
The Arg108Cys Variant of Methylmalonyl-CoA Mutase: Clinical Implications for the Mexican Population Based on Molecular Dynamics and Docking
 , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
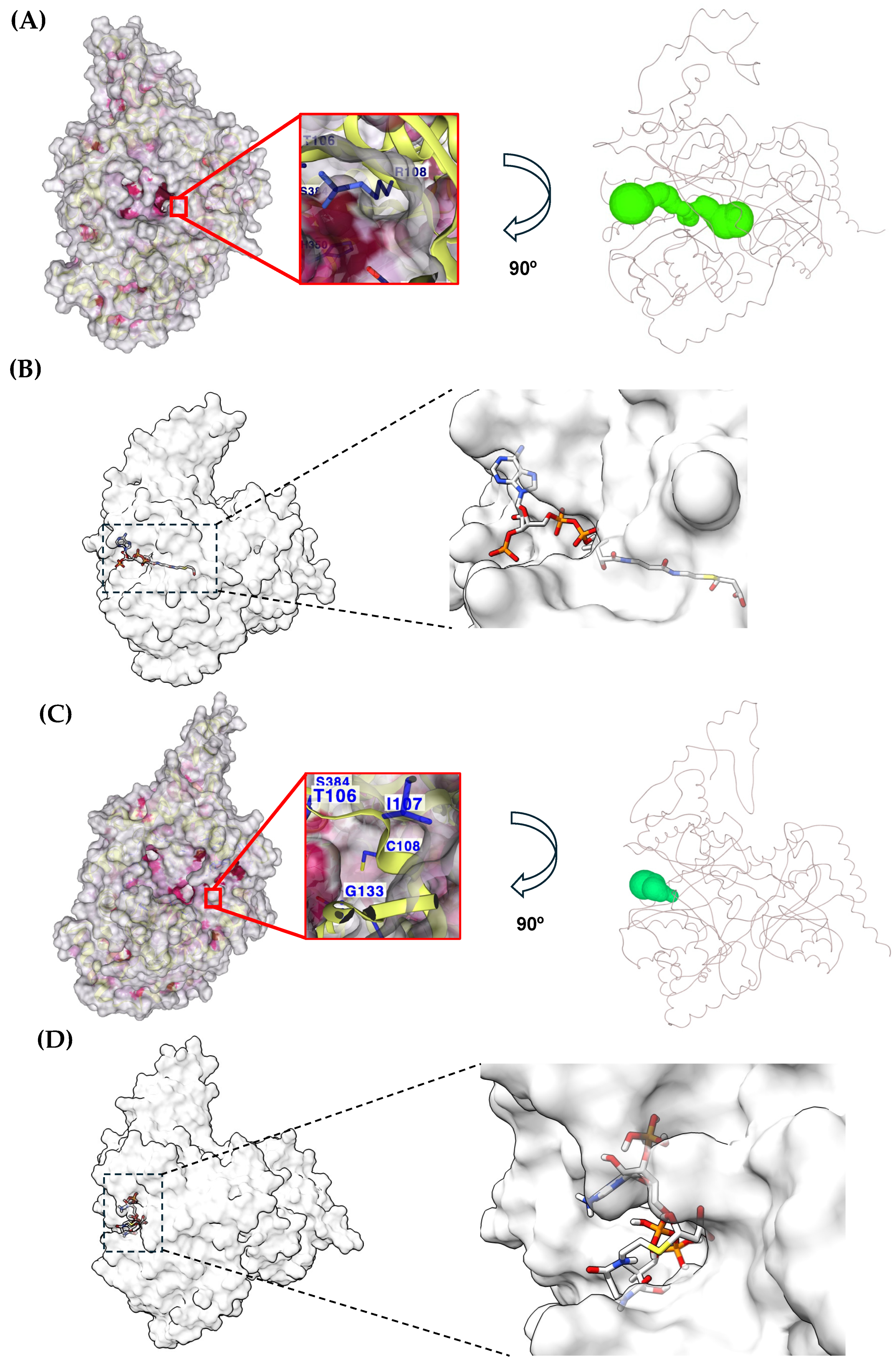
2. Results
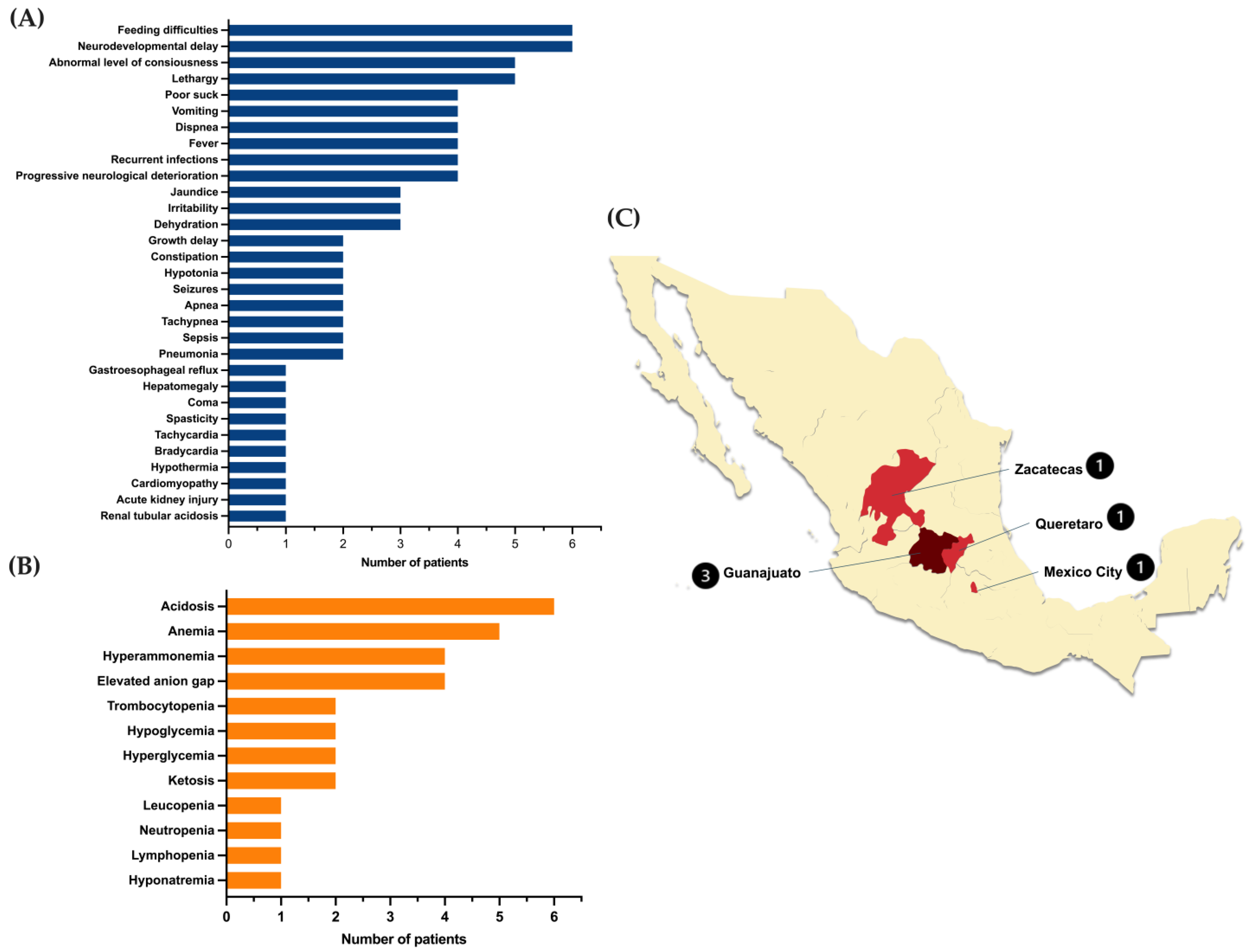
2.1. Clinical and Demographic Characteristics of MUTd Patients
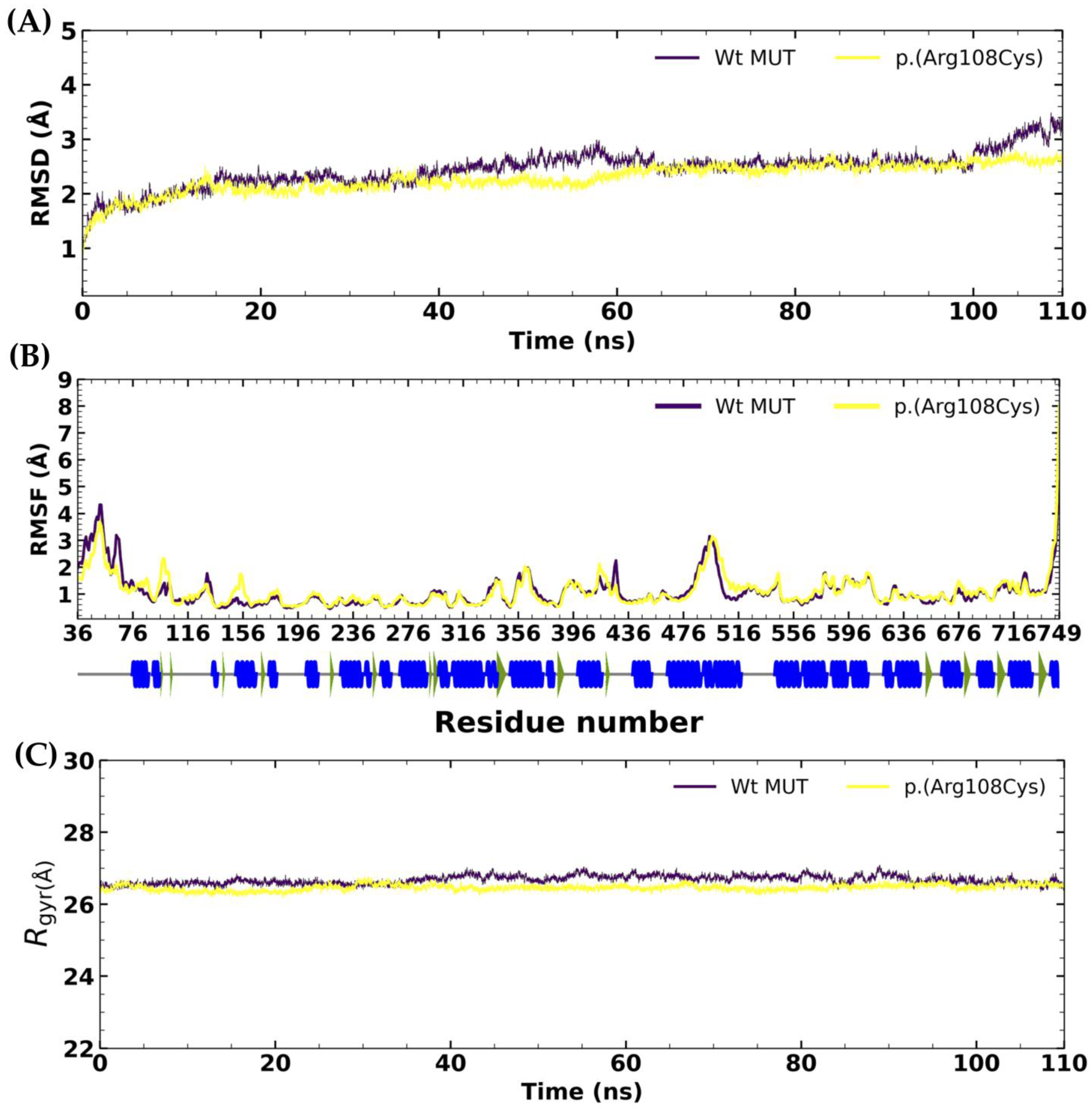
2.2. Molecular Dynamics of Human Methylmalonyl-CoA Mutase After 100 Ns Simulation
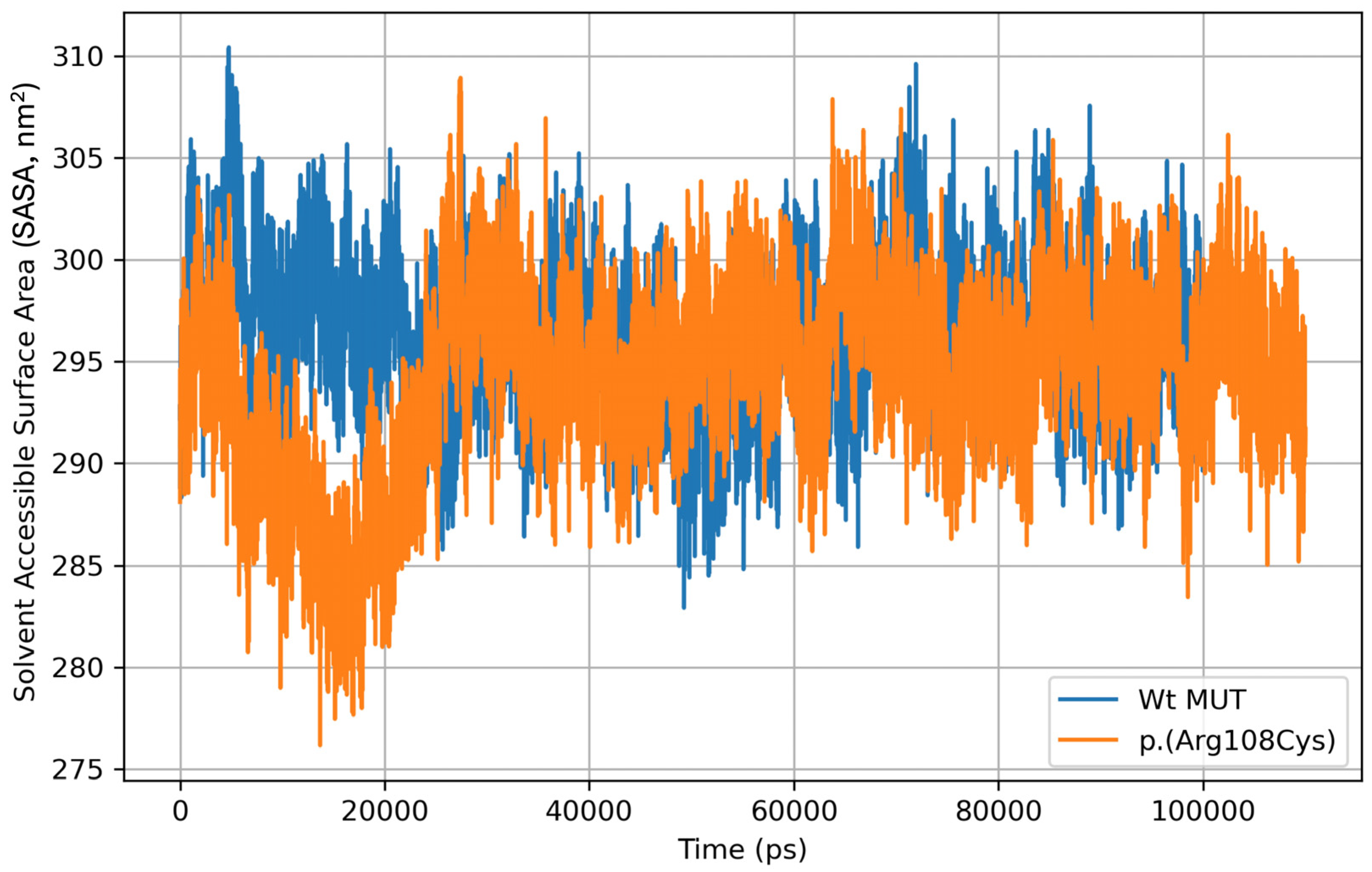
RMSD, RMSF, Rgyr, and SASA Analyses
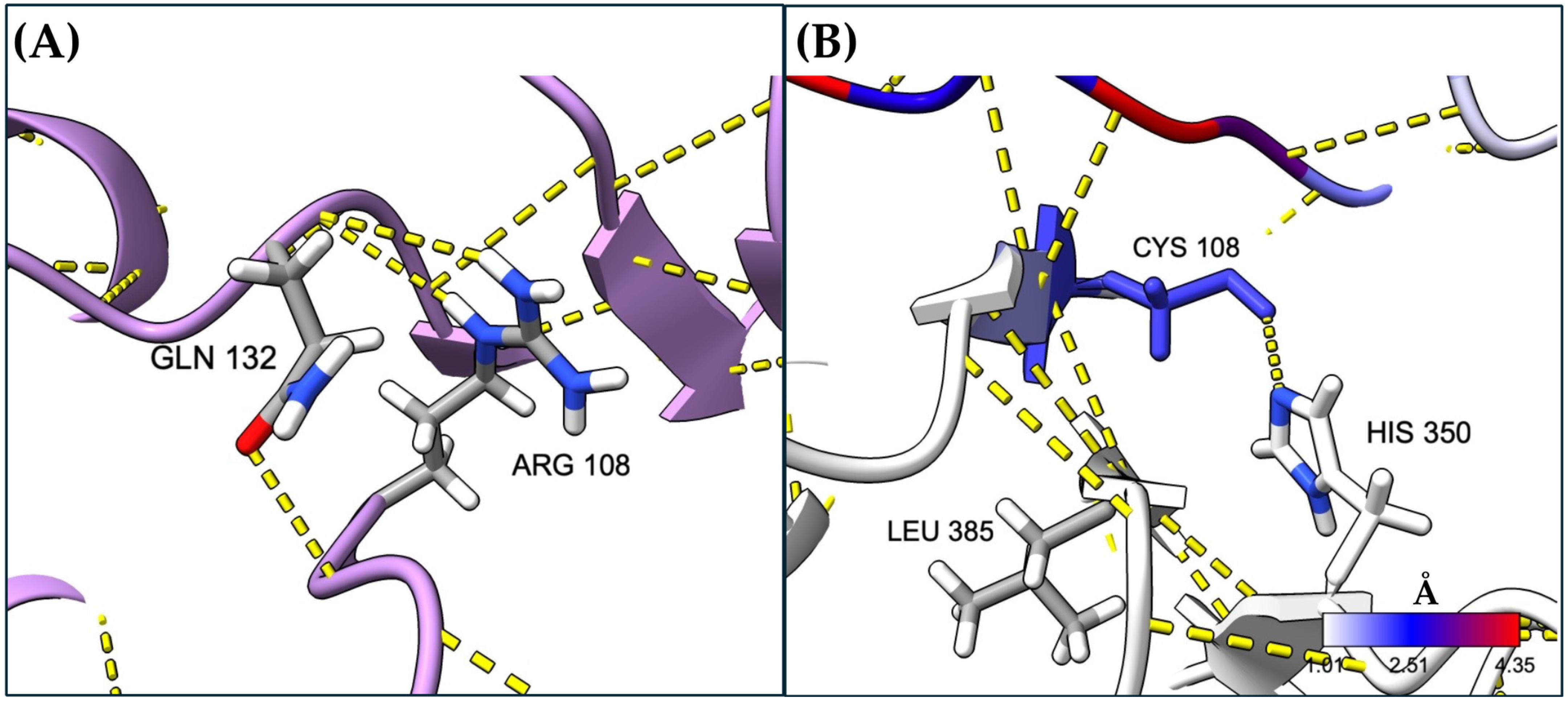
2.3. Long-Range Changes Attributable to the Amino Acid Residue Change at Position 108
2.4. Changes in Secondary Structure Between Wt MUT and p.(Arg108Cys) Variant
2.5. Local Changes in MUT Exerted by Cys Substitution at Position 108
3. Discussion
4. Materials and Methods
4.1. Families and Patients
4.2. In Silico Analyses
4.3. Molecular Dynamics Simulations
4.4. Analysis of Simulated Trajectories
4.5. Molecular Docking
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AdoCbl | adenosylcobalamin |
| C3 | Propionylcarnitine |
| MDS | molecular dynamics simulations |
| MMA | methylmalonic acidemia |
| MMAA | metabolism of cobalamin-associated A |
| MMAB | metabolism of cobalamin-associated B |
| MUT | methylmalonyl-CoA mutase |
| MMUT | methylmalonyl-CoA mutase gene |
| NPT | constant number of particles, pressure at 1 ATM, and temperature |
| NVT | constant number of particles, volume, and temperature |
| OH-2 Cbl | hydroxocobalamin |
| PDB | Protein Data Bank |
| RMSD | root mean square deviation |
| RMSF | root mean square fluctuation |
| Wt | wild-type |
References
- Takahashi-Iñiguez, T.; García-Hernandez, E.; Arreguín-Espinosa, R.; Flores, M.E. Role of vitamin B 12 on methylmalonyl-CoA mutase activity. J. Zhejiang Univ. Sci. B 2012, 13, 423–437. [Google Scholar]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar]
- Frenkel, E.P.; Kitchens, R.L. Intracellular Localization of Hepatic Propionyl-CoA Carboxylase and Methylmalonyl-CoA Mutase in Humans and Normal and Vitamin B12 Deficient Rats. Br. J. Haematol. 1975, 31, 501–513. [Google Scholar] [PubMed]
- Acquaviva, C.; Benoist, J.F.; Pereira, S.; Callebaut, I.; Koskas, T.; Porquet, D.; Elion, J. Molecular basis of methylmalonyl-CoA mutase apoenzyme defect in 40 European patients affected by mut° and mut–forms of methylmalonic acidemia: Identification of 29 novel mutations in the MUT gene. Hum. Mutat. 2005, 25, 167–176. [Google Scholar]
- Mascarenhas, R.; Ruetz, M.; Gouda, H.; Heitman, N.; Yaw, M.; Banerjee, R. Architecture of the human G-protein-methylmalonyl-CoA mutase nanoassembly for B12 delivery and repair. Nat. Commun. 2023, 14, 4332. [Google Scholar] [CrossRef] [PubMed]
- Head, P.E.; Meier, J.L.; Venditti, C.P. New insights into the pathophysiology of methylmalonic acidemia. J. Inherit. Metab. Dis. 2023, 46, 436–449. [Google Scholar] [PubMed]
- Costanzo, M.; Cevenini, A.; Kollipara, L.; Caterino, M.; Bianco, S.; Pirozzi, F.; Scerra, G.; D’Agostino, M.; Pavone, L.M.; Sickmann, A. Methylmalonic acidemia triggers lysosomal-autophagy dysfunctions. Cell Biosci. 2024, 14, 63. [Google Scholar]
- Armstrong, A.J.; Collado, M.S.; Henke, B.R.; Olson, M.W.; Hoang, S.A.; Hamilton, C.A.; Pourtaheri, T.D.; Chapman, K.A.; Summar, M.M.; Johns, B.A. A novel small molecule approach for the treatment of propionic and methylmalonic acidemias. Mol. Genet. Metab. 2021, 133, 71–82. [Google Scholar]
- Manoli, I.; Gebremariam, A.; McCoy, S.; Pass, A.R.; Gagné, J.; Hall, C.; Ferry, S.; Van Ryzin, C.; Sloan, J.L.; Sacchetti, E. Biomarkers to predict disease progression and therapeutic response in isolated methylmalonic acidemia (MMA). J. Inherit. Metab. Dis. 2023, 46, 554–572. [Google Scholar]
- Hörster, F.; Tuncel, A.T.; Gleich, F.; Plessl, T.; Froese, S.D.; Garbade, S.F.; Kölker, S.; Baumgartner, M.R.; Additional Contributors from E-IMD. Delineating the clinical spectrum of isolated methylmalonic acidurias: cblA and mut. J. Inherit. Metab. Dis. 2021, 44, 193–214. [Google Scholar]
- Manoli, I.; Sloan, J.L.; Venditti, C.P. Isolated Methylmalonic Acidemia. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Forny, P.; Hörster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grünert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 2021, 44, 566–592. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.-H.; Chien, Y.-H.; Lin, H.-Y.; Liao, H.-C.; Ho, H.-J.; Lai, C.-J.; Chiang, C.-C.; Lin, N.-C.; Yang, C.-F.; Hwu, W.-L. Methylmalonic acidemia/propionic acidemia–the biochemical presentation and comparing the outcome between liver transplantation versus non-liver transplantation groups. Orphanet J. Rare Dis. 2019, 14, 73. [Google Scholar] [PubMed]
- Minnee, R.C.; Sakamoto, S.; Fukuda, A.; Uchida, H.; Hirukawa, K.; Honda, M.; Okumura, S.; Ito, T.; Yilmaz, T.U.; Fang, Y. Long-Term Outcomes of Living Donor Liver Transplantation for Methylmalonic Acidemia. Pediatr. Transplant. 2024, 28, e14834. [Google Scholar]
- Ibarra-González, I.; Fernández-Lainez, C.; Vela-Amieva, M.; Guillén-López, S.; Belmont-Martínez, L.; López-Mejía, L.; Carrillo-Nieto, R.I.; Guillén-Zaragoza, N.A. A Review of Disparities and Unmet Newborn Screening Needs over 33 Years in a Cohort of Mexican Patients with Inborn Errors of Intermediary Metabolism. Int. J. Neonatal Screen. 2023, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Lainez, C.; Vela-Amieva, M.; Reyna-Fabián, M.; Fernández-Hernández, L.; Guillén-López, S.; López-Mejía, L.; Alcántara-Ortigoza, M.Á.; González-del Angel, A.; Carrillo-Nieto, R.I.; Ortega-Valdez, E. Isolated methylmalonic acidemia in Mexico: Genotypic spectrum, report of two novel MMUT variants and a possible synergistic heterozygosity effect. Mol. Genet. Metab. Rep. 2024, 41, 101155. [Google Scholar]
- Worgan, L.C.; Niles, K.; Tirone, J.C.; Hofmann, A.; Verner, A.; Sammak, A.A.; Kucic, T.; Lepage, P.; Rosenblatt, D.S. Spectrum of mutations in mut methylmalonic acidemia and identification of a common Hispanic mutation and haplotype. Hum. Mutat. 2006, 27, 31–43. [Google Scholar]
- Richardson, J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981, 34, 167–339. [Google Scholar]
- Beyzaei, Z.; Moravej, H.; Imanieh, M.H.; Inaloo, S.; Geramizadeh, B. Clinical spectrum and genetic variation of six patients with methylmalonic aciduria (MMA); a report from Iran. BMC Pediatr. 2024, 24, 795. [Google Scholar]
- Wang, T.; Ma, J.; Zhang, Q.; Gao, A.; Wang, Q.; Li, H.; Xiang, J.; Wang, B. Expanded newborn screening for inborn errors of metabolism by tandem mass spectrometry in Suzhou, China: Disease spectrum, prevalence, genetic characteristics in a Chinese population. Front. Genet. 2019, 10, 1052. [Google Scholar]
- Imtiaz, F.; Al-Mubarak, B.M.; Al-Mostafa, A.; Al-Hamed, M.; Allam, R.; Al-Hassnan, Z.; Al-Owain, M.; Al-Zaidan, H.; Rahbeeni, Z.; Qari, A. Spectrum of mutations in 60 saudi patients with mut methylmalonic acidemia. JIMD Rep. 2016, 29, 39–46. [Google Scholar]
- Lu, F.; Zhang, B.; Yang, Y.; Shi, Y.; Zheng, F.; Zhou, Q.; Chen, Y.; Zhou, L.; Yu, B. Mutation spectrum and genotype-phenotype correlation of pediatric patients with methylmalonic acidemia. Pediatr. Res. 2024, 1–8. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, C.; Dong, R.; Zhang, X.; Song, Y.; Hao, S.; Gai, Z.; Zhou, B.; Hui, L.; Wang, S. Improving methylmalonic acidemia (MMA) screening and MMA genotype prediction using random forest classifier in two Chinese populations. Eur. J. Med. Res. 2024, 29, 540. [Google Scholar] [PubMed]
- Narang, A.; Uppilli, B.; Vivekanand, A.; Naushin, S.; Yadav, A.; Singhal, K.; Shamim, U.; Sharma, P.; Zahra, S.; Mathur, A. Frequency spectrum of rare and clinically relevant markers in multiethnic Indian populations (ClinIndb): A resource for genomic medicine in India. Hum. Mutat. 2020, 41, 1833–1847. [Google Scholar]
- Vela-Amieva, M.; Abreu-Gonzalez, M.; Gonzalez-del Angel, A.; Ibarra-Gonzalez, I.; Fernandez-Lainez, C.; Barrientos-Rios, R.; Monroy-Santoyo, S.; Guillén-López, S.; Alcántara-Ortigoza, M. Phenylalanine hydroxylase deficiency in Mexico: Genotype–phenotype correlations, BH4 responsiveness and evidence of a founder effect. Clin. Genet. 2015, 88, 62–67. [Google Scholar] [PubMed]
- Mendiola-Vidal, N.G.; Contreras-Cubas, C.; Barajas-Olmos, F.; Villafan-Bernal, J.R.; Yañez-Felix, A.L.; García-Ortiz, H.; Centeno-Cruz, F.; Mendoza-Caamal, E.; Alaez-Verson, C.; Jiménez-Ruíz, J.L. Effect of the Complex Allele p.[Ile148Thr; Ile1023_Val1024del] in Cystic Fibrosis and Tracing of a Founder Effect in Mexican Families. Life 2024, 14, 1445. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Kochan, G.; Muniz, J.R.; Wu, X.; Gileadi, C.; Ugochukwu, E.; Krysztofinska, E.; Gravel, R.A.; Oppermann, U.; Yue, W.W. Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation. J. Biol. Chem. 2010, 285, 38204–38213. [Google Scholar]
- Chu, J.; Pupavac, M.; Watkins, D.; Tian, X.; Feng, Y.; Chen, S.; Fenter, R.; Zhang, V.W.; Wang, J.; Wong, L.-J. Next generation sequencing of patients with mut methylmalonic aciduria: Validation of somatic cell studies and identification of 16 novel mutations. Mol. Genet. Metab. 2016, 118, 264–271. [Google Scholar]
- Kirchner, B.; Di Dio, P.J.; Hutter, J. Real-world predictions from ab initio molecular dynamics simulations. In Multiscale Molecular Methods in Applied Chemistry; Springer: Berlin/Heidelberg, Germany, 2012; pp. 109–153. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Lemkul, J. From proteins to perturbed Hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- González-González, A.; Méndez-Álvarez, D.; Vázquez-Jiménez, L.K.; Delgado-Maldonado, T.; Ortiz-Pérez, E.; Paz-González, A.D.; Bandyopadhyay, D.; Rivera, G. Molecular docking and dynamic simulations of quinoxaline 1, 4-di-N-oxide as inhibitors for targets from Trypanosoma cruzi, Trichomonas vaginalis, and Fasciola hepatica. J. Mol. Model. 2023, 29, 180. [Google Scholar]
- Delgado-Maldonado, T.; Gonzalez-Morales, L.D.; Juarez-Saldivar, A.; Lara-Ramírez, E.E.; Rojas-Verde, G.; Moreno-Rodriguez, A.; Bandyopadhyay, D.; Rivera, G. Structure-based Virtual Screening from Natural Products as Inhibitors of SARS-CoV-2 Spike Protein and ACE2 Receptor Binding and their Biological Evaluation In vitro. Med. Chem. 2024, 20, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, J.; Pei, J.; Lai, L. CavityPlus 2022 update: An integrated platform for comprehensive protein cavity detection and property analyses with user-friendly tools and cavity databases. J. Mol. Biol. 2023, 435, 168141. [Google Scholar] [CrossRef]
- Pravda, L.; Sehnal, D.; Toušek, D.; Navrátilová, V.; Bazgier, V.; Berka, K.; Svobodová Vařeková, R.; Koča, J.; Otyepka, M. MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins: Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE, 2005; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wt | Arg108Cys | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Structural Motif | Start | End | Type of b-Sheet | Number of Residues | Start | End | Type of Sheet | Number of Residues | |
| Catalytic domain | Strand | Thr106 | Ala111 | B | 6 | Thr106 | Cys108 | B | 3 |
| Tyr110 | Ala11 | C | 2 | ||||||
| Helix | Val116 | Lys128 | H | 13 | Val116 | Ile127 | H | 12 | |
| Strand | Gly133 | Val136 | B | 4 | Ser135 | Val136 | C | 2 | |
| Helix | Pro151 | Val153 | G | 3 | Lost | ||||
| Strand | Val184 | Met186 | B | 3 | Ser185 | Met186 | C | 2 | |
| Helix | Lys210 | Lys212 | G | 3 | Lost | ||||
| Strand | Gly215 | Ile217 | B | 3 | Gly215 | Thr216 | D | 2 | |
| Helix | Leu222 | Met226 | H | 5 | Leu22 | Phe225 | G | 4 | |
| Strand | Ile259 | Ser262 | B | 4 | Ile259 | Ile261 | B | 3 | |
| Helix | Ala302 | Arg304 | G | 3 | Lost | ||||
| Phe315 | Phe337 | H | 23 | Phe315 | Met336 | H | 22 | ||
| Strand | Ala349 | Thr353 | B | 5 | Ala349 | Gln352 | B | 4 | |
| Helix | Asn365 | Phe379 | H | 15 | Pro363 | Asn365 | G | 3 | |
| Ile367 | Phe379 | H | 13 | ||||||
| Absent | Glu392 | Leu394 | G | 3 | |||||
| Linker domain | Helix | Met455 | Glu461 | H | 7 | Gly454 | Glu461 | H | 8 |
| Asn507 | Ser524 | H | 18 | Asn507 | Lys522 | H | 16 | ||
| Ile548 | Arg557 | H | 10 | Leu549 | Ala558 | H | 10 | ||
| Ala586 | Phe591 | H | 6 | Tyr587 | Phe591 | H | 5 | ||
| AdoCbl-Binding Domain | Strand | Ala668 | Thr673 | C | 6 | Ala668 | Val671 | E | 4 |
| Helix | His678 | Ser691 | H | 14 | Val682 | Ser691 | H | 10 | |
| Strand | Leu698 | Gly703 | C | 6 | Leu698 | Gly702 | E | 5 | |
| Asn720 | Phe722 | C | 3 | Val721 | Phe722 | E | 2 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vela-Amieva, M.; Delgado-Maldonado, T.; Ortega-Valdez, E.; Rivera, G.; López-Velázquez, G.; Fernández-Lainez, C. The Arg108Cys Variant of Methylmalonyl-CoA Mutase: Clinical Implications for the Mexican Population Based on Molecular Dynamics and Docking. Int. J. Mol. Sci. 2025, 26, 2887. https://doi.org/10.3390/ijms26072887
Vela-Amieva M, Delgado-Maldonado T, Ortega-Valdez E, Rivera G, López-Velázquez G, Fernández-Lainez C. The Arg108Cys Variant of Methylmalonyl-CoA Mutase: Clinical Implications for the Mexican Population Based on Molecular Dynamics and Docking. International Journal of Molecular Sciences. 2025; 26(7):2887. https://doi.org/10.3390/ijms26072887
Chicago/Turabian StyleVela-Amieva, Marcela, Timoteo Delgado-Maldonado, Enrique Ortega-Valdez, Gildardo Rivera, Gabriel López-Velázquez, and Cynthia Fernández-Lainez. 2025. "The Arg108Cys Variant of Methylmalonyl-CoA Mutase: Clinical Implications for the Mexican Population Based on Molecular Dynamics and Docking" International Journal of Molecular Sciences 26, no. 7: 2887. https://doi.org/10.3390/ijms26072887
APA StyleVela-Amieva, M., Delgado-Maldonado, T., Ortega-Valdez, E., Rivera, G., López-Velázquez, G., & Fernández-Lainez, C. (2025). The Arg108Cys Variant of Methylmalonyl-CoA Mutase: Clinical Implications for the Mexican Population Based on Molecular Dynamics and Docking. International Journal of Molecular Sciences, 26(7), 2887. https://doi.org/10.3390/ijms26072887