_Kim.png)

Synergistic Anti-Cancer Activity of Melittin and Erlotinib in Non-Small Cell Lung Cancer


Abstract
1. Introduction
2. Results
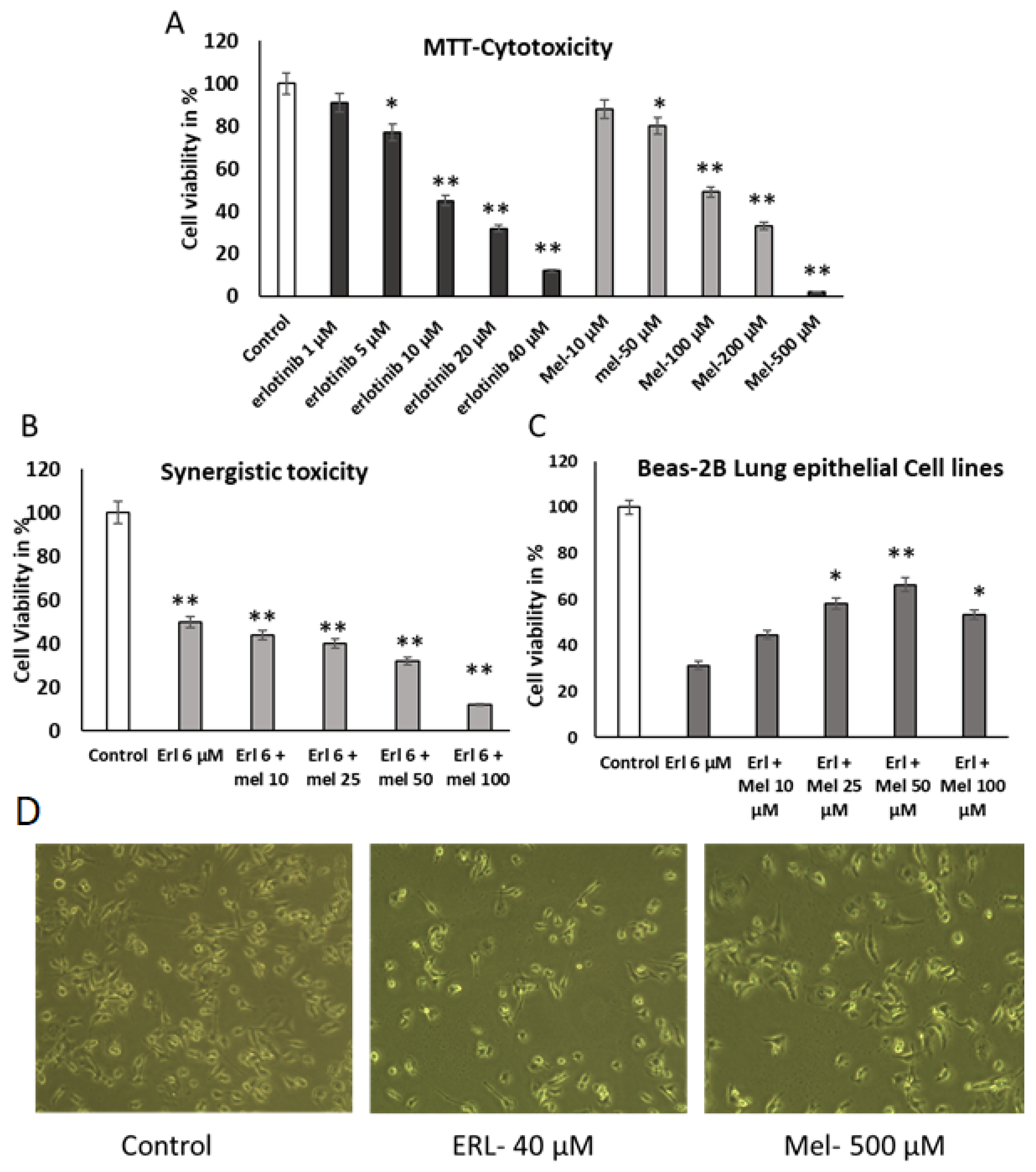
2.1. Evaluation of the Effect of Melittin and Erlotinib on A549 Lung Cancer Cells
2.2. Melittin Abrogates A549 Migration Using Scratch Wound Assay
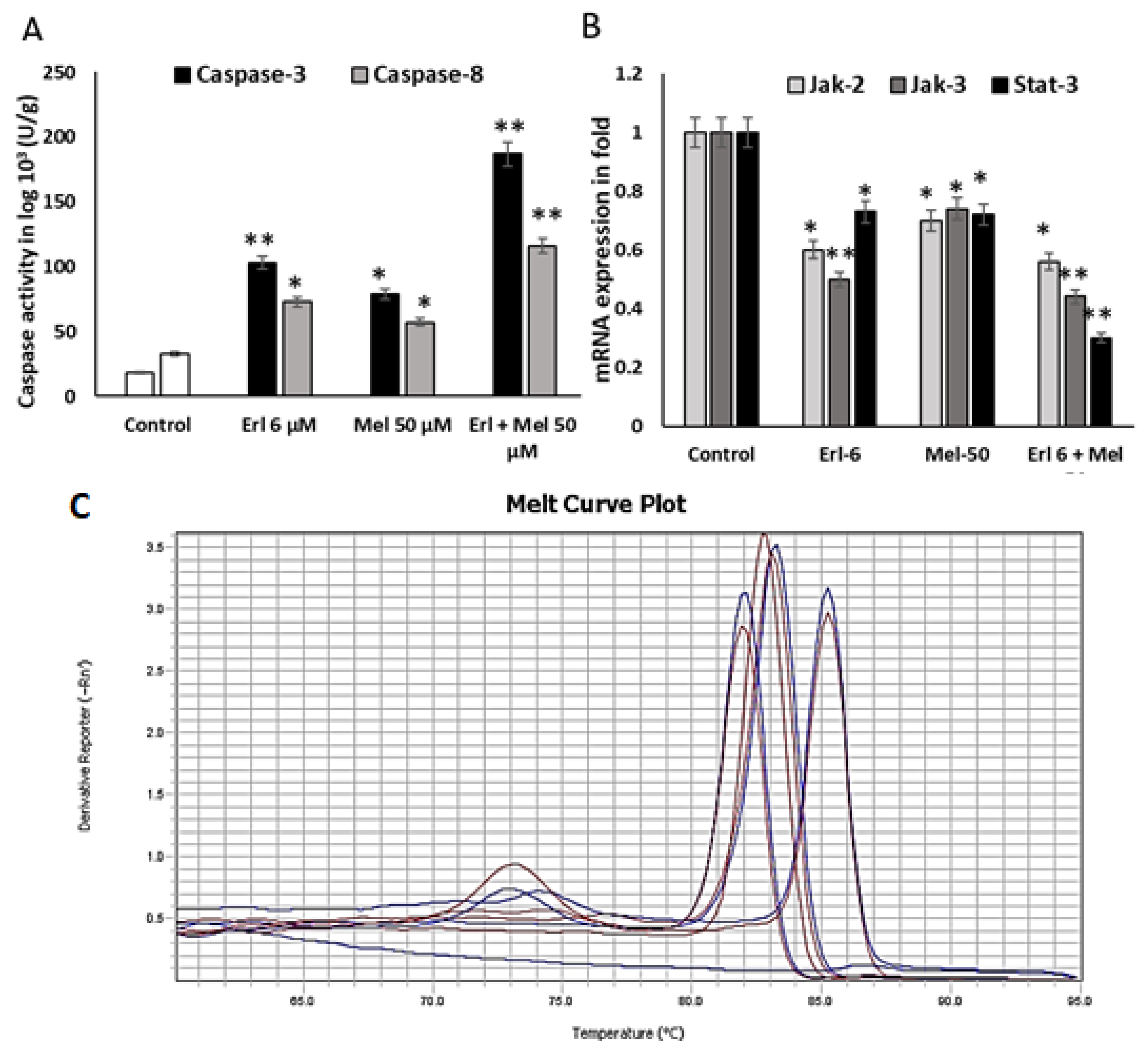
2.3. Melittin Upregulates Mitochondrial Mediated Apoptotic Markers
2.4. Melittin Downregulates Cancer Progressive Genes
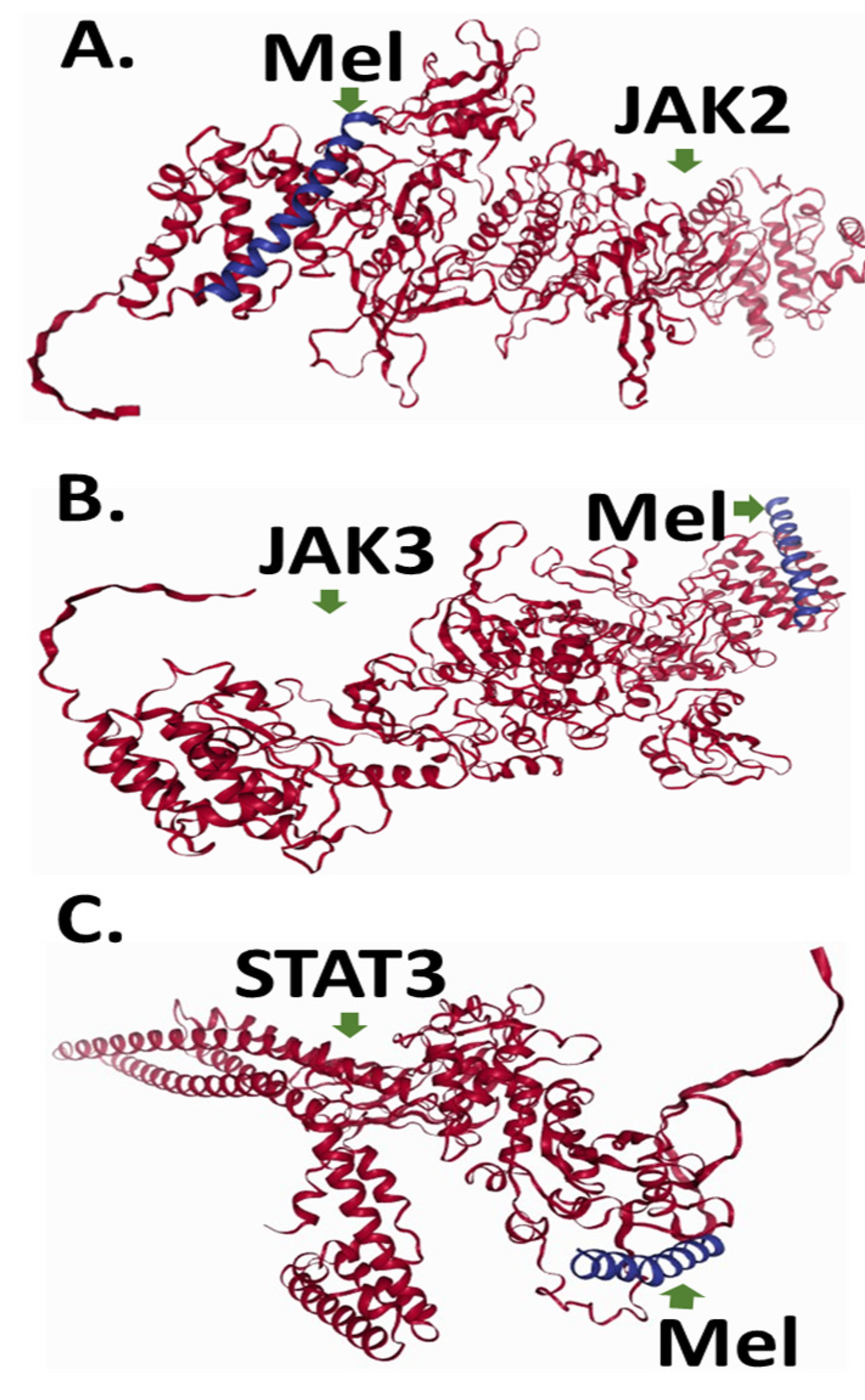
2.5. To Elucidate Potential Molecular Targets of Melittin, We Conducted In Silico Protein–Protein Docking Simulations Focusing on Key Regulators of Apoptotic Signaling Pathways, Specifically JAK2, JAK3, and STAT3
3. Discussion
4. Materials and Methods
4.1. Cell Line Sources
4.2. Wound Healing Assay (Scratch Wound Assay)
4.3. Caspase-3 and Caspase-8 Activity Detection
4.4. Gene Expression Analysis by Real-Time PCR
4.5. Quantitative Real-Time PCR
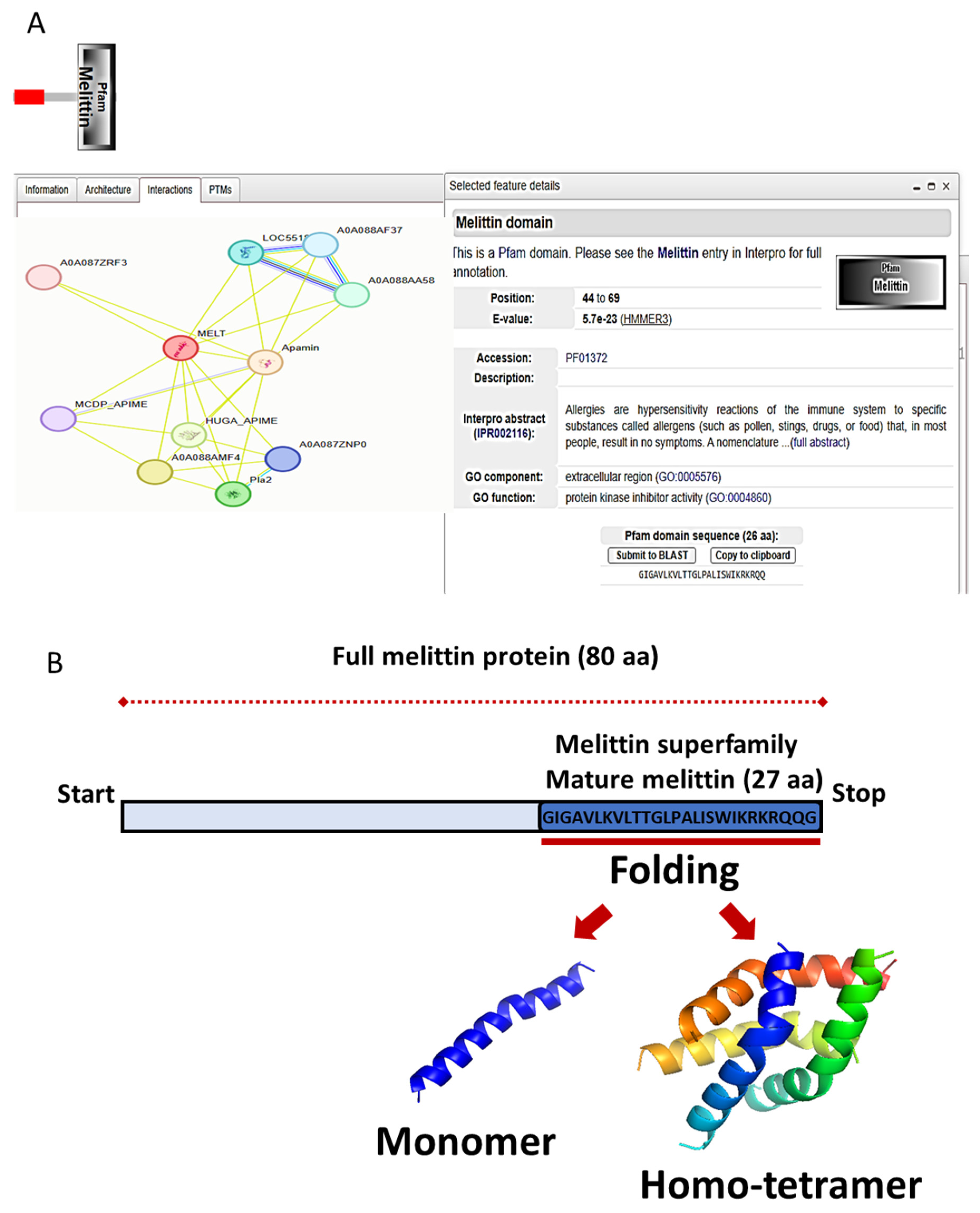
4.6. Protein Collection and Secondary Structure Prediction
4.7. Protein Docking Prediction
4.8. Statistical Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCLC | Non-Small Cell Lung Cancer |
| EGFR | Epidermal Growth Factor Receptor |
| µM | micromolar |
| JAK2 | Janus kinase 2 |
| JAK3 | Janus kinase 3 |
References
- WHO (World Health Organization). Fact Sheet: Lung Cancer. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/lung-cancer (accessed on 26 October 2024).
- Pandey, P.; Khan, F.; Khan, M.A.; Kumar, R.; Upadhyay, T.K. An updated review summarizing the anticancer efficacy of melittin from bee venom in several models of human cancers. Nutrients 2023, 15, 3111. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A. Exploring the Therapeutic Promise of Melittin: Anti-Inflammatory and Anti-Cancer Applications of Bee Venom. J. Intern. Med. Pharmacol. 2024, 1, 01–094. [Google Scholar]
- Misra, S.K.; Ye, M.; Kim, S.; Pan, D. Defined nanoscale chemistry influences delivery of peptido-toxins for cancer therapy. PLoS ONE 2015, 10, e0125908. [Google Scholar]
- Rehman, H.M.; Rehman, H.M.; Ahmed, N.; IMRAN, M. In silico Design and Evaluation of Novel Cell Targeting Melittin-Interleukin-24 Fusion Protein: A Potential Drug Candidate Against Breast Cancer. Sains Malaysiana 2023, 52, 3223–3237. [Google Scholar]
- Laurindo, L.F.; de Lima, E.P.; Laurindo, L.F.; Rodrigues, V.D.; Chagas, E.F.B.; Araújo, A.C.; Guiguer, E.L.; Pomini, K.T.; Rici, R.E.G.; Maria, D.A.; et al. The Therapeutic Potential of Bee Venom-Derived Apamin and Melittin Conjugates in Cancer Treatment: A Systematic Review. Pharmacol. Res. 2024, 209, 107430. [Google Scholar] [PubMed]
- Jafari, Z.; Sadeghi, S.; Dehaghi, M.M.; Bigham, A.; Honarmand, S.; Tavasoli, A.; Hoseini, M.H.M.; Varma, R.S. Immunomodulatory activities and biomedical applications of melittin and its recent advances. Arch. Pharm. 2024, 357, 2300569. [Google Scholar]
- Ceremuga, M.; Stela, M.; Janik, E.; Gorniak, L.; Synowiec, E.; Sliwinski, T.; Sitarek, P.; Saluk-Bijak, J.; Bijak, M. Melittin-A Natural Peptide from Bee Venom Which Induces Apoptosis in Human Leukaemia Cells. Biomolecules 2020, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar]
- Guha, S.; Ferrie, R.P.; Ghimire, J.; Ventura, C.R.; Wu, E.; Sun, L.; Kim, S.Y.; Wiedman, G.R.; Hristova, K.; Wimley, W.C. Applications and evolution of melittin, the quintessential membrane active peptide. Biochem. Pharmacol. 2021, 193, 114769. [Google Scholar] [CrossRef]
- Bajpai, P.; Chandra, P. Relevance of Conventional Herbal Remedies in the Prevention and Treatment of Malignant Tumors: Looking Toward the Future. Curr. Cancer Ther. Rev. 2025, 21, 54–75. [Google Scholar]
- Zhou, M.; Niu, H.; Lu, D.; Zhang, H.; Luo, D.; Yu, Z.; Huang, G.; Li, J.; Xiong, C.; Tang, Q.; et al. Wu Mei Wan suppresses colorectal cancer stemness by regulating Sox9 expression via JAK2/STAT3 pathway. J. Ethnopharmacol. 2025, 338, 118998. [Google Scholar]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [PubMed]
- Zhang, H.Q.; Sun, C.; Xu, N.; Liu, W. The current landscape of the antimicrobial peptide melittin and its therapeutic potential. Front. Immunol. 2024, 15, 1326033. [Google Scholar] [CrossRef]
- Liu, W.W.; Liu, Y.; Liang, S.; Wu, J.H.; Wang, Z.C.; Gong, S.L. Hypoxia- and radiation-induced overexpression of Smac by an adenoviral vector and its effects on cell cycle and apoptosis in MDA-MB-231 human breast cancer cells. Exp. Ther. Med. 2013, 6, 1560–1564. [Google Scholar] [PubMed]
- Zhang, F.-Q.; Yang, W.-T.; Duan, S.-Z.; Xia, Y.-C.; Zhu, R.-Y.; Chen, Y.-B. JAK2 inhibitor TG101348 overcomes erlotinib-resistance in non-small cell lung carcinoma cells with mutated EGF receptor. Oncotarget 2015, 6, 14329–14343. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Yang, L.; Zhao, W.; Gong, X.; Yue, D.; Liu, Y.; Tian, Y.; Dong, K. Exploring potential network pharmacology-and molecular docking-based mechanism of melittin in treating rheumatoid arthritis. Medicine 2023, 102, e34728. [Google Scholar] [CrossRef]
- Gouda, M.; Ibrahim, H.-I.M.; Negm, A. Chitosan Containing Nano Zn-Organic Framework: Synthesis, Characterization and Biological Activity. Polymers 2022, 14, 1276. [Google Scholar] [CrossRef]
- Balthazar, J.D.; Soosaimanickam, M.P.; Emmanuel, C.; Krishnaraj, T.; Sheikh, A.; Alghafis, S.F.; Ibrahim, H.I.M. 8-Hydroxyquinoline a natural chelating agent from Streptomyces spp. inhibits A549 lung cancer cell lines via BCL2/STAT3 regulating pathways. World J. Microbiol. Biotechnol. 2022, 38, 182. [Google Scholar] [CrossRef]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [PubMed]
- Venkatraman, V.; Yang, Y.D.; Sael, L.; Kihara, D. Protein-protein docking using region-based 3D Zernike descriptors. BMC Bioinform. 2009, 10, 407. [Google Scholar]
- Zhou, H.; Skolnick, J. GOAP: A Generalized Orientation-Dependent, All-Atom Statistical Potential for Protein Structure Prediction. Biophys. J. 2011, 101, 2043–2052. [Google Scholar]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2009, 11, 2714–2726. [Google Scholar]
- Huang, S.-Y.; Zou, X. Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins Struct. Funct. Bioinform. 2011, 79, 2648–2661. [Google Scholar]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 2.0; Schrodinger, LLC: San Carlos, CA, USA, 2000.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Template | % Identity | Oligo-state | Folding | Coverage |
|---|---|---|---|---|---|
| Mel | P0DPR9.1.A | 96.67% | Monomer | AlphaFold v2 | 100% |
| JAK2 | O60674.1.A | 100% | Monomer | AlphaFold v2 | 100% |
| JAK3 | P52333.1.A | 99.38% | Monomer | AlphaFold v2 | 100% |
| STAT3 | P42227.1.A | 99.87% | Monomer | AlphaFold v2 | 100% |
| P_P Interaction | GOAP Score | GOAP Rank | DFIRE Score | DFIRE Rank | ITScore Score | ITScore Rank | Ranksum Score | RMSD |
|---|---|---|---|---|---|---|---|---|
| Mel-JAK3 | −150,685 | 1 | −107,543 | 9 | −54,966 | 9 | 19 | 5.3 |
| Mel-JAK2 | −157,299 | 15 | −110,823 | 24 | −57,837 | 15 | 54 | 3.2 |
| Mel-STAT3 | −118,943 | 41 | −72,978 | 88 | −38,027 | 15 | 144 | 4.1 |
| S. No. | Primer Name | Forward Sequence | Reverse Sequence | PCR Products |
|---|---|---|---|---|
| 1 | Jak-2 | AACGCTGAGGGGGATTATCT; | TGGTTGGGTGGATACCAGA | 156 |
| 2 | Jak-3 | GGGAGATCCAGATCCTCAAAG; | GCAGATCTGTGAGGCGTAGAG | 194 |
| 3 | STAT-3 | CTTTGAGACCGAGGTGTATCACC | GTCAGCATGTTGTACCACAGG | 187 |
| 4 | GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA | 166 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, H.M.; Alessa, J.; Khalil, H.B.; Bekhet, G.A.; Khalifa, A. Synergistic Anti-Cancer Activity of Melittin and Erlotinib in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2025, 26, 2903. https://doi.org/10.3390/ijms26072903
Ibrahim HM, Alessa J, Khalil HB, Bekhet GA, Khalifa A. Synergistic Anti-Cancer Activity of Melittin and Erlotinib in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2025; 26(7):2903. https://doi.org/10.3390/ijms26072903
Chicago/Turabian StyleIbrahim, Hairulislam M., Jihad Alessa, Hala Badr Khalil, Gamal A. Bekhet, and Ashraf Khalifa. 2025. "Synergistic Anti-Cancer Activity of Melittin and Erlotinib in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 26, no. 7: 2903. https://doi.org/10.3390/ijms26072903
APA StyleIbrahim, H. M., Alessa, J., Khalil, H. B., Bekhet, G. A., & Khalifa, A. (2025). Synergistic Anti-Cancer Activity of Melittin and Erlotinib in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 26(7), 2903. https://doi.org/10.3390/ijms26072903