

BIRC3 RNA Editing Modulates Lipopolysaccharide-Induced Liver Inflammation: Potential Implications for Animal Health


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
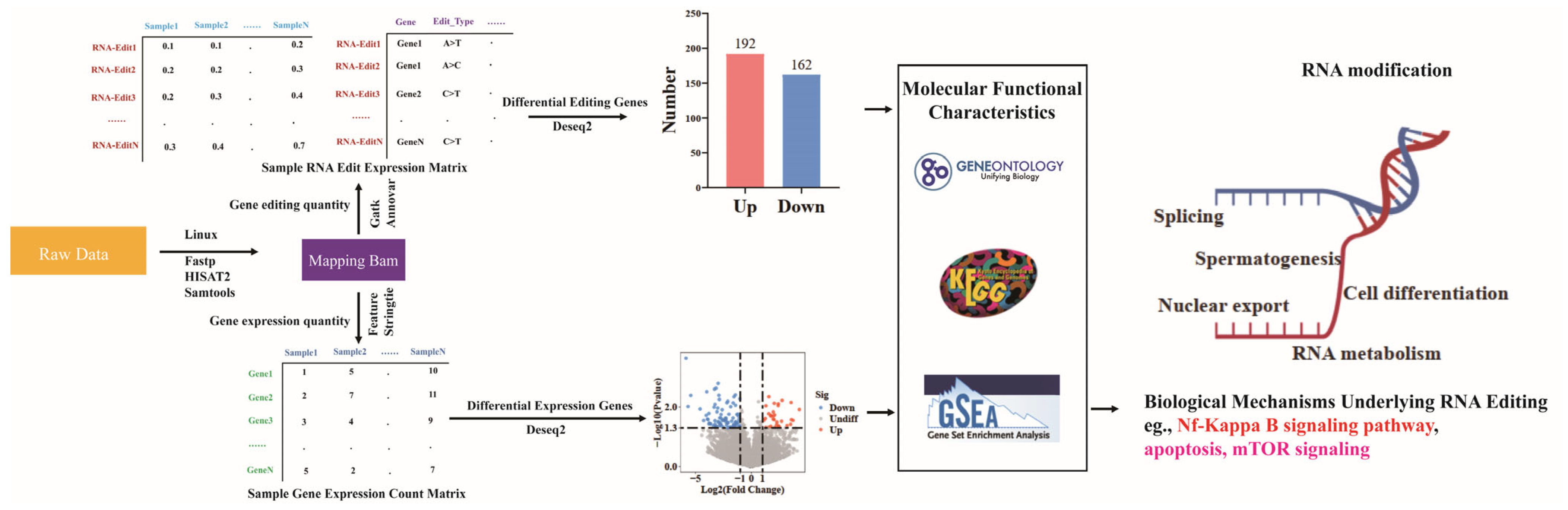
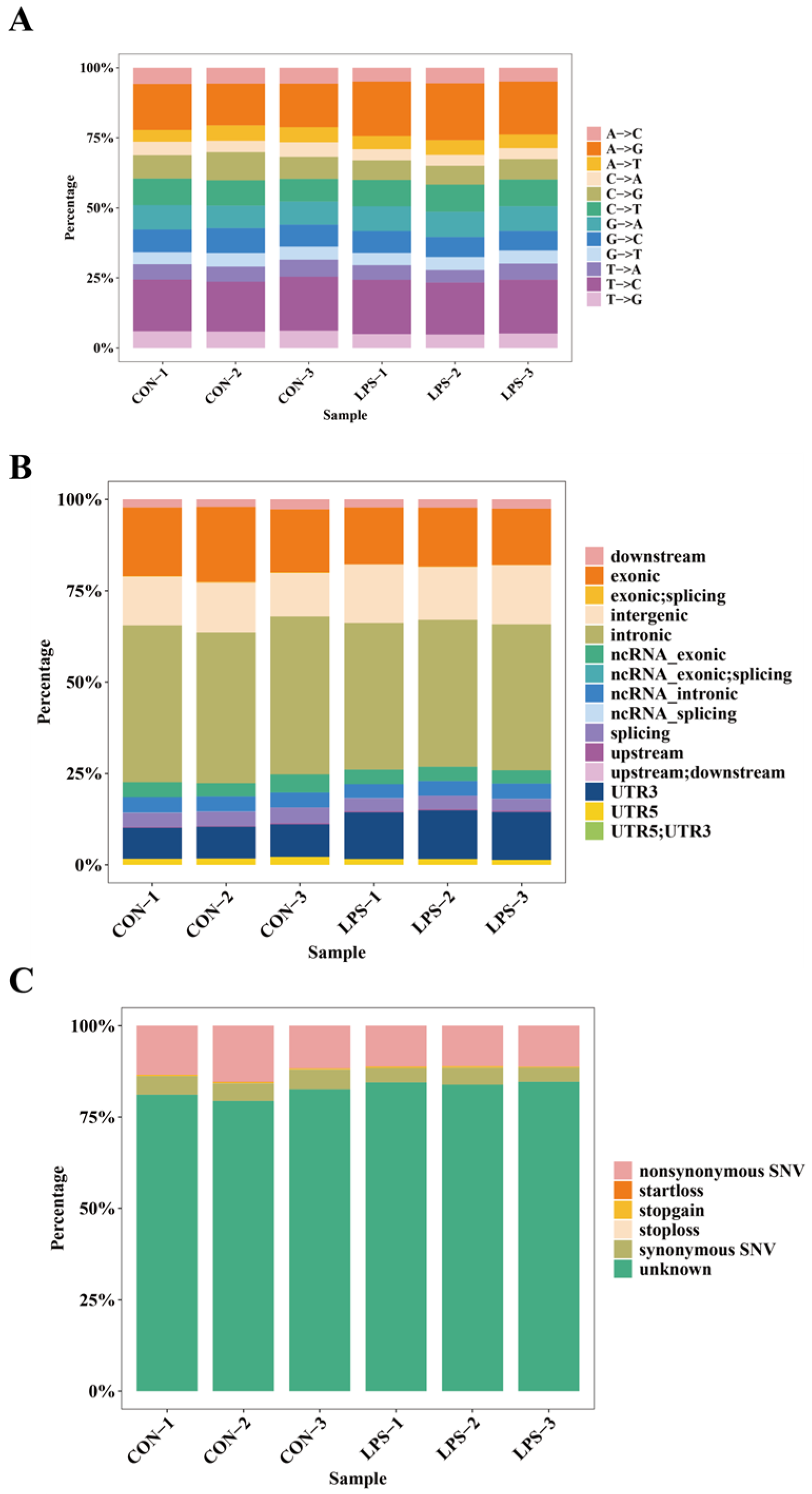
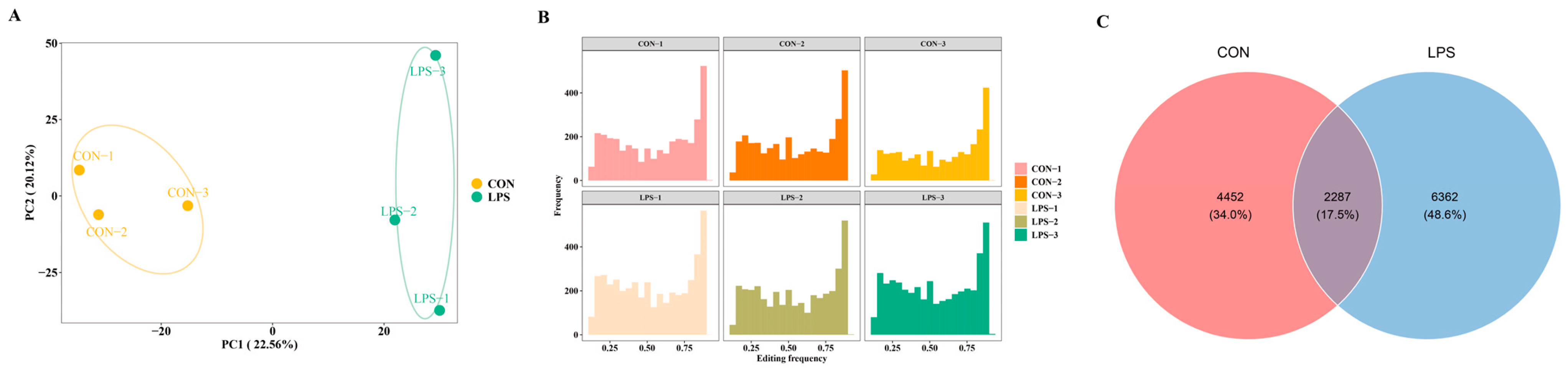
2.1. Comprehensive Map of RNA Editing Events
2.2. Differential Analysis of RNA Editing Events
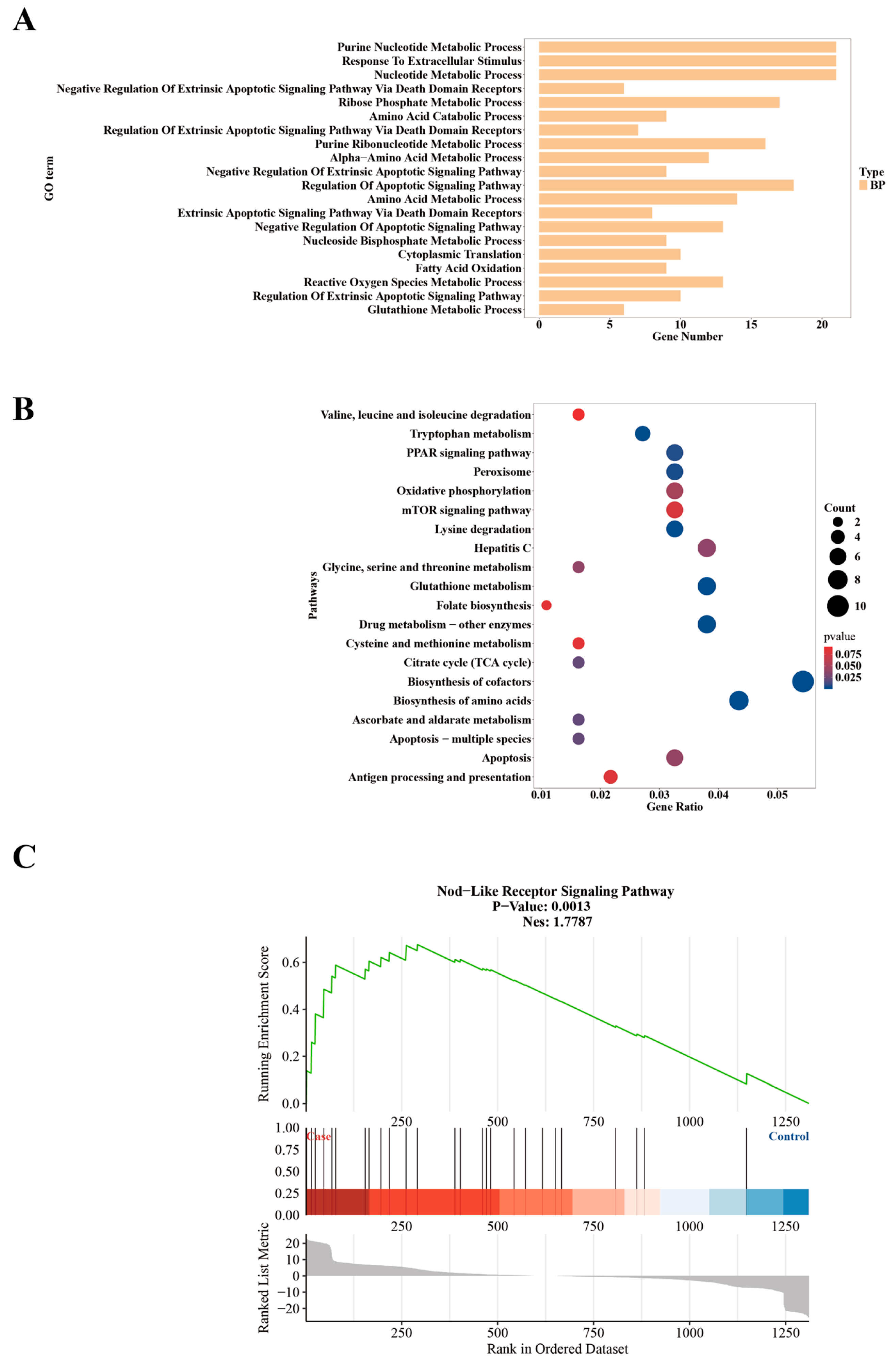
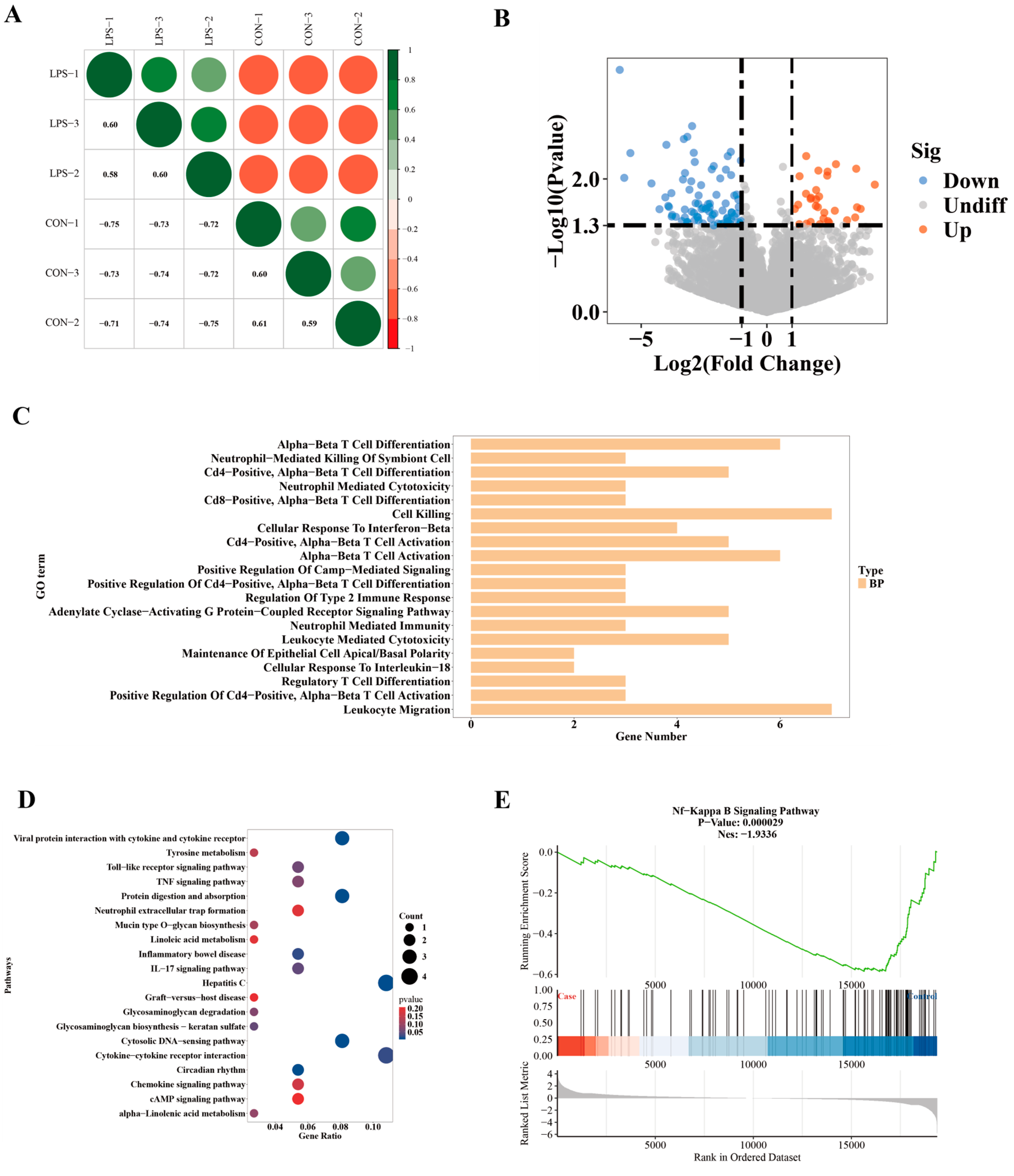
2.3. Functional Enrichment Analysis of Differential Editing Gene Sets
2.4. LPS Alters Gene Expression at the Transcriptional Level
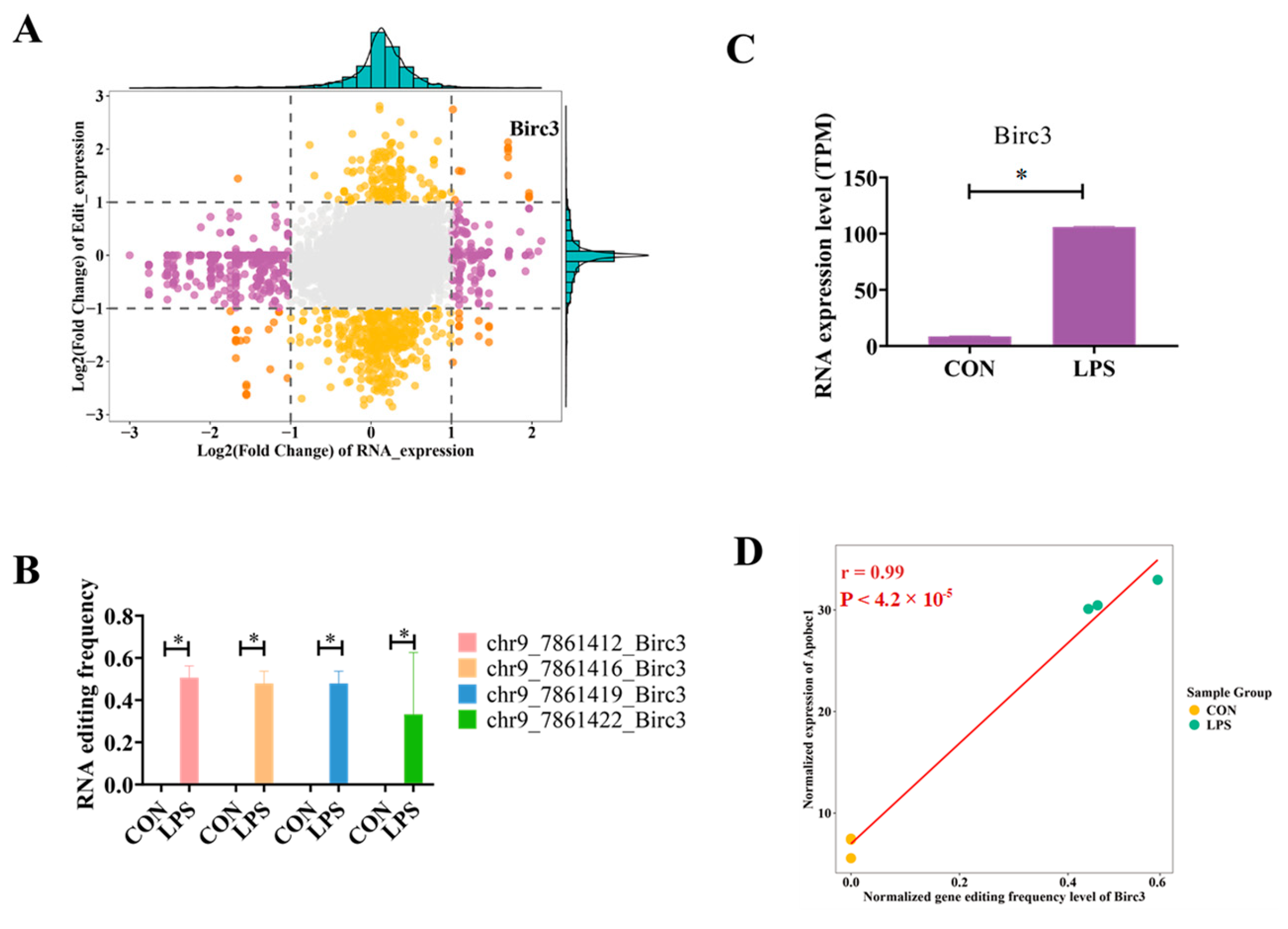
2.5. Integrated Analysis of RNA Editing and RNA Expression
3. Materials and Methods
3.1. Ethics Statement
3.2. Sample Collection and Sequencing
3.3. RNA-Seq Data Preprocessing
3.4. Alignments and RNA Editing Event Identification
3.5. Differential Editing Analysis of Differential Editing Events
3.6. RNA Expression Quantification and Analysis
3.7. Correlation Analysis Between RNA Editing and Gene Expression
3.8. Pathway Enrichment
3.9. Statistical Analysis
4. Discussion
4.1. LPS-Induced Alterations in RNA Editing and Gene Expression: Implications for Liver Inflammation and Damage
4.2. Birc3 Editing May Be a Potential Inflammatory Mechanism Induced by LPS
4.3. Strategies for Preventing and Managing LPS-Induced Liver Damage
4.4. Limitations and Future Research Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Rapala, J.; Lahti, K.; Räsänen, L.A.; Esala, A.L.; Niemelä, S.I.; Sivonen, K. Endotoxins associated with cyanobacteria and their removal during drinking water treatment. Water Res. 2002, 36, 2627–2635. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Miyauchi, K.; Shirasuna, K.; Bollwein, H.; Magata, F.; Murayama, C.; Miyamoto, A. Effects of lipopolysaccharide (LPS) and peptidoglycan (PGN) on estradiol production in bovine granulosa cells from small and large follicles. Toxicol. Vitr. 2012, 26, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, F.; Zhang, M.; Zhang, Z.; Bai, M.; Guo, G.; Wang, L. Endotoxin contamination, a potentially important inflammation factor in water and wastewater: A review. Sci. Total Environ. 2019, 681, 365–378. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Guo, H.; Gong, Y.; Zhao, R. Lipopolysaccharide-induced mitochondrial dysfunction in boar sperm is mediated by activation of oxidative phosphorylation. Theriogenology 2017, 87, 1–8. [Google Scholar] [CrossRef]
- Huang, J.; Liu, J.; Chang, G.; Wang, Y.; Ma, N.; Roy, A.C.; Shen, X. Glutamine supplementation attenuates the inflammation caused by LPS-induced acute lung injury in mice by regulating the TLR4/MAPK signaling pathway. Inflammation 2021, 44, 2180–2192. [Google Scholar] [CrossRef]
- Salliou, N. Quitting livestock farming: Transfarmation pathways and factors of change from post-livestock farmers’ accounts. Front. Sustain. Food Syst. 2023, 7, 1122992. [Google Scholar] [CrossRef]
- Tullo, E.; Finzi, A.; Guarino, M. Environmental impact of livestock farming and Precision Livestock Farming as a mitigation strategy. Sci. Total Environ. 2019, 650, 2751–2760. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Li, W.; Yan, K.; Liu, Y.; Xu, H.; Yang, X. Allicin protects porcine oocytes against LPS-induced defects during maturation in vitro. Theriogenology 2022, 182, 138–147. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Cheng, J.; Cheng, J.; Zhao, L.; Dai, C.; Deng, Y. Lipopolysaccharide accelerates tryptophan degradation in the ovary and the derivative kynurenine disturbs hormone biosynthesis and reproductive performance. J. Hazard. Mater. 2023, 458, 131988. [Google Scholar]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in health and disease. Adv. Clin. Chem. 2020, 95, 165–218. [Google Scholar]
- Schaub, M.; Keller, W. RNA Editing by Adenosine Deaminases Generates RNA and Protein Diversity. Biochimie 2002, 84, 791–803. [Google Scholar] [PubMed]
- Tang, W.; Fei, Y.; Page, M. Biological Significance of RNA Editing in Cells. Mol. Biotechnol. 2012, 52, 91–100. [Google Scholar] [PubMed]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Härtel, B.; Brennicke, A. RNA Editing in Plants and Its Evolution. Annu. Rev. Genet. 2013, 47, 335–352. [Google Scholar]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K. Dynamic Landscape and Regulation of RNA Editing in Mammals. Nature 2017, 550, 249–254. [Google Scholar]
- Liao, W.; Nie, W.; Ahmad, I.; Chen, G.; Zhu, B. The Occurrence, Characteristics, and Adaptation of A-to-I RNA Editing in Bacteria: A Review. Front. Microbiol. 2023, 14, 1143929. [Google Scholar]
- Hu, Y.-X.; Huang, A.; Li, Y.; Molloy, D.P.; Huang, C. Emerging Roles of the C-to-U RNA Editing in Plant Stress Responses. Plant Sci. 2024, 349, 112263. [Google Scholar]
- Yablonovitch, A.L.; Deng, P.; Jacobson, D.; Li, J.B. The Evolution and Adaptation of A-to-I RNA Editing. PLoS Genet. 2017, 13, e1007064. [Google Scholar]
- Kung, C.P.; Maggi, L.B., Jr.; Weber, J.D. The role of RNA editing in cancer development and metabolic disorders. Front. Endocrinol. 2018, 9, 762. [Google Scholar]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar]
- Brümmer, A.; Yang, Y.; Chan, T.W.; Xiao, X. Structure-mediated modulation of mRNA abundance by A-to-I editing. Nat. Commun. 2017, 8, 1255. [Google Scholar]
- Liu, Z.; Zhang, J. Human C-to-U coding RNA editing is largely nonadaptive. Mol. Biol. Evol. 2018, 35, 963–969. [Google Scholar]
- Keegan, L.P.; Hajji, K.; O’Connell, M.A. Adenosine deaminase acting on RNA (ADAR) enzymes: A journey from weird to wondrous. Acc. Chem. Res. 2023, 56, 3165–3174. [Google Scholar]
- Yang, B.; Li, X.; Lei, L.; Chen, J. APOBEC: From mutator to editor. J. Genet. Genom. 2017, 44, 423–437. [Google Scholar]
- Huang, S.; Liu, H.; Xu, R.; Li, W.; Yang, H.; Bao, X.; Zhao, Y. RNA Editing Analysis Reveals Methyl Jasmonic Acid Regulation of Fucoxanthin and Fatty Acid Metabolism in Phaeodactylum tricornutum. Mar. Drugs 2025, 23, 66. [Google Scholar] [CrossRef]
- Vucic, D.; Fairbrother, W.J. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin. Cancer Res. 2007, 13, 5995–6000. [Google Scholar]
- Yang, X.D.; Sun, S.C. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol. Rev. 2015, 266, 56–71. [Google Scholar]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-κB signaling: Balancing life and death–a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar]
- Tang, C.; Zhuang, H.; Wang, W.; Wang, Q.; Ma, X.; Wang, B.; Shang, C. CircNUP54 promotes hepatocellular carcinoma progression via facilitating HuR cytoplasmic export and stabilizing BIRC3 mRNA. Cell Death Dis. 2024, 15, 191. [Google Scholar]
- Aissa, A.F.; Amaral, C.L.D.; Venancio, V.P.; Machado, C.D.S.; Hernandes, L.C.; Santos, P.W.D.S.; Curi, R.; Bianchi, M.D.L.P.; Antunes, L.M.G. Methionine-supplemented diet affects the expression of cardiovascular disease-related genes and increases inflammatory cytokines in mice heart and liver. J. Toxicol. Environ. Health 2017, 80, 1116–1128. [Google Scholar]
- Fu, P.Y.; Hu, B.; Ma, X.L.; Yang, Z.F.; Yu, M.C.; Sun, H.X.; Zhou, J. New insight into BIRC3: A novel prognostic indicator and a potential therapeutic target for liver cancer. J. Cell. Biochem. 2019, 120, 6035–6045. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar]
- Pham, M.; Tu, Y.; Lv, X. Accelerating BWA-MEM Read Mapping on GPUs. Int. Conf. Supercomput. 2023, 2023, 155–166. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar]
- Love, M.; Anders, S.; Huber, W. Differential Analysis of Count Data–the DESeq2 Package. Genome Biol. 2014, 15, 10–1186. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar]
- Joly, J.H.; Lowry, W.E.; Graham, N.A. Differential Gene Set Enrichment Analysis: A statistical approach to quantify the relative enrichment of two gene sets. Bioinformatics 2020, 36, 5247–5254. [Google Scholar]
- Zhe, Q.; Sulei, W.; Weiwei, T.; Hongyan, L.; Jianwei, W. Effects of Jiaotaiwan on depressive-like behavior in mice after lipopolysaccharide administration. Metab. Brain Dis. 2017, 32, 415–426. [Google Scholar]
- Lv, Y.; Xiang, X.; Jiang, Y.; Tang, L.; Zhou, Y.; Zhong, H.; Yan, J. Identification and characterization of lipopolysaccharide induced TNFα factor from blunt snout bream, Megalobrama amblycephala. Int. J. Mol. Sci. 2017, 18, 233. [Google Scholar] [CrossRef]
- Dai, Y.J.; Liu, W.B.; Abasubong, K.P.; Zhang, D.D.; Li, X.F. The Mechanism of Lipopolysaccharide Escaping the Intestinal Barrier in Megalobrama amblycephala Fed a High-Fat Diet. Front. Nutr. 2022, 9, 853409. [Google Scholar]
- Wang, K.; Lai, W.; Min, T.; Wei, J.; Bai, Y.; Cao, H.; Su, Z. The Effect of Enteric-Derived Lipopolysaccharides on Obesity. Int. J. Mol. Sci. 2024, 25, 4305. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huang, D.; Nie, S.; Xie, M. Polysaccharide from the seeds of Plantago asiatica L. protect against lipopolysaccharide-induced liver injury. J. Med. Food 2019, 22, 1058–1066. [Google Scholar] [PubMed]
- Chen, Y.; Sheppard, D.; Dong, X.; Hu, X.; Chen, M.; Chen, R.; Chen, L.F. H. pylori infection confers resistance to apoptosis via Brd4-dependent BIRC3 eRNA synthesis. Cell Death Dis. 2020, 11, 667. [Google Scholar]
- Thorne, A.; Bansal, A.; Necker-Brown, A.; Mostafa, M.M.; Gao, A.; Georgescu, A.; Newton, R. Differential regulation of BIRC2 and BIRC3 expression by inflammatory cytokines and glucocorticoids in pulmonary epithelial cells. PLoS ONE 2023, 18, e0286783. [Google Scholar]
- Zhang, S.; Yang, Y.; Weng, W.; Guo, B.; Cai, G.; Ma, Y.; Cai, S. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 14. [Google Scholar]
- Lee, S.H.; Mayr, C. Gain of additional BIRC3 protein functions through 3-UTR-mediated protein complex formation. Mol. Cell 2019, 74, 701–712. [Google Scholar]
- Mendoza-Rodríguez, M.; Romero, H.A.; Fuentes-Panana, E.M.; Ayala-Sumuano, J.T.; Meza, I. IL-1β induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017, 390, 39–44. [Google Scholar]
- Wang, L.; Wei, Y.; Yan, Y.; Wang, H.; Yang, J.; Zheng, Z.; Ge, L. CircDOCK1 suppresses cell apoptosis via inhibition of miR-196a-5p by targeting BIRC3 in OSCC. Oncol. Rep. 2018, 39, 951–966. [Google Scholar]
- Asslaber, D.; Wacht, N.; Leisch, M.; Qi, Y.; Maeding, N.; Hufnagl, C.; Egle, A. BIRC3 expression predicts CLL progression and defines treatment sensitivity via enhanced NF-κB nuclear translocation. Clin. Cancer Res. 2019, 25, 1901–1912. [Google Scholar]
- Gupta, U.; Dey, P. Rise of the guardians: Gut microbial maneuvers in bacterial infections. Life Sci. 2023, 330, 121993. [Google Scholar] [PubMed]
- Munteanu, C.; Schwartz, B. Interactions between dietary antioxidants, dietary Fiber and the gut microbiome: Their putative role in inflammation and Cancer. Int. J. Mol. Sci. 2024, 25, 8250. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Zhang, J.; Zeng, Z.; Zhou, R.; Zhao, J.; Yan, W.; Zhu, L. Ubiquitin-specific protease 8 ameliorates lipopolysaccharide-induced spleen injury via suppression of NF-κB and MAPK signaling pathways. Mol. Med. Rep. 2022, 26, 370. [Google Scholar]
- Song, Y.D.; Song, Y.H.; Liu, S.X.; Huang, H.R.; Zhong, Y.Q. Inhibition of inflammatory cell infiltration by antagonistic peptides that specifically bind to CCR5 via regulation of the NF-κB signaling pathway in rats with colitis. Arch. Med. Sci. 2021, 20, 2022. [Google Scholar]
- Li, Q.; Gloudemans, M.J.; Geisinger, J.M.; Fan, B.; Aguet, F.; Sun, T.; Ramaswami, G.; Li, Y.I.; Ma, J.-B.; Pritchard, J.K.; et al. RNA editing underlies genetic risk of common inflammatory diseases. Nature 2022, 608, 569–577. [Google Scholar]
- Nichols, C.E.; Kendrick, K.R.; Macander, B.M.; Rivas, C.; Ulbricht, R.J. Tissue and Sex-Specific Changes in RNA Editing During Induced Acute Inflammation. FASEB J. 2022, 36. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Cao, D.; Shi, M.; Yang, X. BIRC3 RNA Editing Modulates Lipopolysaccharide-Induced Liver Inflammation: Potential Implications for Animal Health. Int. J. Mol. Sci. 2025, 26, 2941. https://doi.org/10.3390/ijms26072941
Li W, Cao D, Shi M, Yang X. BIRC3 RNA Editing Modulates Lipopolysaccharide-Induced Liver Inflammation: Potential Implications for Animal Health. International Journal of Molecular Sciences. 2025; 26(7):2941. https://doi.org/10.3390/ijms26072941
Chicago/Turabian StyleLi, Wangchang, Duming Cao, Meiyi Shi, and Xiaogan Yang. 2025. "BIRC3 RNA Editing Modulates Lipopolysaccharide-Induced Liver Inflammation: Potential Implications for Animal Health" International Journal of Molecular Sciences 26, no. 7: 2941. https://doi.org/10.3390/ijms26072941
APA StyleLi, W., Cao, D., Shi, M., & Yang, X. (2025). BIRC3 RNA Editing Modulates Lipopolysaccharide-Induced Liver Inflammation: Potential Implications for Animal Health. International Journal of Molecular Sciences, 26(7), 2941. https://doi.org/10.3390/ijms26072941