
The Spectrum of Small Heat Shock Protein B8 (HSPB8)-Associated Neuromuscular Disorders

Abstract
1. Introduction
2. Distal Hereditary Motor Neuropathy (dHMN)
2.1. Clinical Features
2.2. Serology Studies
2.3. Electrophysiology Studies
2.4. Radiology Studies
2.5. Pathology Findings
3. Charcot–Marie–Tooth Disease Type 2L (CMT2L)
3.1. Clinical Features
3.2. Electrophysiology Studies
3.3. Radiology Studies
3.4. Nerve Pathology Findings
4. HSPB8-Myopathy
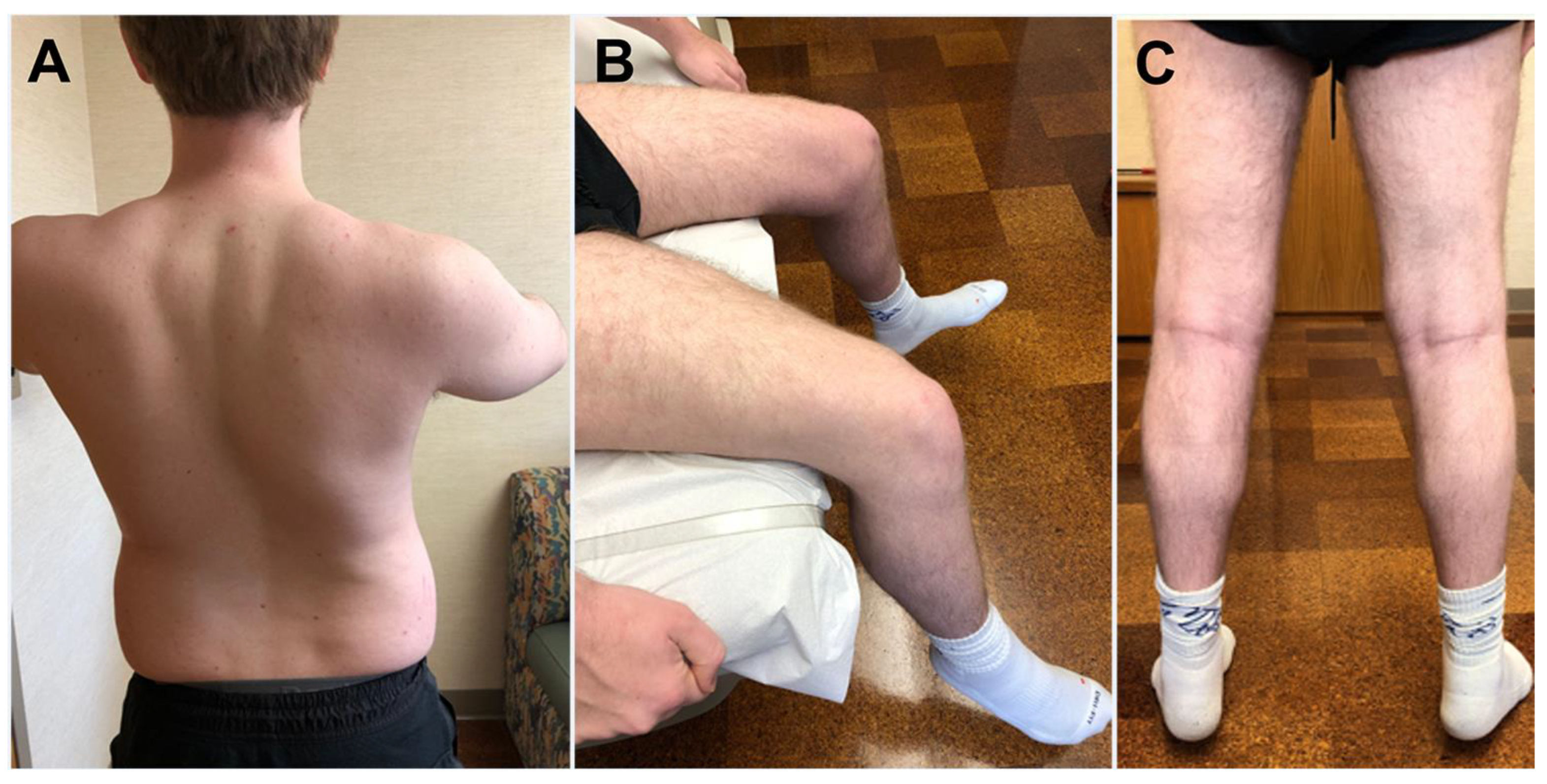
4.1. Clinical Features
4.2. Serology Studies
4.3. Electrophysiology Studies
4.4. Radiology Studies
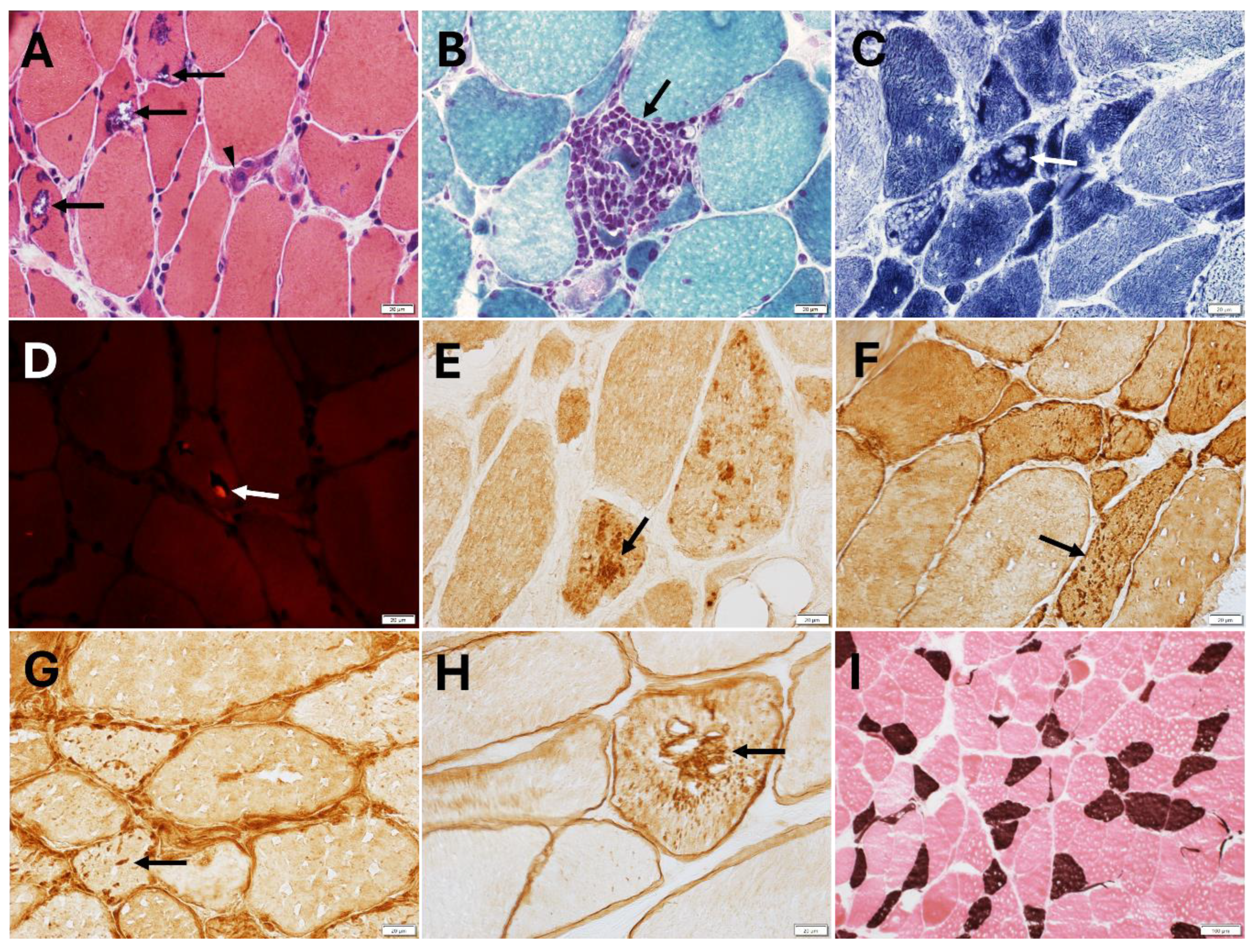
4.5. Muscle Pathology Findings
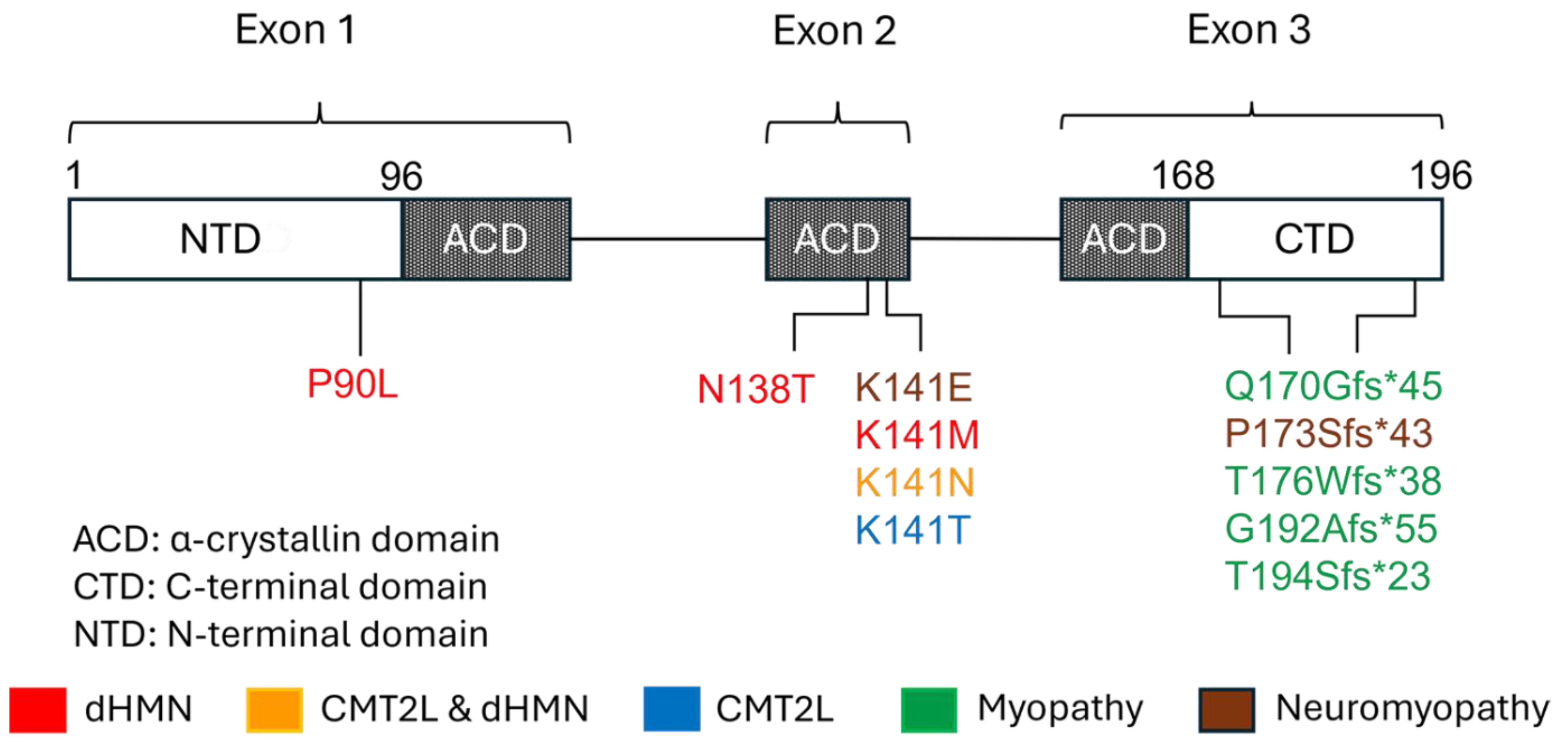
5. Gene and Protein Structure and Function
6. Genetic Aspects and Pathomechanisms
7. Animal and In Vitro Models
8. Therapeutic Strategies
9. Potential Limitations of Current Studies
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| ACD | α-crystallin domain |
| CASA | Chaperone-assisted selective autophagy |
| CMT | Charcot–Marie–Tooth |
| CRYAB | Alpha-B crystallin |
| CTD | C-terminal domain |
| CK | Creatinine kinase |
| dHMN | Distal hereditary motor neuropathy |
| GFP | Green fluorescent protein |
| HSP | Heat shock protein |
| HSPB | Small heat shock protein |
| KI | Knock-in |
| KO | Knock-out |
| LC3B | Microtubule-associated protein 1 light chain 3B |
| MFM | Myofibrillar myopathy |
| MTOR | Mechanistic target of rapamycin kinase |
| NADH | Nicotinamide adenine dinucleotide tetrazolium reductase |
| SQSTM1 | Sequestosome 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| TFEB | Transcription factor EB |
| TIA1 | T-cell intracellular antigen 1 |
References
- Benndorf, R.; Velazquez, R.; Zehr, J.D.; Pond, S.L.K.; Martin, J.L.; Lucaci, A.G. Human HspB1, HspB3, HspB5 and HspB8: Shaping these disease factors during vertebrate evolution. Cell Stress Chaperones 2022, 27, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Makhoba, X.H. Two sides of the same coin: Heat shock proteins as biomarkers and therapeutic targets for some complex diseases. Front. Mol. Biosci. 2025, 12, 1491227. [Google Scholar] [CrossRef]
- Carra, S.; Sivilotti, M.; Chavez Zobel, A.T.; Lambert, H.; Landry, J. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum. Mol. Genet. 2005, 14, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef]
- Harding, A.E.; Thomas, P.K. Hereditary distal spinal muscular atrophy. A report on 34 cases and a review of the literature. J. Neurol. Sci. 1980, 45, 337–348. [Google Scholar] [CrossRef]
- Irobi, J.; Tissir, F.; De Jonghe, P.; De Vriendt, E.; Van Broeckhoven, C.; Timmerman, V.; Beuten, J. A clone contig of 12q24.3 encompassing the distal hereditary motor neuropathy type II gene. Genomics 2000, 65, 34–43. [Google Scholar] [CrossRef]
- Frasquet, M.; Rojas-Garcia, R.; Argente-Escrig, H.; Vazquez-Costa, J.F.; Muelas, N.; Vilchez, J.J.; Sivera, R.; Millet, E.; Barreiro, M.; Diaz-Manera, J.; et al. Distal hereditary motor neuropathies: Mutation spectrum and genotype-phenotype correlation. Eur. J. Neurol. 2021, 28, 1334–1343. [Google Scholar] [CrossRef]
- Irobi, J.; Dierick, I.; Jordanova, A.; Claeys, K.G.; De Jonghe, P.; Timmerman, V. Unraveling the genetics of distal hereditary motor neuronopathies. Neuromol. Med. 2006, 8, 131–146. [Google Scholar] [CrossRef]
- Tazir, M.; Nouioua, S. Distal hereditary motor neuropathies. Rev. Neurol. 2024, 180, 1031–1036. [Google Scholar] [CrossRef]
- Timmerman, V.; Raeymaekers, P.; Nelis, E.; De Jonghe, P.; Muylle, L.; Ceuterick, C.; Martin, J.J.; Van Broeckhoven, C. Linkage analysis of distal hereditary motor neuropathy type II (distal HMN II) in a single pedigree. J. Neurol. Sci. 1992, 109, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, V.; De Jonghe, P.; Simokovic, S.; Lofgren, A.; Beuten, J.; Nelis, E.; Ceuterick, C.; Martin, J.J.; Van Broeckhoven, C. Distal hereditary motor neuropathy type II (distal HMN II): Mapping of a locus to chromosome 12q24. Hum. Mol. Genet. 1996, 5, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Geuens, T.; Petiot, P.; Pereon, Y.; Adriaenssens, E.; Haidar, M.; Capponi, S.; Maisonobe, T.; Fournier, E.; Dubourg, O.; et al. Axonal Neuropathies due to Mutations in Small Heat Shock Proteins: Clinical, Genetic, and Functional Insights into Novel Mutations. Hum. Mutat. 2017, 38, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Dierick, I.; Baets, J.; Irobi, J.; Jacobs, A.; De Vriendt, E.; Deconinck, T.; Merlini, L.; Van den Bergh, P.; Rasic, V.M.; Robberecht, W.; et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: A genotype-phenotype correlation study. Brain 2008, 131, 1217–1227. [Google Scholar] [CrossRef]
- Bansagi, B.; Griffin, H.; Whittaker, R.G.; Antoniadi, T.; Evangelista, T.; Miller, J.; Greenslade, M.; Forester, N.; Duff, J.; Bradshaw, A.; et al. Genetic heterogeneity of motor neuropathies. Neurology 2017, 88, 1226–1234. [Google Scholar] [CrossRef]
- Timmerman, V.; Beuten, J.; Irobi, J.; De Jonghe, P.; Martin, J.J.; Van Broeckhoven, C. Distal hereditary motor neuropathy type II (distal HMN type II): Phenotype and molecular genetics. Ann. N. Y. Acad. Sci. 1999, 883, 60–64. [Google Scholar] [CrossRef]
- Bird, T.D. Charcot-Marie-Tooth Hereditary Neuropathy Overview. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2025; (last revision 01-23-2025). [Google Scholar]
- Tang, B.S.; Luo, W.; Xia, K.; Xiao, J.F.; Jiang, H.; Shen, L.; Tang, J.G.; Zhao, G.H.; Cai, F.; Pan, Q.; et al. A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2L) maps to chromosome 12q24. Hum. Genet. 2004, 114, 527–533. [Google Scholar] [CrossRef]
- Tang, B.S.; Zhao, G.H.; Luo, W.; Xia, K.; Cai, F.; Pan, Q.; Zhang, R.X.; Zhang, F.F.; Liu, X.M.; Chen, B.; et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 2005, 116, 222–224. [Google Scholar] [CrossRef]
- Nakhro, K.; Park, J.M.; Kim, Y.J.; Yoon, B.R.; Yoo, J.H.; Koo, H.; Choi, B.O.; Chung, K.W. A novel Lys141Thr mutation in small heat shock protein 22 (HSPB8) gene in Charcot-Marie-Tooth disease type 2L. Neuromuscul. Disord. 2013, 23, 656–663. [Google Scholar] [CrossRef]
- Bienfait, H.M.; Baas, F.; Koelman, J.H.; de Haan, R.J.; van Engelen, B.G.; Gabreels-Festen, A.A.; Ongerboer de Visser, B.W.; Meggouh, F.; Weterman, M.A.; De Jonghe, P.; et al. Phenotype of Charcot-Marie-Tooth disease Type 2. Neurology 2007, 68, 1658–1667. [Google Scholar] [CrossRef]
- Ghaoui, R.; Palmio, J.; Brewer, J.; Lek, M.; Needham, M.; Evila, A.; Hackman, P.; Jonson, P.H.; Penttila, S.; Vihola, A.; et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 2016, 86, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Lornage, X.; Lannes, B.; Schneider, R.; Bierry, G.; Dondaine, N.; Boland, A.; Deleuze, J.F.; Bohm, J.; Thompson, J.; et al. HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy. Acta Neuropathol. 2017, 134, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Laura, M.; Casali, C.; Nishino, I.; Hayashi, Y.K.; Magri, S.; Taroni, F.; Stuani, C.; Saveri, P.; Moggio, M.; et al. Altered TDP-43-dependent splicing in HSPB8-related distal hereditary motor neuropathy and myofibrillar myopathy. Eur. J. Neurol. 2018, 25, 154–163. [Google Scholar] [CrossRef]
- Al-Tahan, S.; Weiss, L.; Yu, H.; Tang, S.; Saporta, M.; Vihola, A.; Mozaffar, T.; Udd, B.; Kimonis, V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 2019, 5, e349. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, S.; Liewluck, T.; Elliott, J.L.; Engel, A.G.; Milone, M. A novel heterozygous mutation in the C-terminal region of HSPB8 leads to limb-girdle rimmed vacuolar myopathy. Neuromuscul. Disord. 2020, 30, 236–240. [Google Scholar] [CrossRef]
- Inoue-Shibui, A.; Niihori, T.; Kobayashi, M.; Suzuki, N.; Izumi, R.; Warita, H.; Hara, K.; Shirota, M.; Funayama, R.; Nakayama, K.; et al. A novel deletion in the C-terminal region of HSPB8 in a family with rimmed vacuolar myopathy. J. Hum. Genet. 2021, 66, 965–972. [Google Scholar] [CrossRef]
- Tan, J.; Duong, H.; Milone, M.; Gibbs, L.; Tedesco, B.; Poletti, A.; Kimonis, V. A novel c.515delC HSPB8-multisystem proteinopathy associated with inclusion body myopathy with cardiomyopathy. medRxiv 2024. [Google Scholar] [CrossRef]
- Yang, G.; Lv, X.; Yang, M.; Feng, Y.; Wang, G.; Yan, C.; Lin, P. Expanding the spectrum of HSPB8-related myopathy: A novel mutation causing atypical pediatric-onset axial and limb-girdle involvement with autophagy abnormalities and molecular dynamics studies. J. Hum. Genet. 2024. [Google Scholar] [CrossRef]
- Kim, K.K.; Kim, R.; Kim, S.H. Crystal structure of a small heat-shock protein. Nature 1998, 394, 595–599. [Google Scholar] [CrossRef]
- Van Montfort, R.; Slingsby, C.; Vierling, E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv. Protein Chem. 2001, 59, 105–156. [Google Scholar] [CrossRef]
- Shemetov, A.A.; Seit-Nebi, A.S.; Gusev, N.B. Structure, properties, and functions of the human small heat-shock protein HSP22 (HspB8, H11, E2IG1): A critical review. J. Neurosci. Res. 2008, 86, 264–269. [Google Scholar] [CrossRef]
- Sudnitsyna, M.V.; Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. The role of intrinsically disordered regions in the structure and functioning of small heat shock proteins. Curr. Protein Pept. Sci. 2012, 13, 76–85. [Google Scholar] [CrossRef]
- Cristofani, R.; Piccolella, M.; Crippa, V.; Tedesco, B.; Montagnani Marelli, M.; Poletti, A.; Moretti, R.M. The Role of HSPB8, a Component of the Chaperone-Assisted Selective Autophagy Machinery, in Cancer. Cells 2021, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurro, M.R.; et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 2019, 9, e028486. [Google Scholar] [CrossRef]
- Tedesco, B.; Cristofani, R.; Ferrari, V.; Cozzi, M.; Rusmini, P.; Casarotto, E.; Chierichetti, M.; Mina, F.; Galbiati, M.; Piccolella, M.; et al. Insights on Human Small Heat Shock Proteins and Their Alterations in Diseases. Front. Mol. Biosci. 2022, 9, 842149. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, B.; Vendredy, L.; Timmerman, V.; Poletti, A. The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy 2023, 19, 1619–1641. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Cristofani, R.; Crippa, V.; Ferrari, V.; Tedesco, B.; Casarotto, E.; Chierichetti, M.; Galbiati, M.; Piccolella, M.; Messi, E.; et al. Autophagic and Proteasomal Mediated Removal of Mutant Androgen Receptor in Muscle Models of Spinal and Bulbar Muscular Atrophy. Front. Endocrinol. 2019, 10, 569. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Tedesco, B.; Mediani, L.; Asselbergh, B.; Crippa, V.; Antoniani, F.; Carra, S.; Poletti, A.; Timmerman, V. BAG3 Pro209 mutants associated with myopathy and neuropathy relocate chaperones of the CASA-complex to aggresomes. Sci. Rep. 2020, 10, 8755. [Google Scholar] [CrossRef]
- Heimdal, K.; Sanchez-Guixe, M.; Aukrust, I.; Bollerslev, J.; Bruland, O.; Jablonski, G.E.; Erichsen, A.K.; Gude, E.; Koht, J.A.; Erdal, S.; et al. STUB1 mutations in autosomal recessive ataxias-evidence for mutation-specific clinical heterogeneity. Orphanet J. Rare Dis. 2014, 9, 146. [Google Scholar] [CrossRef]
- Mukherjee, T.; Ramaglia, V.; Abdel-Nour, M.; Bianchi, A.A.; Tsalikis, J.; Chau, H.N.; Kalia, S.K.; Kalia, L.V.; Chen, J.J.; Arnoult, D.; et al. The eIF2alpha kinase HRI triggers the autophagic clearance of cytosolic protein aggregates. J. Biol. Chem. 2021, 296, 100050. [Google Scholar] [CrossRef]
- Tang, B.; Liu, X.; Zhao, G.; Luo, W.; Xia, K.; Pan, Q.; Cai, F.; Hu, Z.; Zhang, C.; Chen, B.; et al. Mutation analysis of the small heat shock protein 27 gene in chinese patients with Charcot-Marie-Tooth disease. Arch. Neurol. 2005, 62, 1201–1207. [Google Scholar] [CrossRef]
- Houlden, H.; Laura, M.; Wavrant-De Vrieze, F.; Blake, J.; Wood, N.; Reilly, M.M. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology 2008, 71, 1660–1668. [Google Scholar] [CrossRef]
- Bugiardini, E.; Khan, A.M.; Phadke, R.; Lynch, D.S.; Cortese, A.; Feng, L.; Gang, Q.; Pittman, A.M.; Morrow, J.M.; Turner, C.; et al. Genetic and phenotypic characterisation of inherited myopathies in a tertiary neuromuscular centre. Neuromuscul. Disord. 2019, 29, 747–757. [Google Scholar] [CrossRef]
- Muelas, N.; Carretero-Vilarroig, L.; Marti, P.; Azorin, I.; Frasquet, M.; Poyatos-Garcia, J.; Portela, S.; Martinez-Vicente, L.; Argente-Escrig, H.; Sivera, R.; et al. Clinical features, mutation spectrum and factors related to reaching molecular diagnosis in a cohort of patients with distal myopathies. J. Neurol. 2025, 272, 97. [Google Scholar] [CrossRef]
- Kolb, S.J.; Snyder, P.J.; Poi, E.J.; Renard, E.A.; Bartlett, A.; Gu, S.; Sutton, S.; Arnold, W.D.; Freimer, M.L.; Lawson, V.H.; et al. Mutant small heat shock protein B3 causes motor neuropathy: Utility of a candidate gene approach. Neurology 2010, 74, 502–506. [Google Scholar] [CrossRef]
- Nam, D.E.; Nam, S.H.; Lee, A.J.; Hong, Y.B.; Choi, B.O.; Chung, K.W. Small heat shock protein B3 (HSPB3) mutation in an axonal Charcot-Marie-Tooth disease family. J. Peripher. Nerv. Syst. 2018, 23, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.A.S.; Lacene, E.; Brochier, G.; Labasse, C.; Madelaine, A.; Silva, V.G.D.; Corazzini, R.; Papadopoulos, K.; Behin, A.; Laforet, P.; et al. Genetic Mutations and Demographic, Clinical, and Morphological Aspects of Myofibrillar Myopathy in a French Cohort. Genet. Test. Mol. Biomark. 2018, 22, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Curro, R.; Ronco, R.; Blake, J.; Rossor, A.M.; Bugiardini, E.; Laura, M.; Warner, T.; Yousry, T.; Poh, R.; et al. Mutations in alpha-B-crystallin cause autosomal dominant axonal Charcot-Marie-Tooth disease with congenital cataracts. Eur. J. Neurol. 2024, 31, e16063. [Google Scholar] [CrossRef]
- Kim, M.V.; Kasakov, A.S.; Seit-Nebi, A.S.; Marston, S.B.; Gusev, N.B. Structure and properties of K141E mutant of small heat shock protein HSP22 (HspB8, H11) that is expressed in human neuromuscular disorders. Arch. Biochem. Biophys. 2006, 454, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Sisto, A.; van Wermeskerken, T.; Pancher, M.; Gatto, P.; Asselbergh, B.; Assuncao Carreira, A.S.; De Winter, V.; Adami, V.; Provenzani, A.; Timmerman, V. Autophagy induction by piplartine ameliorates axonal degeneration caused by mutant HSPB1 and HSPB8 in Charcot-Marie-Tooth type 2 neuropathies. Autophagy 2024, 1–28. [Google Scholar] [CrossRef]
- Tedesco, B.; Vendredy, L.; Adriaenssens, E.; Cozzi, M.; Asselbergh, B.; Crippa, V.; Cristofani, R.; Rusmini, P.; Ferrari, V.; Casarotto, E.; et al. HSPB8 frameshift mutant aggregates weaken chaperone-assisted selective autophagy in neuromyopathies. Autophagy 2023, 19, 2217–2239. [Google Scholar] [CrossRef]
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 2016, 6, 22827. [Google Scholar] [CrossRef]
- Fontaine, J.M.; Sun, X.; Hoppe, A.D.; Simon, S.; Vicart, P.; Welsh, M.J.; Benndorf, R. Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 2006, 20, 2168–2170. [Google Scholar] [CrossRef]
- Bouhy, D.; Juneja, M.; Katona, I.; Holmgren, A.; Asselbergh, B.; De Winter, V.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Libert, C.; et al. A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol. 2018, 135, 131–148. [Google Scholar] [CrossRef]
- Irobi, J.; Almeida-Souza, L.; Asselbergh, B.; De Winter, V.; Goethals, S.; Dierick, I.; Krishnan, J.; Timmermans, J.P.; Robberecht, W.; De Jonghe, P.; et al. Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 2010, 19, 3254–3265. [Google Scholar] [CrossRef]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Sanbe, A.; Marunouchi, T.; Abe, T.; Tezuka, Y.; Okada, M.; Aoki, S.; Tsumura, H.; Yamauchi, J.; Tanonaka, K.; Nishigori, H.; et al. Phenotype of cardiomyopathy in cardiac-specific heat shock protein B8 K141N transgenic mouse. J. Biol. Chem. 2013, 288, 8910–8921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, F.; Li, X.; Huang, S.; Zi, X.; Liu, T.; Liu, S.; Li, X.; Xia, K.; Pan, Q.; et al. A novel transgenic mouse model of Chinese Charcot-Marie-Tooth disease type 2L. Neural Regen. Res. 2014, 9, 413–419. [Google Scholar] [CrossRef]
- Carra, S.; Boncoraglio, A.; Kanon, B.; Brunsting, J.F.; Minoia, M.; Rana, A.; Vos, M.J.; Seidel, K.; Sibon, O.C.; Kampinga, H.H. Identification of the Drosophila ortholog of HSPB8: Implication of HSPB8 loss of function in protein folding diseases. J. Biol. Chem. 2010, 285, 37811–37822. [Google Scholar] [CrossRef]
- Jablonska, J.; Dubinska-Magiera, M.; Jagla, T.; Jagla, K.; Daczewska, M. Drosophila Hsp67Bc hot-spot variants alter muscle structure and function. Cell Mol. Life Sci. 2018, 75, 4341–4356. [Google Scholar] [CrossRef]
- Silva, M.C.; Nandi, G.A.; Tentarelli, S.; Gurrell, I.K.; Jamier, T.; Lucente, D.; Dickerson, B.C.; Brown, D.G.; Brandon, N.J.; Haggarty, S.J. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun. 2020, 11, 3258. [Google Scholar] [CrossRef]
- Vantaggiato, C.; Orso, G.; Guarato, G.; Brivio, F.; Napoli, B.; Panzeri, E.; Masotti, S.; Santorelli, F.M.; Lamprou, M.; Gumeni, S.; et al. Rescue of lysosomal function as therapeutic strategy for SPG15 hereditary spastic paraplegia. Brain 2023, 146, 1103–1120. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Pupyshev, A.B.; Belichenko, V.M.; Tenditnik, M.V.; Bashirzade, A.A.; Dubrovina, N.I.; Ovsyukova, M.V.; Akopyan, A.A.; Fedoseeva, L.A.; Korolenko, T.A.; Amstislavskaya, T.G.; et al. Combined induction of mTOR-dependent and mTOR-independent pathways of autophagy activation as an experimental therapy for Alzheimer’s disease-like pathology in a mouse model. Pharmacol. Biochem. Behav. 2022, 217, 173406. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Klyushnik, T.P.; Akopyan, A.A.; Singh, S.K.; Tikhonova, M.A. Disaccharide trehalose in experimental therapies for neurodegenerative disorders: Molecular targets and translational potential. Pharmacol. Res. 2022, 183, 106373. [Google Scholar] [CrossRef]
- Vendredy, L.; De Winter, V.; Van Lent, J.; Orije, J.; Authier, T.D.S.; Katona, I.; Asselbergh, B.; Adriaenssens, E.; Weis, J.; Verhoye, M.; et al. RNA Interference Targeting Small Heat Shock Protein B8 Failed to Improve Distal Hereditary Motor Neuropathy in the Mouse Model. J. Gene Med. 2025, 27, e70013. [Google Scholar] [CrossRef] [PubMed]
- Chierichetti, M.; Cristofani, R.; Crippa, V.; Ferrari, V.; Cozzi, M.; Casarotto, E.; Pramaggiore, P.; Cornaggia, L.; Patelli, G.; Mohamed, A.; et al. Small heat shock protein B8: From cell functions to its involvement in diseases and potential therapeutic applications. Neural Regen. Res. 2025, 20, 2872–2886. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | dHMN | CMT2L | Myopathy |
|---|---|---|---|
| Age of onset | Childhood to adulthood | Adolescence to adulthood | Childhood to adulthood |
| Weakness | Distal predominant | Distal predominant | Distal, proximal, or axial predominant |
| Tendon reflexes | Diminished or absent | Diminished or absent | Normal, diminished, or absent |
| Muscle atrophy | Present | Present | Present |
| Cramps | Can be present | Absent | Can be present |
| Fasciculations | Can be present | Absent | Can be present in neuromyopathy |
| Skeletal abnormalities | Pes cavus | Pes cavus and scoliosis | Pes cavus, scoliosis, lumbar lordosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashed, H.R.; Nath, S.R.; Milone, M. The Spectrum of Small Heat Shock Protein B8 (HSPB8)-Associated Neuromuscular Disorders. Int. J. Mol. Sci. 2025, 26, 2905. https://doi.org/10.3390/ijms26072905
Rashed HR, Nath SR, Milone M. The Spectrum of Small Heat Shock Protein B8 (HSPB8)-Associated Neuromuscular Disorders. International Journal of Molecular Sciences. 2025; 26(7):2905. https://doi.org/10.3390/ijms26072905
Chicago/Turabian StyleRashed, Hebatallah R., Samir R. Nath, and Margherita Milone. 2025. "The Spectrum of Small Heat Shock Protein B8 (HSPB8)-Associated Neuromuscular Disorders" International Journal of Molecular Sciences 26, no. 7: 2905. https://doi.org/10.3390/ijms26072905
APA StyleRashed, H. R., Nath, S. R., & Milone, M. (2025). The Spectrum of Small Heat Shock Protein B8 (HSPB8)-Associated Neuromuscular Disorders. International Journal of Molecular Sciences, 26(7), 2905. https://doi.org/10.3390/ijms26072905