
Glatiramer Acetate Modifies the Immune Profiles of Monocyte-Derived Dendritic Cells In Vitro Without Affecting Their Generation

 ,
,  , ,
, ,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
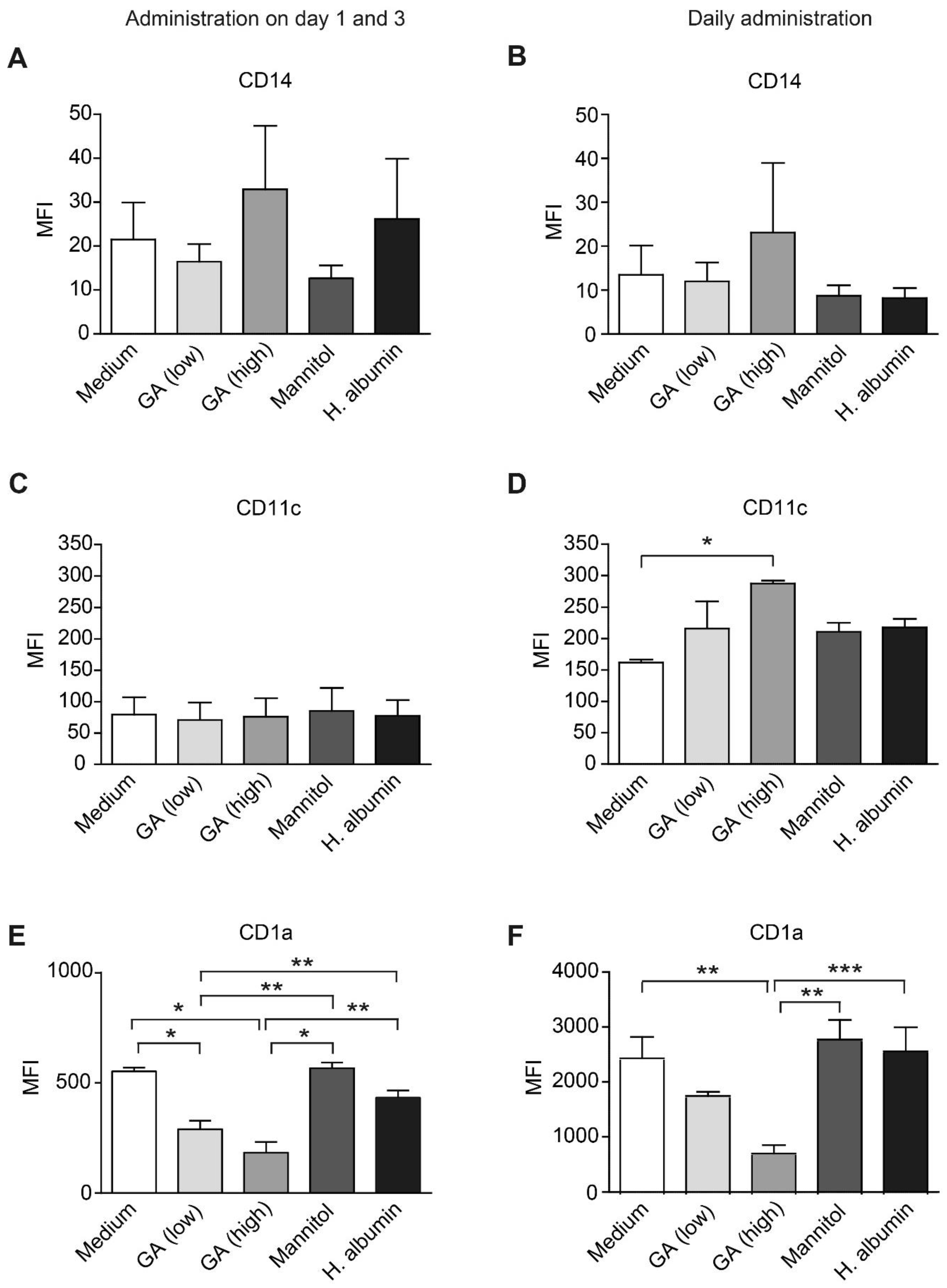
2.1. GA Reduces CD1a Expression on moDCs
2.2. GA Upregulates APC Activation Markers in moDCs
2.3. GA Does Not Influence the Expression of Transcription Factors Essential for the Differentiation of Monocytes into Macrophages or DCs
2.4. GA Upregulates Intracellular CD68 in Monocytes
2.5. Monocytes Enhance the Expression of Adhesion Molecules in Response to GA
3. Materials and Methods
3.1. Isolation of Monocytes
3.2. In Vitro Generation and Treatment of moDCs
3.3. In Vitro Treatment of Monocytes
3.4. Measurement of Cell Viability and Apoptosis
3.5. Immunocytochemistry
3.6. Flow Cytometry
3.7. Gene Expression Analysis
3.8. Statistical Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Yong, V.W. Myeloid cells—Targets of medication in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef]
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687. [Google Scholar] [CrossRef]
- Hettinger, J.; Richards, D.M.; Hansson, J.; Barra, M.M.; Joschko, A.C.; Krijgsveld, J.; Feuerer, M. Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 2013, 14, 821–830. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Blood monocytes and their subsets: Established features and open questions. Front. Immunol. 2015, 6, 423. [Google Scholar] [CrossRef] [PubMed]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- Monaghan, K.L.; Zheng, W.; Hu, G.; Wan, E.C.K. Monocytes and monocyte-derived antigen-presenting cells have distinct gene signatures in experimental model of multiple sclerosis. Front. Immunol. 2019, 10, 2779. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T.; Takahashi, T.; Nakashima, I. Peripheral blood monocyte count at onset may affect the prognosis in multiple sclerosis. J. Neuroimmunol. 2018, 319, 37–40. [Google Scholar] [CrossRef]
- Prineas, J.W.; Parratt, J.D.E. Multiple sclerosis: Microglia, monocytes, and macrophage-mediated demyelination. J. Neuropathol. Exp. Neurol. 2021, 80, 975–996. [Google Scholar] [CrossRef]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J. Rejuvenation of regeneration in the aging central nervous system. Cell Stem. Cell 2012, 10, 96–103. [Google Scholar] [CrossRef]
- Nuyts, A.H.; Lee, W.P.; Bashir-Dar, R.; Berneman, Z.N.; Cools, N. Dendritic cells in multiple sclerosis: Key players in the immunopathogenesis, key players for new cellular immunotherapies? Mult. Scler. 2013, 19, 995–1002. [Google Scholar] [CrossRef]
- Varkony, H.; Weinstein, V.; Klinger, E.; Sterling, J.; Cooperman, H.; Komlosh, T.; Ladkani, D.; Schwartz, R. The glatiramoid class of immunomodulator drugs. Expert. Opin. Pharmacother. 2009, 10, 657–668. [Google Scholar] [CrossRef]
- Johnson, K.P. Glatiramer acetate for treatment of relapsing-remitting multiple sclerosis. Expert. Rev. Neurother. 2012, 12, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T.; Calabrese, P.; Penner, I.K.; Apfel, R. QualiCOP: Real-world effectiveness, tolerability, and quality of life in patients with relapsing-remitting multiple sclerosis treated with glatiramer acetate, treatment-naive patients, and previously treated patients. J. Neurol. 2016, 263, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Prod'homme, T.; Zamvil, S.S. The evolving mechanisms of action of glatiramer acetate. Cold Spring Harb. Perspect. Med. 2019, 9, a029249. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.; Goodman, A.D.; Johnson, K.; Kachuck, N.; Lindsey, J.W.; Lisak, R.; Luzzio, C.; Myers, L.; Panitch, H.; Preiningerova, J.; et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: Results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult. Scler. 2010, 16, 342–350. [Google Scholar] [CrossRef]
- Arnon, R.; Aharoni, R. Glatiramer acetate: From bench to bed and back. Isr. Med. Assoc. J. 2019, 21, 151–157. [Google Scholar]
- Borchard, G.; Crommelin, D.J.A. Equivalence of glatiramer acetate products: Challenges in assessing pharmaceutical equivalence and critical clinical performance attributes. Expert. Opin. Drug. Deliv. 2018, 15, 247–259. [Google Scholar] [CrossRef]
- Aharoni, R.; Teitelbaum, D.; Sela, M.; Arnon, R. Bystander suppression of experimental autoimmune encephalomyelitis by T cell lines and clones of the Th2 type induced by copolymer 1. J. Neuroimmunol. 1998, 91, 135–146. [Google Scholar] [CrossRef]
- Haas, J.; Korporal, M.; Balint, B.; Fritzsching, B.; Schwarz, A.; Wildemann, B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. J. Neuroimmunol. 2009, 216, 113–117. [Google Scholar] [CrossRef]
- Aharoni, R.; Eilam, R.; Stock, A.; Vainshtein, A.; Shezen, E.; Gal, H.; Friedman, N.; Arnon, R. Glatiramer acetate reduces Th-17 inflammation and induces regulatory T-cells in the CNS of mice with relapsing-remitting or chronic EAE. J. Neuroimmunol. 2010, 225, 100–111. [Google Scholar] [CrossRef]
- Oreja-Guevara, C.; Ramos-Cejudo, J.; Aroeira, L.S.; Chamorro, B.; Diez-Tejedor, E. TH1/TH2 Cytokine profile in relapsing-remitting multiple sclerosis patients treated with Glatiramer acetate or Natalizumab. BMC Neurol. 2012, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Kuerten, S.; Jackson, L.J.; Kaye, J.; Vollmer, T.L. Impact of glatiramer acetate on b cell-mediated pathogenesis of multiple sclerosis. CNS Drugs 2018, 32, 1039–1051. [Google Scholar] [CrossRef]
- Sand, K.L.; Knudsen, E.; Rolin, J.; Al-Falahi, Y.; Maghazachi, A.A. Modulation of natural killer cell cytotoxicity and cytokine release by the drug glatiramer acetate. Cell Mol. Life. Sci. 2009, 66, 1446–1456. [Google Scholar] [CrossRef]
- Al-Falahi, Y.; Sand, K.L.; Knudsen, E.; Damaj, B.B.; Rolin, J.; Maghazachi, A.A. Splenic natural killer cell activity in two models of experimental neurodegenerative diseases. J. Cell Mol. Med. 2009, 13, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Hoglund, R.A.; Holmoy, T.; Harbo, H.F.; Maghazachi, A.A. A one year follow-up study of natural killer and dendritic cells activities in multiple sclerosis patients receiving glatiramer acetate (GA). PLoS ONE 2013, 8, e62237. [Google Scholar] [CrossRef] [PubMed]
- Vieira, P.L.; Heystek, H.C.; Wormmeester, J.; Wierenga, E.A.; Kapsenberg, M.L. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J. Immunol. 2003, 170, 4483–4488. [Google Scholar] [CrossRef]
- Kim, H.J.; Ifergan, I.; Antel, J.P.; Seguin, R.; Duddy, M.; Lapierre, Y.; Jalili, F.; Bar-Or, A. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J. Immunol. 2004, 172, 7144–7153. [Google Scholar] [CrossRef]
- Chuluundorj, D.; Harding, S.A.; Abernethy, D.; La Flamme, A.C. Glatiramer acetate treatment normalized the monocyte activation profile in MS patients to that of healthy controls. Immunol. Cell Biol. 2017, 95, 297–305. [Google Scholar] [CrossRef]
- Weber, M.S.; Prod'homme, T.; Youssef, S.; Dunn, S.E.; Rundle, C.D.; Lee, L.; Patarroyo, J.C.; Stuve, O.; Sobel, R.A.; Steinman, L.; et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat. Med. 2007, 13, 935–943. [Google Scholar] [CrossRef]
- Pul, R.; Moharregh-Khiabani, D.; Skuljec, J.; Skripuletz, T.; Garde, N.; Voss, E.V.; Stangel, M. Glatiramer acetate modulates TNF-alpha and IL-10 secretion in microglia and promotes their phagocytic activity. J. Neuroimmune. Pharmacol. 2011, 6, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Pul, R.; Morbiducci, F.; Skuljec, J.; Skripuletz, T.; Singh, V.; Diederichs, U.; Garde, N.; Voss, E.V.; Trebst, C.; Stangel, M. Glatiramer acetate increases phagocytic activity of human monocytes in vitro and in multiple sclerosis patients. PLoS ONE 2012, 7, e51867. [Google Scholar] [CrossRef]
- Weber, M.S.; Starck, M.; Wagenpfeil, S.; Meinl, E.; Hohlfeld, R.; Farina, C. Multiple sclerosis: Glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004, 127, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, M.; Montarolo, F.; Perga, S.; Martire, S.; Brescia, F.; Malucchi, S.; Bertolotto, A. Biological activity of glatiramer acetate on Treg and anti-inflammatory monocytes persists for more than 10years in responder multiple sclerosis patients. Clin. Immunol. 2017, 181, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Bakri, Y.; Sarrazin, S.; Mayer, U.P.; Tillmanns, S.; Nerlov, C.; Boned, A.; Sieweke, M.H. Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood 2005, 105, 2707–2716. [Google Scholar] [CrossRef]
- Singh, V.; Prajeeth, C.K.; Gudi, V.; Benardais, K.; Voss, E.V.; Stangel, M. 2-Chlorodeoxyadenosine (cladribine) induces apoptosis in human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2013, 173, 288–297. [Google Scholar] [CrossRef]
- Kasindi, A.; Fuchs, D.T.; Koronyo, Y.; Rentsendorj, A.; Black, K.L.; Koronyo-Hamaoui, M. Glatiramer Acetate Immunomodulation: Evidence of Neuroprotection and Cognitive Preservation. Cells 2022, 11, 1578. [Google Scholar] [CrossRef]
- Huang, A.; Groer, C.; Lu, R.; Forrest, M.L.; Griffin, J.D.; Berkland, C.J. Glatiramer acetate complexed with CpG as intratumoral immunotherapy in combination with anti-PD-1. Mol. Pharm. 2022, 19, 4357–4369. [Google Scholar] [CrossRef]
- Murphy, R.A.; Coates, M.; Thrane, S.; Sabnis, A.; Harrison, J.; Schelenz, S.; Edwards, A.M.; Vorup-Jensen, T.; Davies, J.C. Synergistic activity of repurposed peptide drug glatiramer acetate with tobramycin against cystic fibrosis Pseudomonas aeruginosa. Microbiol. Spectr. 2022, 10, e0081322. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Mohamed, G.A.; Fahmy, U.A.; Eid, B.G.; Al-Rabia, M.W.; Khedr, A.I.M.; Nasrullah, M.Z.; Ibrahim, S.R.M.; Abdel-Naim, A.B.; Ahmed, O.A.A.; et al. Thioctamer: A novel thioctic acid-glatiramer acetate nanoconjugate expedites wound healing in diabetic rats. Drug Deliv. 2022, 29, 1776–1784. [Google Scholar] [CrossRef]
- Alves, V.; Martins, P.H.; Miranda, B.; de Andrade, I.B.; Pereira, L.; Maeda, C.T.; de Sousa Araujo, G.R.; Frases, S. Assessing the in vitro potential of glatiramer acetate (Copaxone((R))) as a chemotherapeutic candidate for the treatment of cryptococcus neoformans infection. J. Fungi. 2023, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Keating, G.M. Glatiramer acetate: A review of its use in relapsing-remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. Drugs 2010, 70, 1545–1577. [Google Scholar] [CrossRef]
- Miller, A.; Shapiro, S.; Gershtein, R.; Kinarty, A.; Rawashdeh, H.; Honigman, S.; Lahat, N. Treatment of multiple sclerosis with copolymer-1 (Copaxone): Implicating mechanisms of Th1 to Th2/Th3 immune-deviation. J. Neuroimmunol. 1998, 92, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.W.; Schmied, M.C.; Cook, S.L.; Krieger, J.I.; Hafler, D.A. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J. Clin. Invest. 2000, 105, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Farina, C.; Yassouridis, A.; Wiendl, H.; Then Bergh, F.; Dose, T.; Wekerle, H.; Hohlfeld, R. Multiple sclerosis: Comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7452–7457. [Google Scholar] [CrossRef]
- Hussien, Y.; Sanna, A.; Soderstrom, M.; Link, H.; Huang, Y.M. Glatiramer acetate and IFN-beta act on dendritic cells in multiple sclerosis. J. Neuroimmunol. 2001, 121, 102–110. [Google Scholar] [CrossRef]
- Jung, S.; Siglienti, I.; Grauer, O.; Magnus, T.; Scarlato, G.; Toyka, K. Induction of IL-10 in rat peritoneal macrophages and dendritic cells by glatiramer acetate. J. Neuroimmunol. 2004, 148, 63–73. [Google Scholar] [CrossRef]
- Derakhshani, A.; Asadzadeh, Z.; Baradaran, B.; Safarpour, H.; Rahmani, S.; Leone, P.; Abdoli Shadbad, M.; Hosseinkhani, N.; Ghasemigol, M.; Ayromlou, H.; et al. The expression pattern of VISTA in the PBMCs of relapsing-remitting multiple sclerosis patients: A single-cell RNA sequencing-based study. Biomed. Pharmacother. 2022, 148, 112725. [Google Scholar] [CrossRef]
- Sie, C.; Korn, T. Dendritic cells in central nervous system autoimmunity. Semin. Immunopathol. 2017, 39, 99–111. [Google Scholar] [CrossRef]
- Osugi, Y.; Vuckovic, S.; Hart, D.N. Myeloid blood CD11c(+) dendritic cells and monocyte-derived dendritic cells differ in their ability to stimulate T lymphocytes. Blood 2002, 100, 2858–2866. [Google Scholar] [CrossRef]
- Mena, J.; Alloza, I.; Tulloch Navarro, R.; Aldekoa, A.; Diez Garcia, J.; Villanueva Etxebarria, A.; Lindskog, C.; Antiguedad, A.; Boyero, S.; Mendibe-Bilbao, M.D.M.; et al. Genomic multiple sclerosis risk variants modulate the expression of the ANKRD55-IL6ST gene region in immature dendritic cells. Front. Immunol. 2021, 12, 816930. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef] [PubMed]
- Kouwenhoven, M.; Ozenci, V.; Tjernlund, A.; Pashenkov, M.; Homman, M.; Press, R.; Link, H. Monocyte-derived dendritic cells express and secrete matrix-degrading metalloproteinases and their inhibitors and are imbalanced in multiple sclerosis. J. Neuroimmunol. 2002, 126, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Hesske, L.; Vincenzetti, C.; Heikenwalder, M.; Prinz, M.; Reith, W.; Fontana, A.; Suter, T. Induction of inhibitory central nervous system-derived and stimulatory blood-derived dendritic cells suggests a dual role for granulocyte-macrophage colony-stimulating factor in central nervous system inflammation. Brain 2010, 133, 1637–1654. [Google Scholar] [CrossRef]
- McMahon, E.J.; Bailey, S.L.; Castenada, C.V.; Waldner, H.; Miller, S.D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005, 11, 335–339. [Google Scholar] [CrossRef]
- Parsa, R.; Lund, H.; Tosevski, I.; Zhang, X.M.; Malipiero, U.; Beckervordersandforth, J.; Merkler, D.; Prinz, M.; Gyllenberg, A.; James, T.; et al. TGFbeta regulates persistent neuroinflammation by controlling Th1 polarization and ROS production via monocyte-derived dendritic cells. Glia 2016, 64, 1925–1937. [Google Scholar] [CrossRef]
- Raiotach-Regue, D.; Grau-Lopez, L.; Naranjo-Gomez, M.; Ramo-Tello, C.; Pujol-Borrell, R.; Martinez-Caceres, E.; Borras, F.E. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur. J. Immunol. 2012, 42, 771–782. [Google Scholar] [CrossRef]
- Sun, H.; Zhi, K.; Hu, L.; Fan, Z. The activation and regulation of beta2 integrins in phagocytes and phagocytosis. Front. Immunol. 2021, 12, 633639. [Google Scholar] [CrossRef]
- Cheong, C.; Matos, I.; Choi, J.H.; Dandamudi, D.B.; Shrestha, E.; Longhi, M.P.; Jeffrey, K.L.; Anthony, R.M.; Kluger, C.; Nchinda, G.; et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 2010, 143, 416–429. [Google Scholar] [CrossRef]
- Leslie, D.S.; Dascher, C.C.; Cembrola, K.; Townes, M.A.; Hava, D.L.; Hugendubler, L.C.; Mueller, E.; Fox, L.; Roura-Mir, C.; Moody, D.B.; et al. Serum lipids regulate dendritic cell CD1 expression and function. Immunology 2008, 125, 289–301. [Google Scholar] [CrossRef]
- Gogolak, P.; Rethi, B.; Szatmari, I.; Lanyi, A.; Dezso, B.; Nagy, L.; Rajnavolgyi, E. Differentiation of CD1a- and CD1a+ monocyte-derived dendritic cells is biased by lipid environment and PPARgamma. Blood 2007, 109, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Capello, E.; Mancardi, G.L.; Aloisi, F. Dendritic cells in multiple sclerosis lesions: Maturation stage, myelin uptake, and interaction with proliferating T cells. J. Neuropathol. Exp. Neurol. 2006, 65, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Bine, S.; Haziot, A.; Malikova, I.; Pelletier, J.; Charron, D.; Boucraut, J.; Mooney, N.; Gelin, C. Alteration of CD1 expression in multiple sclerosis. Clin. Exp. Immunol. 2012, 169, 10–16. [Google Scholar] [CrossRef]
- Hussien, Y.; Sanna, A.; Soderstrom, M.; Link, H.; Huang, Y.M. Multiple sclerosis: Expression of CD1a and production of IL-12p70 and IFN-gamma by blood mononuclear cells in patients on combination therapy with IFN-beta and glatiramer acetate compared to monotherapy with IFN-beta. Mult. Scler. 2004, 10, 16–25. [Google Scholar] [CrossRef]
- Fridkis-Hareli, M.; Strominger, J.L. Promiscuous binding of synthetic copolymer 1 to purified HLA-DR molecules. J. Immunol. 1998, 160, 4386–4397. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, N.; Ohnishi, H.; Kondo, N.; Strominger, J.L. The role of dendritic cells in the generation of CD4(+) CD25(HI) Foxp3(+) T cells induced by amino acid copolymers. Int. Immunol. 2013, 25, 53–65. [Google Scholar] [CrossRef]
- Hausler, D.; Hajiyeva, Z.; Traub, J.W.; Zamvil, S.S.; Lalive, P.H.; Bruck, W.; Weber, M.S. Glatiramer acetate immune modulates B-cell antigen presentation in treatment of MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e698. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Invest. 2017, 97, 4–13. [Google Scholar] [CrossRef]
- Kurushima, H.; Ramprasad, M.; Kondratenko, N.; Foster, D.M.; Quehenberger, O.; Steinberg, D. Surface expression and rapid internalization of macrosialin (mouse CD68) on elicited mouse peritoneal macrophages. J. Leukoc. Biol. 2000, 67, 104–108. [Google Scholar] [CrossRef]
- Nissen, J.C.; Thompson, K.K.; West, B.L.; Tsirka, S.E. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp. Neurol. 2018, 307, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Warren, M.K.; Haskill, S. Colony-stimulating factor-induced monocyte survival and differentiation into macrophages in serum-free cultures. J. Immunol. 1987, 139, 3703–3709. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skuljec, J.; Sardari, M.; Su, C.; Müller-Dahlke, J.; Singh, V.; Janjic, M.M.; Kleinschnitz, C.; Pul, R. Glatiramer Acetate Modifies the Immune Profiles of Monocyte-Derived Dendritic Cells In Vitro Without Affecting Their Generation. Int. J. Mol. Sci. 2025, 26, 3013. https://doi.org/10.3390/ijms26073013
Skuljec J, Sardari M, Su C, Müller-Dahlke J, Singh V, Janjic MM, Kleinschnitz C, Pul R. Glatiramer Acetate Modifies the Immune Profiles of Monocyte-Derived Dendritic Cells In Vitro Without Affecting Their Generation. International Journal of Molecular Sciences. 2025; 26(7):3013. https://doi.org/10.3390/ijms26073013
Chicago/Turabian StyleSkuljec, Jelena, Maryam Sardari, Chuanxin Su, Julia Müller-Dahlke, Vikramjeet Singh, Marija M. Janjic, Christoph Kleinschnitz, and Refik Pul. 2025. "Glatiramer Acetate Modifies the Immune Profiles of Monocyte-Derived Dendritic Cells In Vitro Without Affecting Their Generation" International Journal of Molecular Sciences 26, no. 7: 3013. https://doi.org/10.3390/ijms26073013
APA StyleSkuljec, J., Sardari, M., Su, C., Müller-Dahlke, J., Singh, V., Janjic, M. M., Kleinschnitz, C., & Pul, R. (2025). Glatiramer Acetate Modifies the Immune Profiles of Monocyte-Derived Dendritic Cells In Vitro Without Affecting Their Generation. International Journal of Molecular Sciences, 26(7), 3013. https://doi.org/10.3390/ijms26073013