

Early Cardiac Dysfunction in Duchenne Muscular Dystrophy: A Case Report and Literature Update
 ,
,  ,
, 
Abstract
1. Introduction
2. Detailed Case Description
2.1. Methodology
2.2. Clinical Presentation and Diagnosis
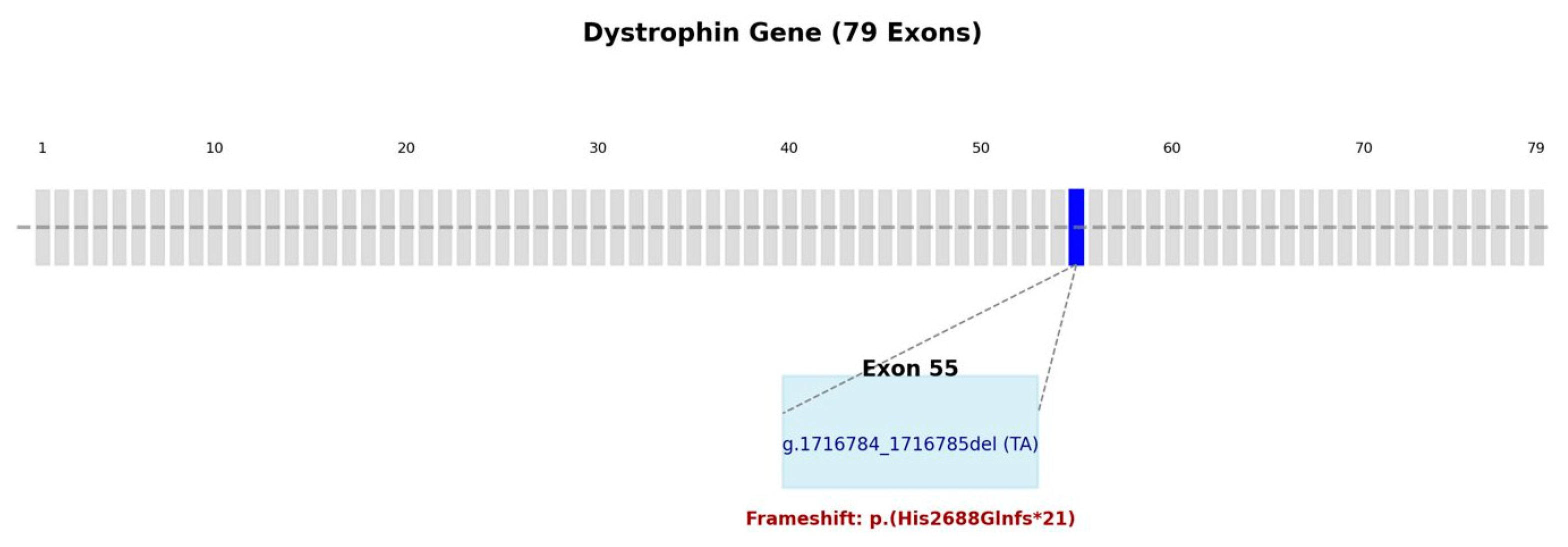
2.3. Genetic Testing
2.4. Multidisciplinary Team and Initial Findings
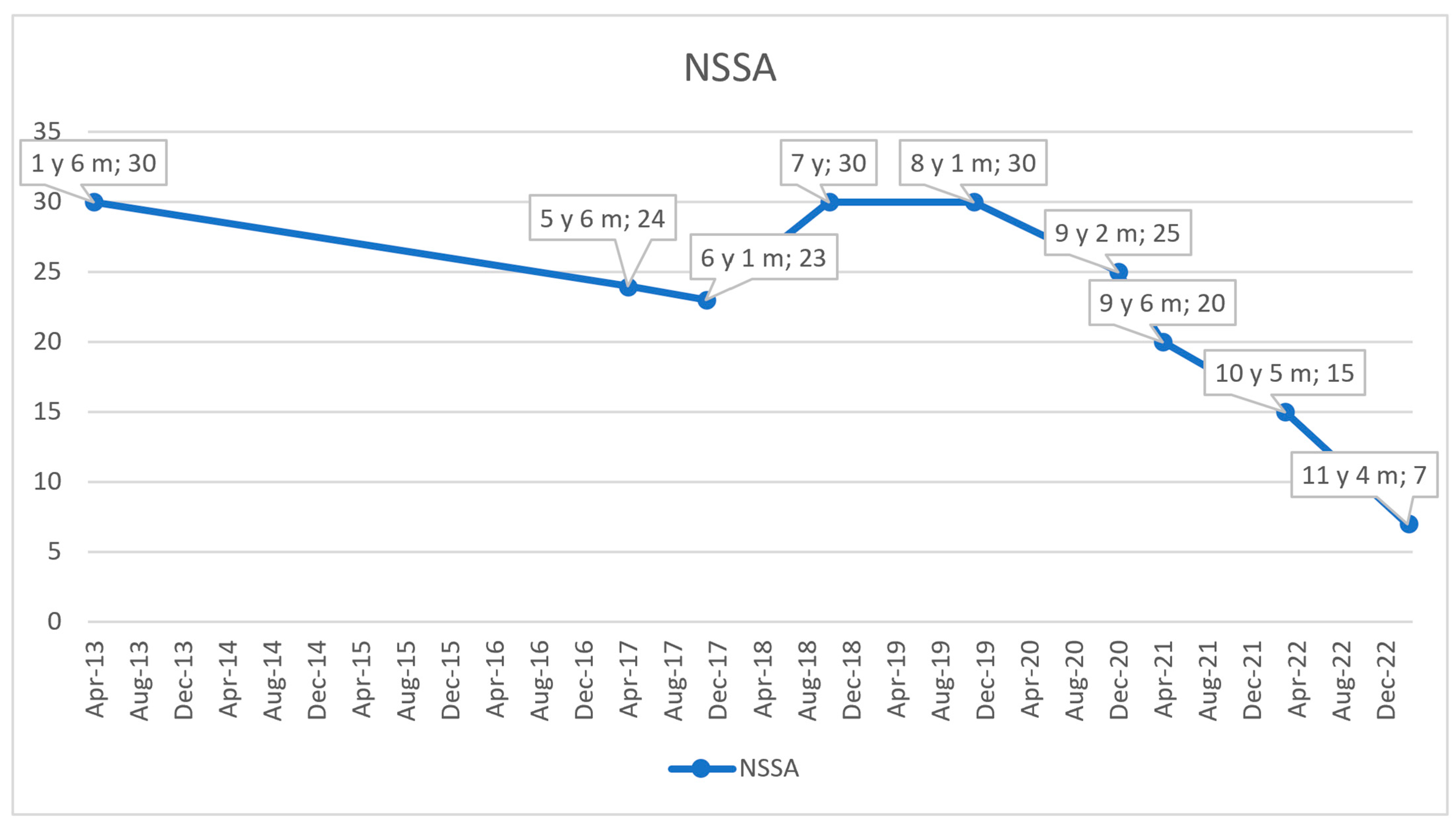
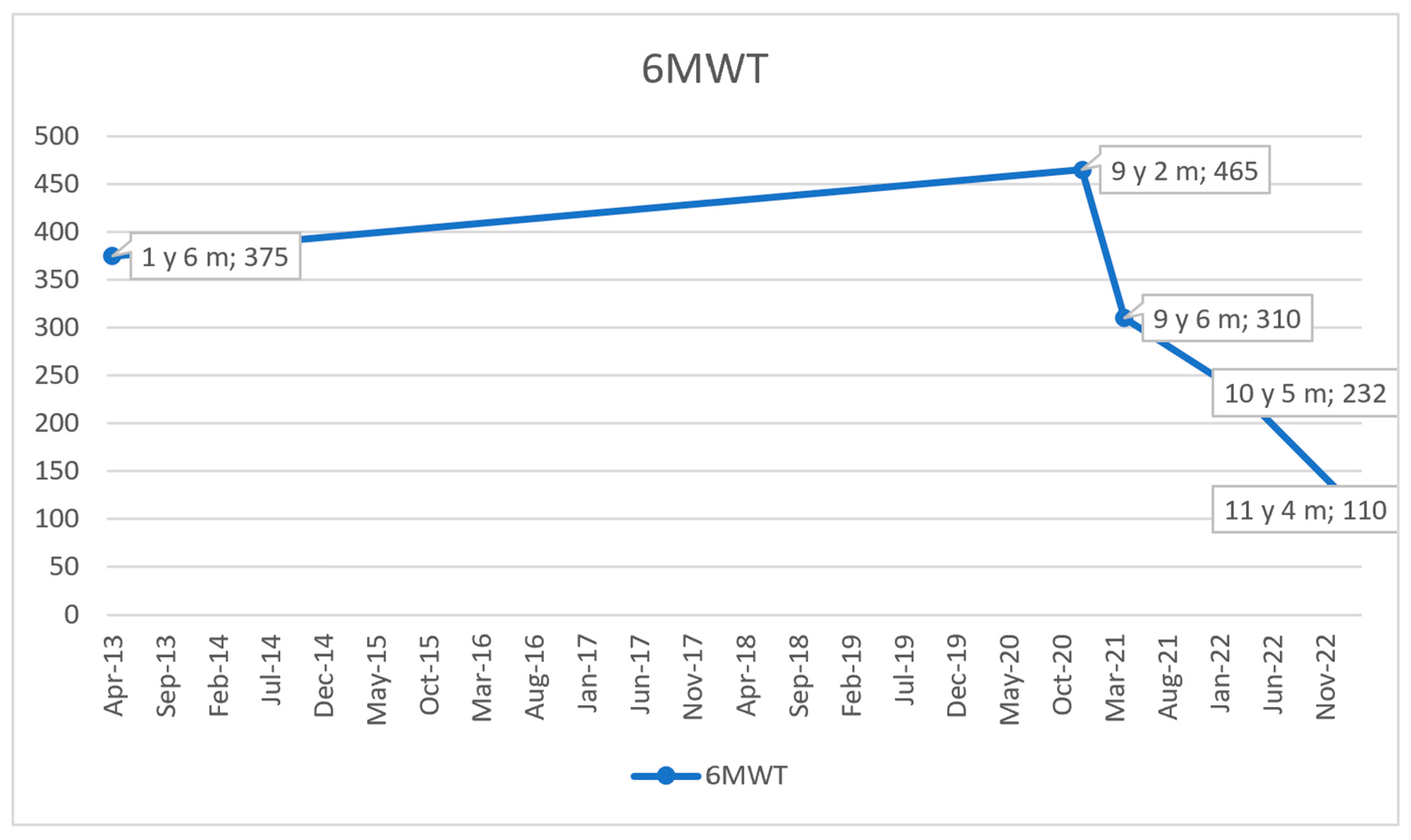
2.5. Cardiac Monitoring and Follow-Ups
2.6. Treatment and Disease Progression
3. Discussion
3.1. Cardiac Complications
3.2. Genetic and Molecular Insights
3.3. Diagnostic and Therapeutic Approaches
3.4. Advanced Cardiac Treatment
3.5. Emerging Therapies
3.6. Care Coordination and Transition from Pediatric to Adult Care
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.I.; Markousis-Mavrogenis, G.; Papavasiliou, A.; Papadopoulos, G.; Kolovou, G. Cardiac involvement in duchenne muscular dystrophy and related dystrophinopathies. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; pp. 31–42. [Google Scholar]
- Salari, N.; Fatahi, B.; Valipour, E.; Kazeminia, M.; Fatahian, R.; Kiaei, A.; Shohaimi, S.; Mohammadi, M. Global prevalence of Duchenne and Becker muscular dystrophy: A systematic review and meta-analysis. J. Orthop. Surg. Res. 2022, 17, 96. [Google Scholar] [CrossRef]
- Old, J.M.; Davies, K.E. Prenatal diagnosis of Duchenne muscular dystrophy by DNA analysis. J. Med. Genet. 1986, 23, 556–559. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D.W. Cardiac pathophysiology and the future of cardiac therapies in duchenne muscular dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Morena, C.; Cavero-Redondo, I.; Saz-Lara, A.; Sequí-Domínguez, I.; Lucerón-Lucas-torres, M.; Martínez-Vizcaíno, V. Genetic modifiers and phenotype of Duchenne muscular dystrophy: A systematic review and meta-analysis. Pharmaceuticals 2021, 14, 798. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, N.; Dan, B. Pathophysiology of Duchenne Muscular Dystrophy: Current Hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in Duchenne muscular dystrophy: Pathogenesis and therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef]
- Matsumura, T.; Saito, T.; Fujimura, H.; Shinno, S. Cardiac troponin I for accurate evaluation of cardiac status in myopathic patients. Brain Dev. 2007, 29, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [CrossRef]
- Zhuang, X.; Luo, S.; Fan, H.; Zhang, J.; Chen, H.; Yan, P.; Bao, L. Duchenne muscular dystrophy and dilated cardiomyopathy with deletion of exon 45 and 49. Cardiol Plus 2022, 7, 97–101. [Google Scholar] [CrossRef]
- Schultz, T.I.; Raucci, F.J.; Salloum, F.N. Cardiovascular Disease in Duchenne Muscular Dystrophy: Overview and Insight into Novel Therapeutic Targets. Basic Transl. Sci. 2022, 7, 608–625. [Google Scholar]
- Buzin, C.H.; Feng, J.; Yan, J.; Scaringe, W.; Liu, Q.; den Dunnen, J.; Mendell, J.R.; Sommer, S.S. Mutation rates in the dystrophin gene: A hotspot of mutation at a CpG dinucleotide. Hum Mutat. 2005, 25, 177–188. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- ClinVar–NCBI. VCV000094785.12. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/94785/?term=((NM_004006.3)+AND+DMD%3A+g.1716784_1716785del)+OR+p (accessed on 7 February 2025).
- Ricci, G.; Bello, L.; Torri, F.; Schirinzi, E.; Pegoraro, E.; Siciliano, G. Therapeutic opportunities and clinical outcome measures in Duchenne muscular dystrophy. Neurol. Sci. 2022, 43, 625–633. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Review Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 77, 77–93. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, P.L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Bushby, K.; Bourke, J.; Bullock, R.; Eagle, M.; Gibson, M.; Quinby, J. The multidisciplinary management of Duchenne muscular dystrophy. Curr. Paediatr. 2005, 15, 292–300. [Google Scholar] [CrossRef]
- Bianchi, M.L.; Biggar, D.; Bushby, K.; Rogol, A.D.; Rutter, M.M.; Tseng, B. Endocrine Aspects of Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2011, 21, 298–303. [Google Scholar] [CrossRef]
- Pane, M.; Lombardo, M.E.; Alfieri, P.; D’Amico, A.; Bianco, F.; Vasco, G.; Dpsych, G.P.; Dpsych, M.M.; Romeo, D.M.; Ricotti, V.; et al. Attention deficit hyperactivity disorder and cognitive function in duchenne muscular dystrophy: Phenotype-genotype correlation. J. Pediatr. 2012, 161, 705–709.e1. [Google Scholar] [CrossRef] [PubMed]
- Perumal, A.R.; Rajeswaran, J.; Nalini, A. Neuropsychological Profile of Duchenne Muscular Dystrophy. Appl. Neuropsychol. Child 2015, 4, 49–57. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Woodrow Benson, D.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Sumanaru, D.; Josseran, L.; Essid, A.; Mbieleu, B.; Haegy, I.; Bergounioux, J. Levosimendan as Rescue Therapy for Acute Heart Failure in a Patient with Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2019, 40, 668–670. [Google Scholar] [CrossRef]
- Spurney, C.F. Cardiomyopathy of duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve 2011, 44, 8–19. [Google Scholar] [CrossRef]
- Adorisio, R.; Mencarelli, E.; Cantarutti, N.; Calvieri, C.; Amato, L.; Cicenia, M.; Silvetti, M.; D’Amico, A.; Grandinetti, M.; Drago, F.; et al. Duchenne dilated cardiomyopathy: Cardiac management from prevention to advanced cardiovascular therapies. J. Clin. Med. 2020, 9, 3186. [Google Scholar] [CrossRef]
- Prakash, N.; Suthar, R.; Sihag, B.K.; Debi, U.; Kumar, R.M.; Sankhyan, N. Cardiac MRI and Echocardiography for Early Diagnosis of Cardiomyopathy Among Boys with Duchenne Muscular Dystrophy: A Cross-Sectional Study. Front. Pediatr. 2022, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. In Nature Reviews Disease Primers; Nature Research: Berlin, Germany, 2021; Volume 4. [Google Scholar]
- Anwar, S.; He, M.; Lim, K.R.Q.; Maruyama, R.; Yokota, T. A genotype-phenotype correlation study of exon skip-equivalent in-frame deletions and exon skip-amenable out-of-frame deletions across the dmd gene to simulate the effects of exon-skipping therapies: A meta-analysis. J. Pers. Med. 2021, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, K.; Dwianingsih, E.K.; Pratiwi, L.; Kalim, A.S.; Mardhiah, H.; Putranti, A.H.; Nurputra, D.K.; Triono, A.; Herini, E.S.; Malueka, R.G.; et al. The analysis of DMD gene deletions by multiplex PCR in Indonesian DMD/BMD patients: The era of personalized medicine. BMC Res. Notes 2019, 12, 704. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple exon skipping in the duchenne muscular dystrophy hot spots: Prospects and challenges. J. Pers. Med. 2018, 8, 41. [Google Scholar] [CrossRef]
- Yamamoto, T.; Awano, H.; Zhang, Z.; Sakuma, M.; Kitaaki, S.; Matsumoto, M.; Nagai, M.; Sato, I.; Imanishi, T.; Hayashi, N.; et al. Cardiac Dysfunction in Duchenne Muscular Dystrophy Is Less Frequent in Patients with Mutations in the Dystrophin Dp116 Coding Region Than in Other Regions. Circ. Genom. Precis. Med. 2018, 11, E001782. [Google Scholar] [CrossRef]
- Zhou, H.; Fu, M.; Mao, B.; Yuan, L. Cardiac Phenotype–Genotype Associations in DMD/BMD: A Meta-Analysis and Systematic Review. Pediatr. Cardiol. 2021, 42, 189–198. [Google Scholar] [CrossRef]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Borodni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Bourke, J.; Turner, C.; Bradlow, W.; Chikermane, A.; Coats, C.; Fenton, M.; Ilina, M.; Johnson, A.; Kapetanakis, S.; Kuhwald, L.; et al. Cardiac care of children with dystrophinopathy and females carrying DMD-gene variations. Open Heart 2022, 9, e001977. [Google Scholar] [CrossRef]
- Feingold, B.; Mahle, W.T.; Auerbach, S.; Clemens, P.; Domenighetti, A.A.; Jefferies, J.L.; Judge, D.P.; Lal, A.K.; Markham, L.W.; Parks, W.J.; et al. Management of cardiac involvement associated with neuromuscular diseases: A scientific statement from the American Heart Association. Circulation 2017, 136, e200–e231. [Google Scholar] [CrossRef] [PubMed]
- Palladino, A.; D’ambrosio, P.; Papa, A.A.; Petillo, R.; Orsini, C.; Scutifero, M.; Nigro, G.; Politano, L. Management of cardiac involvement in muscular dystrophies: Paediatric versus adult forms. Acta Myol. 2016, 35, 128–134. [Google Scholar]
- Soslow, J.H.; Hall, M.; Bryan Burnette, W.; Hor, K.; Chisolm, J.; Spurney, C.; Godown, J.; Xu, M.; Slaughter, J.C.; Markahm, L.W. Creation of a novel algorithm to identify patients with Becker and Duchenne muscular dystrophy within an administrative database and application of the algorithm to assess cardiovascular morbidity. Cardiol. Young 2019, 29, 290–296. [Google Scholar] [CrossRef]
- Bennett, J.; Kertesz, N.J. Management of rhythm disorders in Duchenne muscular dystrophy: Is sudden death a cardiac or pulmonary problem? Pediatr. Pulmonol. 2021, 56, 760–765. [Google Scholar] [CrossRef]
- McCulloch, M.A.; Lal, A.K.; Knecht, K.; Butts, R.J.; Villa, C.R.; Johnson, J.N.; Conway, J.; Bock, M.J.; Schumacher, K.R.; Law, S.P.; et al. Implantable Cardioverter Defibrillator Use in Males with Duchenne Muscular Dystrophy and Severe Left Ventricular Dysfunction. Pediatr. Cardiol. 2020, 41, 925–931. [Google Scholar] [CrossRef]
- Dang, U.J.; Damsker, J.M.; Guglieri, M.; Clemens, P.R.; Perlman, S.J.; Smith, E.C.; Horrocks, I.; Finkel, R.S.; Mah, J.K.; Deconinck, N.; et al. Efficacy and Safety of Vamorolone Over 48 Weeks in Boys with Duchenne Muscular Dystrophy. Neurology 2024, 102, e208112. [Google Scholar] [CrossRef]
- Łoboda, A.; Dulak, J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: Past, present, and future. Pharmacol. Rep. 2020, 72, 1227–1263. [Google Scholar] [CrossRef]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef]
- Sheikh, O.; Yokota, T. Advances in genetic characterization and genotype–phenotype correlation of duchenne and becker muscular dystrophy in the personalized medicine era. J. Pers. Med. 2020, 10, 111. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Manini, A.; Abati, E.; Nuredini, A.; Corti, S.; Comi, G.P. Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence. Front. Neurol. 2022, 12, 814174. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, T.; Qiao, C.; Zhou, L.; Wang, B.; Zhang, J.; Chen, C.; Li, J.; Xiao, X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005, 23, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Signorovitch, J.; Sajeev, G.; Lane, H.; Jenkins, M.; Dieye, I.; Ward, S.J.; McDonald, C.; Goemans, N.; Niks, E.H.; et al. DMD Genotypes and Motor Function in Duchenne Muscular Dystrophy: A Multi-institution Meta-analysis With Implications for Clinical Trials. Neurology 2023, 100, e1540–e1554. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, P.L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transi-tions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Trout, C.J.; Case, L.E.; Clemens, P.R.; Mcarthur, A.; Noritz, G.; Ritzo, M.; Wagner, K.R.; Vroom, E.; Kennedy, A. A Transition Toolkit for Duchenne Muscular Dystrophy. Pediatrics 2018, 142, S110–S117. [Google Scholar] [CrossRef]
- Wasilewska, E.; Małgorzewicz, S.; Sobierajska-Rek, A.; Jabłońska-Brudło, J.; Górska, L.; Śledzińska, K.; Bautembach-Minkowska, J.; Wierzba, J. Transition from childhood to adulthood in patients with duchenne muscular dystrophy. Medicina 2020, 56, 426. [Google Scholar] [CrossRef] [PubMed]
- Lupu, M.; Ioghen, M.; Perjoc, R.-Ș.; Scarlat, A.M.; Vladâcenco, O.A.; Roza, E.; Epure, D.A.-M.; Teleanu, R.I.; Severin, E.M. The Importance of Implementing a Transition Strategy for Patients with Muscular Dystrophy: From Child to Adult—Insights from a Tertiary Centre for Rare Neurological Diseases. Children 2023, 10, 959. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Cardiac Evaluation |
|---|---|
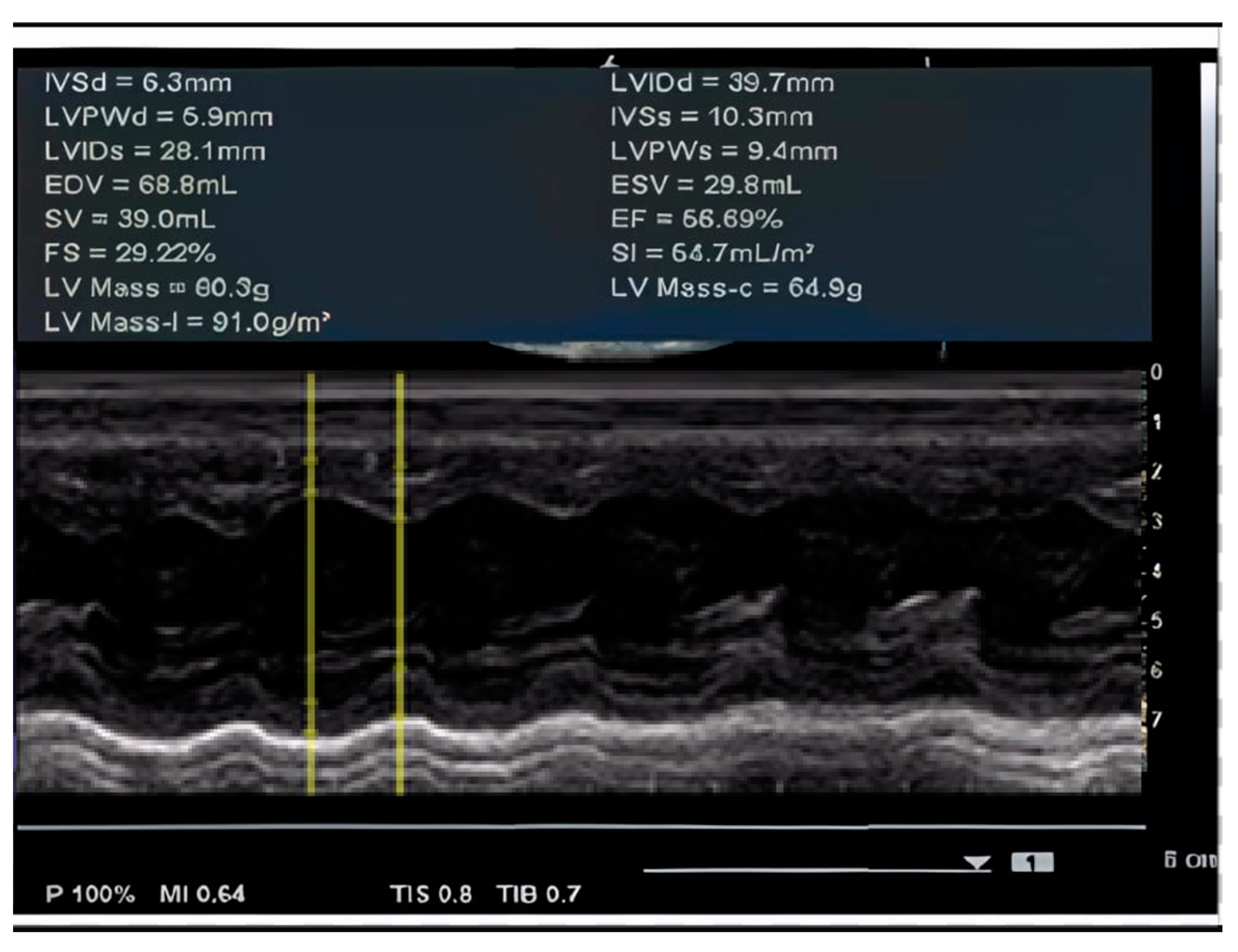
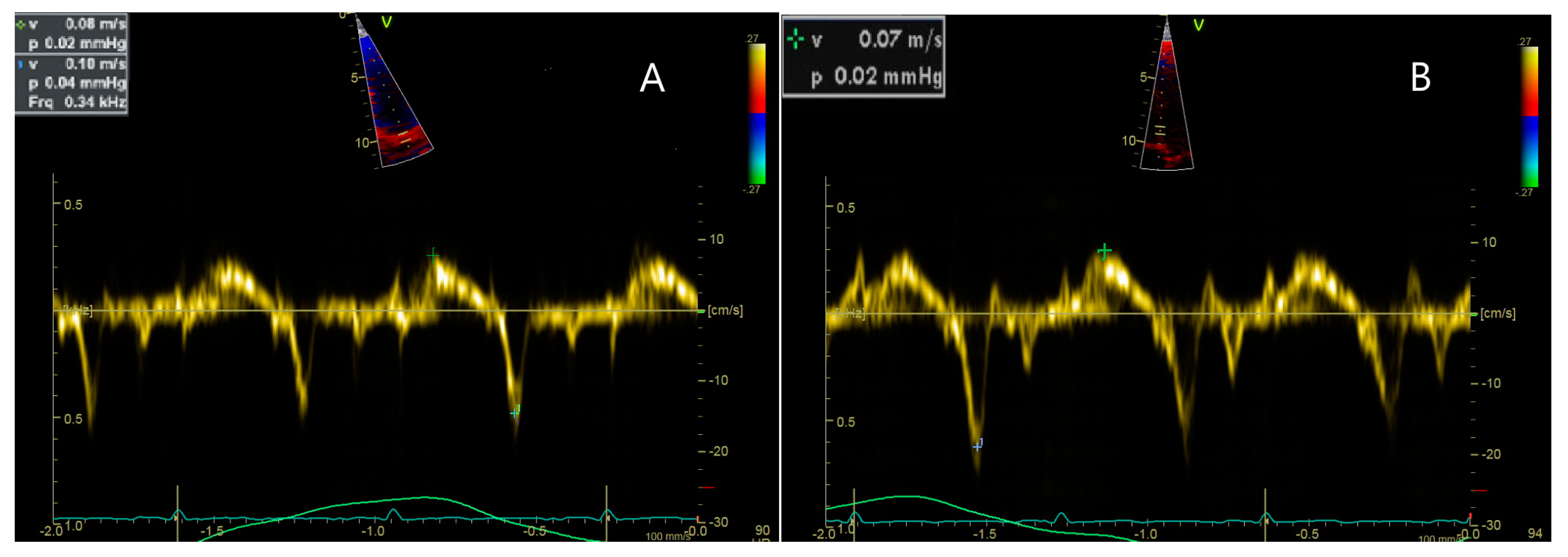
| 3 y 8 m | Cardiac dimensions: 17 mm at the ascending aorta, 30 mm at both LV and RV. Doppler flow velocities of 0.90 m/s Ao and 0.90 m/s at the PV. Mitral and aortic valves appear normal with normal motility. EF = 56.69%. Dilative cardiomegaly in an incipient form in the context of DMD. EKG: Sinus tachycardia with HR = 100 bpm, QRS axis at 0 degrees, juvenile ST-T pattern. |
| 6 y 3 m | Left ventricle dilated with normal systolic–diastolic function. |
| 8 y 1 m | Longitudinal dysfunction of the LV and RV. EKG: Sinus tachycardia with HR = 100 bpm, QRS axis at 0 degrees, juvenile ST-T pattern. |
| 9 y | LV slightly globular with normal global systolic function. Normal diastolic function. No valvulopathies. No pulmonary hypertension. BP = 100/60 mmHg; HR = 75 bpm; no murmurs. EKG: Sinus rhythm with HR = 90 bpm, QRS axis = +45 degrees, PR interval = 0.12 s, juvenile ST-T pattern. |
| 10 y 5 m | LV globular. Longitudinal dysfunction of the LV. BP = 100/60 mmHg, HR = 90 bpm, grade I/VI systolic murmur heard at the left parasternal border. EKG: RBBB. |
| 11 y 4 m | LV globular with preserved ejection fraction. Longitudinal dysfunction of the LV. EKG: Sinus tachycardia HR = 110 bpm, QRS axis +60 degrees, PR interval: 0.18 s, RBBB with secondary ST-T changes. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lupu, M.; Pintilie, I.M.; Teleanu, R.I.; Marin, G.G.; Vladâcenco, O.A.; Severin, E.M. Early Cardiac Dysfunction in Duchenne Muscular Dystrophy: A Case Report and Literature Update. Int. J. Mol. Sci. 2025, 26, 1685. https://doi.org/10.3390/ijms26041685
Lupu M, Pintilie IM, Teleanu RI, Marin GG, Vladâcenco OA, Severin EM. Early Cardiac Dysfunction in Duchenne Muscular Dystrophy: A Case Report and Literature Update. International Journal of Molecular Sciences. 2025; 26(4):1685. https://doi.org/10.3390/ijms26041685
Chicago/Turabian StyleLupu, Maria, Iustina Mihaela Pintilie, Raluca Ioana Teleanu, Georgiana Gabriela Marin, Oana Aurelia Vladâcenco, and Emilia Maria Severin. 2025. "Early Cardiac Dysfunction in Duchenne Muscular Dystrophy: A Case Report and Literature Update" International Journal of Molecular Sciences 26, no. 4: 1685. https://doi.org/10.3390/ijms26041685
APA StyleLupu, M., Pintilie, I. M., Teleanu, R. I., Marin, G. G., Vladâcenco, O. A., & Severin, E. M. (2025). Early Cardiac Dysfunction in Duchenne Muscular Dystrophy: A Case Report and Literature Update. International Journal of Molecular Sciences, 26(4), 1685. https://doi.org/10.3390/ijms26041685