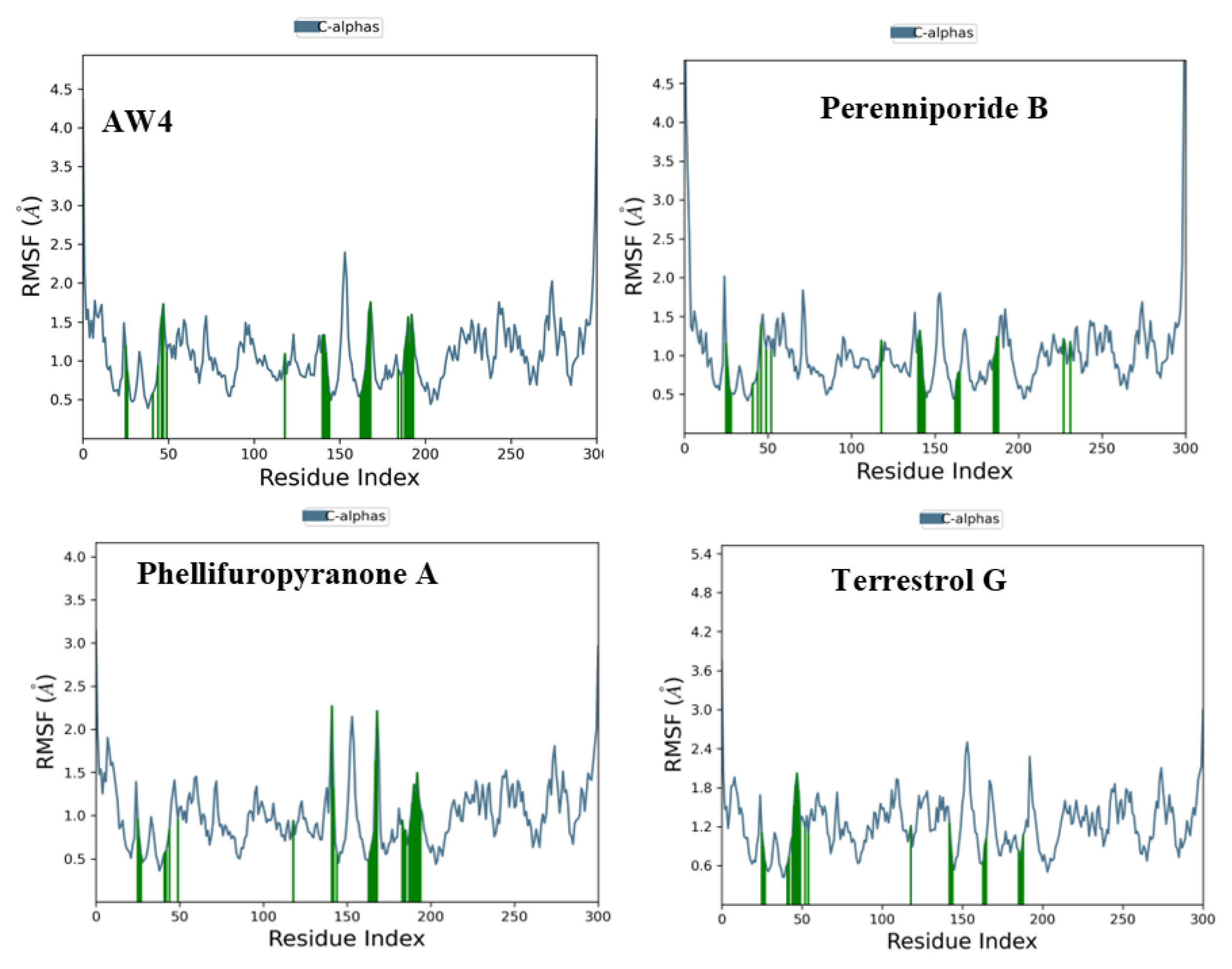
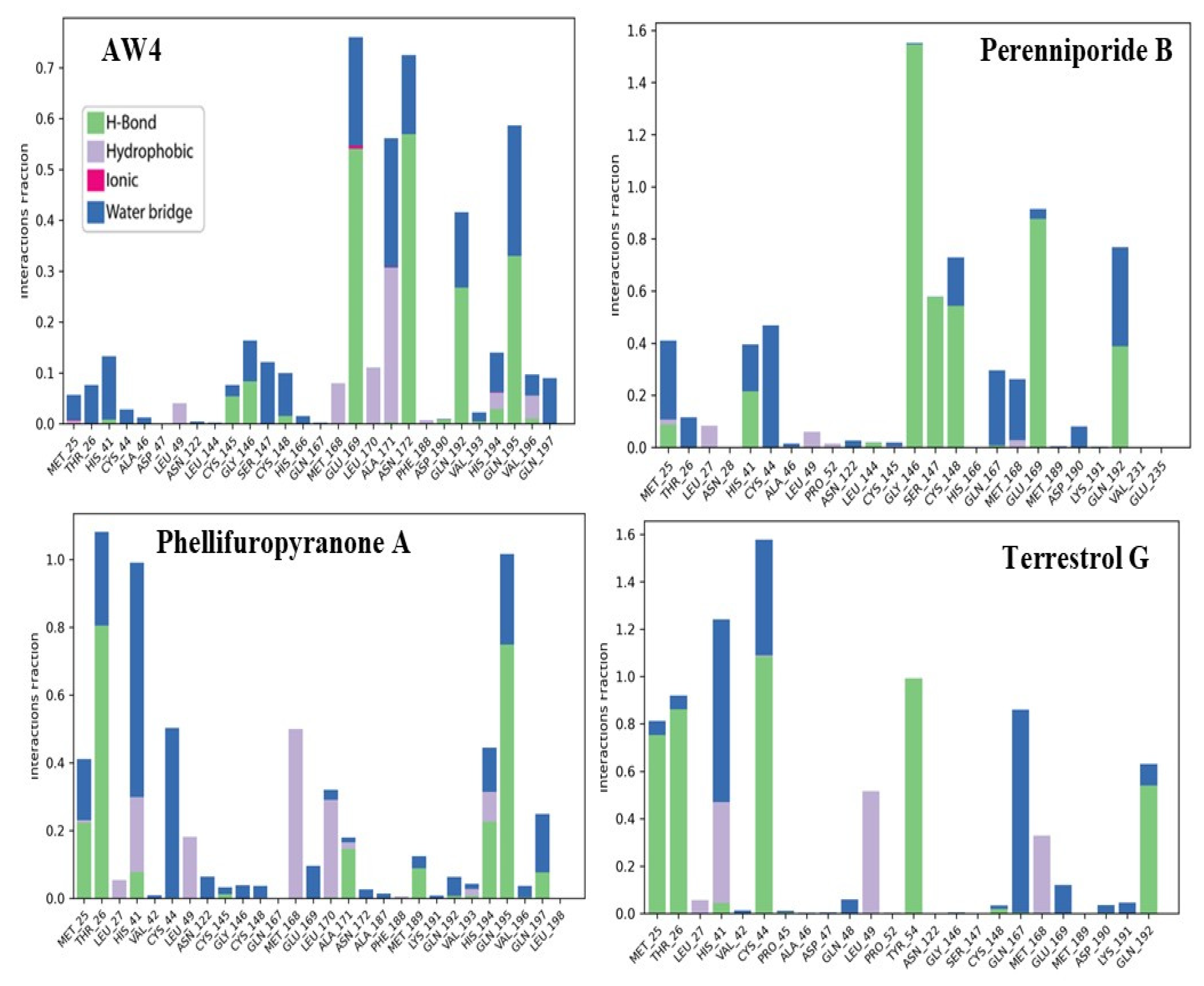
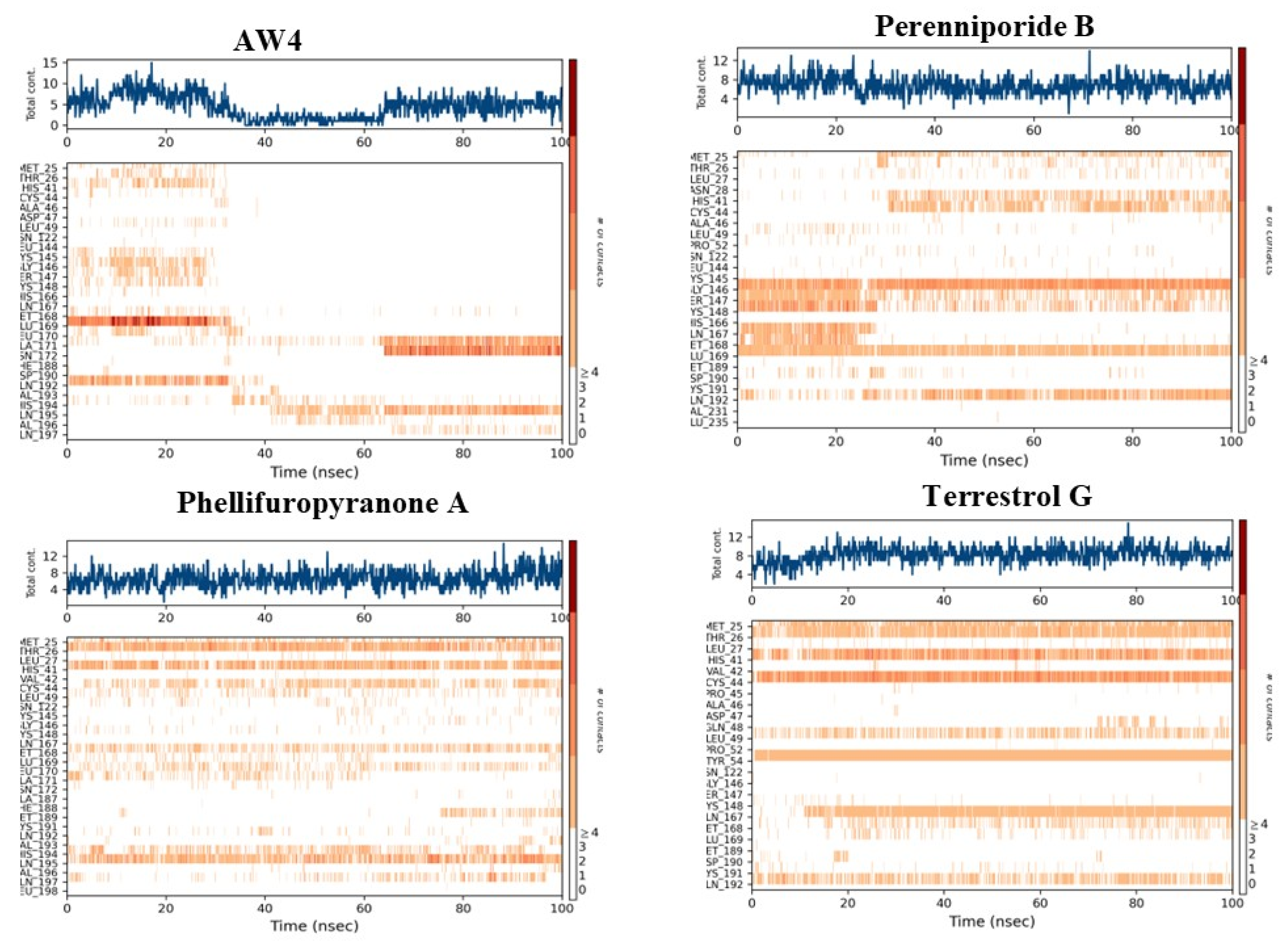
2.5. Molecular Dynamics Simulation Analysis
3. Discussion
4. Materials and Methods
4.1. Construction of Predictive Models
4.2. Molecular Docking Simulation
4.2.1. Ligand Preparation
4.2.2. Receptor Preparation
4.2.3. Active Site Definition
4.2.4. Docking Simulation
4.3. ADMET Analysis
4.4. Molecular Dynamics (MD) Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramadan, N.; Shaib, H. Middle East respiratory syndrome coronavirus (MERS-CoV): A review. Germs 2019, 9, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sharif-Yakan, A.; Kanj, S.S. Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives. PLoS Pathog. 2014, 10, e1004457. [Google Scholar] [CrossRef]
- Lu, X.; Whitaker, B.; Sakthivel, S.K.K.; Kamili, S.; Rose, L.E.; Lowe, L.; Mohareb, E.; Elassal, E.M.; Al-sanouri, T.; Haddadin, A.; et al. Real-Time Reverse Transcription-PCR Assay Panel for Middle East Respiratory Syndrome Coronavirus. J. Clin. Microbiol. 2014, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kandwal, S.; Fayne, D.; Stevenson, N.J. MERS-CoV-nsp5 expression in human epithelial BEAS 2b cells attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol. Life Sci. CMLS 2024, 81, 433. [Google Scholar] [CrossRef]
- Xue, W.; Ding, C.; Qian, K.; Liao, Y. The Interplay Between Coronavirus and Type I IFN Response. Front. Microbiol. 2022, 12, 805472. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses 2016, 8, 82. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve RD productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [PubMed]
- Adeshina, Y.O.; Deeds, E.J.; Karanicolas, J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. USA 2020, 117, 18477–18488. [Google Scholar] [CrossRef] [PubMed]
- Ellingson, S.R.; Davis, B.; Allen, J. Machine learning and ligand binding predictions: A review of data, methods, and obstacles. Biochim. Biophys. Acta BBA-Gen. Subj. 2020, 1864, 129545. [Google Scholar] [CrossRef]
- Biswas, S.; Mita, M.A.; Afrose, S.; Hasan, M.R.; Shimu, M.S.S.; Zaman, S.; Saleh, M.A. An in silico approach to develop potential therapies against Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Heliyon 2024, 10, e25837. [Google Scholar] [CrossRef]
- Tripathi, S.M.; Akash, S.; Rahman, M.A.; Sundriyal, S. Identification of synthetically tractable MERS-CoV main protease inhibitors using structure-based virtual screening and molecular dynamics potential of mean force (PMF) calculations. J. Biomol. Struct. Dyn. 2025, 43, 787–797. [Google Scholar] [CrossRef]
- Zatla, I. Computational Screening of Natural Compounds as Antiviral Candidates Targeting the SARS-CoV-2 Main Protease. J. Integr. OMICS 2024, 14, 1–12. [Google Scholar] [CrossRef]
- Song, J.; Gao, Y.; Yin, P.; Li, Y.; Li, Y.; Zhang, J.; Su, Q.; Fu, X.; Pi, H. The Random Forest Model Has the Best Accuracy Among the Four Pressure Ulcer Prediction Models Using Machine Learning Algorithms. Risk Manag. Healthc. Policy 2021, 14, 1175–1187. [Google Scholar] [CrossRef]
- Skoreński, M.; Sieńczyk, M. Viral proteases as targets for drug design. Curr. Pharm. Des. 2013, 19, 1126–1153. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Tasneem, A.; Rai, G.P.; Reyaz, S.; Bairagya, H.R. The Possible Molecular Mechanism of SARS-CoV-2 Main Protease: New Structural Insights from Computational Methods. SciMedicine J. 2021, 2, 108–126. [Google Scholar] [CrossRef]
- Parise, A.; Cresca, S.; Magistrato, A. Molecular dynamics simulations for the structure-based drug design: Targeting small-GTPases proteins. Expert Opin. Drug Discov. 2024, 19, 1259–1279. [Google Scholar] [CrossRef] [PubMed]
- Paladino, A.; Morra, G.; Colombo, G. Structural Stability and Flexibility Direct the Selection of Activating Mutations in Epidermal Growth Factor Receptor Kinase. J. Chem. Inf. Model. 2015, 55, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Dimasi, A.; Moi, D.; Passarella, D.; Scala, A.; Piperno, A.; Micale, N. Recent Advances in SARS-CoV-2 Main Protease Inhibitors: From Nirmatrelvir to Future Perspectives. Biomolecules 2023, 13, 1339. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and COVID-19 Therapeutics. ChemMedChem 2020, 15, 907–932. [Google Scholar] [CrossRef]
- Charan, J.; Kaur, R.J.; Bhardwaj, P.; Haque, M.; Sharma, P.; Misra, S.; Godman, B. Rapid review of suspected adverse drug events due to remdesivir in the WHO database; findings and implications. Expert Rev. Clin. Pharmacol. 2020, 14, 95–103. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65. [Google Scholar] [CrossRef]
- Wolkove, N.; Baltzan, M. Amiodarone pulmonary toxicity. Can. Respir. J. J. Can. Thorac. Soc. 2009, 16, 43–48. [Google Scholar]
- Eusébio, S.; Soares, A.R.; Fiúza, P.; Garcia, T. Amiodarone-Induced Interstitial Pneumonia: A Cause of Respiratory Failure. Cureus 2024, 16, e74253. [Google Scholar] [CrossRef]
- Greene, N.; Song, M. Predicting in vivo safety characteristics using physiochemical properties and in vitro assays. Future Med. Chem. 2011, 3, 1503–1511. [Google Scholar] [CrossRef]
- Rocha, R.E.O.; Chaves, E.J.F.; Fischer, P.H.C.; Costa, L.S.C.; Grillo, I.B.; da Cruz, L.E.G.; Guedes, F.C.; da Silveira, C.H.; Scotti, M.T.; Camargo, A.D.; et al. A higher flexibility at the SARS-CoV-2 main protease active site compared to SARS-CoV and its potentialities for new inhibitor virtual screening targeting multi-conformers. J. Biomol. Struct. Dyn. 2021, 40, 9214–9234. [Google Scholar] [CrossRef] [PubMed]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobotics 2013, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Guido, R.; Ferrisi, S.; Lofaro, D.; Conforti, D. An Overview on the Advancements of Support Vector Machine Models in Healthcare Applications: A Review. Information 2024, 15, 235. [Google Scholar] [CrossRef]
- Ozturk Kiyak, E.; Ghasemkhani, B.; Birant, D. High-Level K-Nearest Neighbors (HLKNN): A Supervised Machine Learning Model for Classification Analysis. Electronics 2023, 12, 3828. [Google Scholar] [CrossRef]
- Domínguez-Almendros, S.; Benítez-Parejo, N.; Gonzalez-Ramirez, A.R. Logistic regression models. Allergol. Immunopathol. (Madr.) 2011, 39, 295–305. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Capecchi, A.; Probst, D.; Reymond, J.-L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. J. Cheminformatics 2020, 12, 43. [Google Scholar] [CrossRef]
- Refaeilzadeh, P.; Tang, L.; Liu, H. Cross-Validation. In Encyclopedia of Database Systems; Liu, L., Özsu, M.T., Eds.; Springer: Boston, MA, USA, 2009; pp. 532–538. ISBN 978-0-387-39940-9. [Google Scholar]
- Almutairi, G.O.; Malik, A.; Alonazi, M.; Khan, J.M.; Alhomida, A.S.; Khan, M.S.; Alenad, A.M.; Altwaijry, N.; Alafaleq, N.O. Expression, purification, and biophysical characterization of recombinant MERS-CoV main (Mpro) protease. Int. J. Biol. Macromol. 2022, 209, 984–990. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Characterization of the non-covalent interaction between the PF-07321332 inhibitor and the SARS-CoV-2 main protease. J. Mol. Graph. Model. 2022, 110, 108042. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Logistic Regression Model | Support Vector Machine Model | Gradient Boosting Model | K-Nearest Neighbors Model | Random Forest Model | |
|---|---|---|---|---|---|
| Accuracy | 0.81 | 0.78 | 0.84 | 0.59 | 0.97 |
| ROC-AUC Score | 0.90 | 0.90 | 0.93 | 0.82 | 0.98 |
| F1-Score | 0.8 | 0.77 | 0.82 | 0.68 | 0.98 |
| Macro Avg | 0.82 | 0.79 | 0.85 | 0.64 | 0.98 |
| Weighted Avg | 0.81 | 0.78 | 0.84 | 0.59 | 0.98 |
| Compounds | CID PubChem | Source | XP Score (kcal/mol) |
|---|---|---|---|
| Perenniporide B | 60199564 | Tropicoporus linteus | −9.17 |
| Phellifuropyranone A | 24770409 | Tropicoporus linteus | −9.081 |
| Terrestrol G | 24761035 | Penicillium terrestre | −8.71 |
| AW4 | 137348956 | /// | −7.34 |
| Compounds | H-Bond | Distance | Number | Hydrophobic | Distance | Number | Sulfur | Distance |
|---|---|---|---|---|---|---|---|---|
| Perenniporide B | Asp190 Gln167 Gln192 Glu169 Gly146 Gly146 His41 Leu144 Leu49 | 2,488 1,944 1,769 2,697 2,915 1,983 2,579 1,709 2,722 | 9 | Cys148 His41 Leu49 | 5,132 4,569 4,488 | 3 | ||
| Phellifuropyranone A | Gly146 His41 His194 Tht29 Thr29 Val193 | 3,351 3,1879 2,7442 1,7356 2,0224 1,9672 | 6 | Cys148 Cys148 His194 Leu49 Leu49 Met25 Met168 | 5,150 5,003 5,659 5,117 4,937 4,391 5,491 | 7 | Cys145 Met25 | 4,583 3,007 |
| Terrestrol G | Gln167 Leu49 Lys191 Thr26 | 1,7358 2,1533 3,7527 1,7978 | 4 | Leu49 Met25 | 4,876 5,001 | 2 | Cys145 Cys148 | 5,780 4,357 |
| AW4 | Cys148 Gln167 Gln192 Gln192 Glu169 Glu169 Leu49 Tyr54 Val193 | 2.428 2,080 2,405 3,394 1,884 2,723 2,472 2,635 2,674 | 9 | His41 His194 Leu49 | 4,178 4,549 3,655 | 3 |
| Compounds | MW (g/mol) | nRot | nHet | Flexibility | TPSA (Å2) | nRing | Log S | Log P |
|---|---|---|---|---|---|---|---|---|
| Perenniporide B | 366.13 | 7 | 8 | 0.538 | 122.52 | 2 | −3.061 | 1.554 |
| Phellifuropyranone A | 378.07 | 3 | 7 | 0.125 | 124.27 | 4 | −4.103 | 3.562 |
| Terrestrol G | 296.05 | 3 | 6 | 0.250 | 101.15 | 2 | −1.399 | 2.191 |
| AW4 | 533.16 | 15 | 13 | 0.938 | 171.13 | 2 | −1.945 | 1.025 |
| Compounds | QED | SAscore | Pfizer Rule | Lipinski Rule | Golden Triangle |
|---|---|---|---|---|---|
| Perenniporide B | 0.632 | 3.953 | Accepted | Accepted | Accepted |
| Phellifuropyranone A | 0.397 | 2.808 | Accepted | Accepted | Accepted |
| Terrestrol G | 0.559 | 2.716 | Accepted | Accepted | Accepted |
| AW4 | 0.25 | 3.988 | Accepted | Rejected | Rejected |
| Compounds | Caco-2 Permeability | HIA% | P-gp Inhibitor | PPB | Vd (L/kg) |
|---|---|---|---|---|---|
| Perenniporide B | −4.922 | 65.62 | Excellent | 80.876 | 1.259 |
| Phellifuropyranone A | −4.985 | 63.39 | Excellent | 96.695 | 0.355 |
| Terrestrol G | −5.168 | 67.33 | Excellent | 95.148 | 0.511 |
| AW4 | −5.607 | 36.24 | Excellent | 76.47 | 0.341 |
| Compounds | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | CL (ml/min/Kg) | T1/2 (H) |
|---|---|---|---|---|---|---|---|
| Perenniporide B | 0.493 | 0.04 | 0.067 | 0.012 | 0.059 | 6.159 | 0.545 |
| Phellifuropyranone A | 0.934 | 0.152 | 0.447 | 0.156 | 0.498 | 7.691 | 0.814 |
| Terrestrol G | 0.418 | 0.05 | 0.181 | 0.444 | 0.074 | 13.677 | 0.966 |
| AW4 | 0.014 | 0.061 | 0.166 | 0.006 | 0.16 | 2.121 | 0.626 |
| Compounds | hERG Blockers | AMES Toxicity | Skin Sensitization | Carcinogenicity | Respiratory Toxicity |
|---|---|---|---|---|---|
| Perenniporide B | 0.039 | 0.347 | 0.173 | 0.157 | 0.669 |
| Phellifuropyranone A | 0.081 | 0.036 | 0.951 | 0.358 | 0.106 |
| Terrestrol G | 0.165 | 0.524 | 0.952 | 0.128 | 0.134 |
| AW4 | 0.069 | 0.014 | 0.062 | 0.016 | 0.025 |
| Compounds | Hepatotoxicity | Mutagenicity | Cytotoxicity | Ecotoxicity | Ld50 |
|---|---|---|---|---|---|
| Perenniporide B | Inactive | Inactive | Inactive | Inactive | 220 |
| Phellifuropyranone A | Inactive | Inactive | Inactive | Inactive | 800 |
| Terrestrol G | Inactive | Inactive | Inactive | Inactive | 2500 |
| AW4 | Inactive | Inactive | Inactive | Inactive | 3000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouassaf, M.; Mazri, R.; Khan, S.U.; Rengasamy, K.R.R.; Alhatlani, B.Y. Machine Learning-Guided Screening and Molecular Docking for Proposing Naturally Derived Drug Candidates Against MERS-CoV 3CL Protease. Int. J. Mol. Sci. 2025, 26, 3047. https://doi.org/10.3390/ijms26073047
Ouassaf M, Mazri R, Khan SU, Rengasamy KRR, Alhatlani BY. Machine Learning-Guided Screening and Molecular Docking for Proposing Naturally Derived Drug Candidates Against MERS-CoV 3CL Protease. International Journal of Molecular Sciences. 2025; 26(7):3047. https://doi.org/10.3390/ijms26073047
Chicago/Turabian StyleOuassaf, Mebarka, Radhia Mazri, Shafi Ullah Khan, Kannan R. R. Rengasamy, and Bader Y. Alhatlani. 2025. "Machine Learning-Guided Screening and Molecular Docking for Proposing Naturally Derived Drug Candidates Against MERS-CoV 3CL Protease" International Journal of Molecular Sciences 26, no. 7: 3047. https://doi.org/10.3390/ijms26073047
APA StyleOuassaf, M., Mazri, R., Khan, S. U., Rengasamy, K. R. R., & Alhatlani, B. Y. (2025). Machine Learning-Guided Screening and Molecular Docking for Proposing Naturally Derived Drug Candidates Against MERS-CoV 3CL Protease. International Journal of Molecular Sciences, 26(7), 3047. https://doi.org/10.3390/ijms26073047