

Comprehensive Transcriptomic Analyses of Silk-Associated Genes and Functional Characterization of Key Silk Fibroins in Plutella xylostella
 ,
, 
Abstract

1. Introduction
2. Results
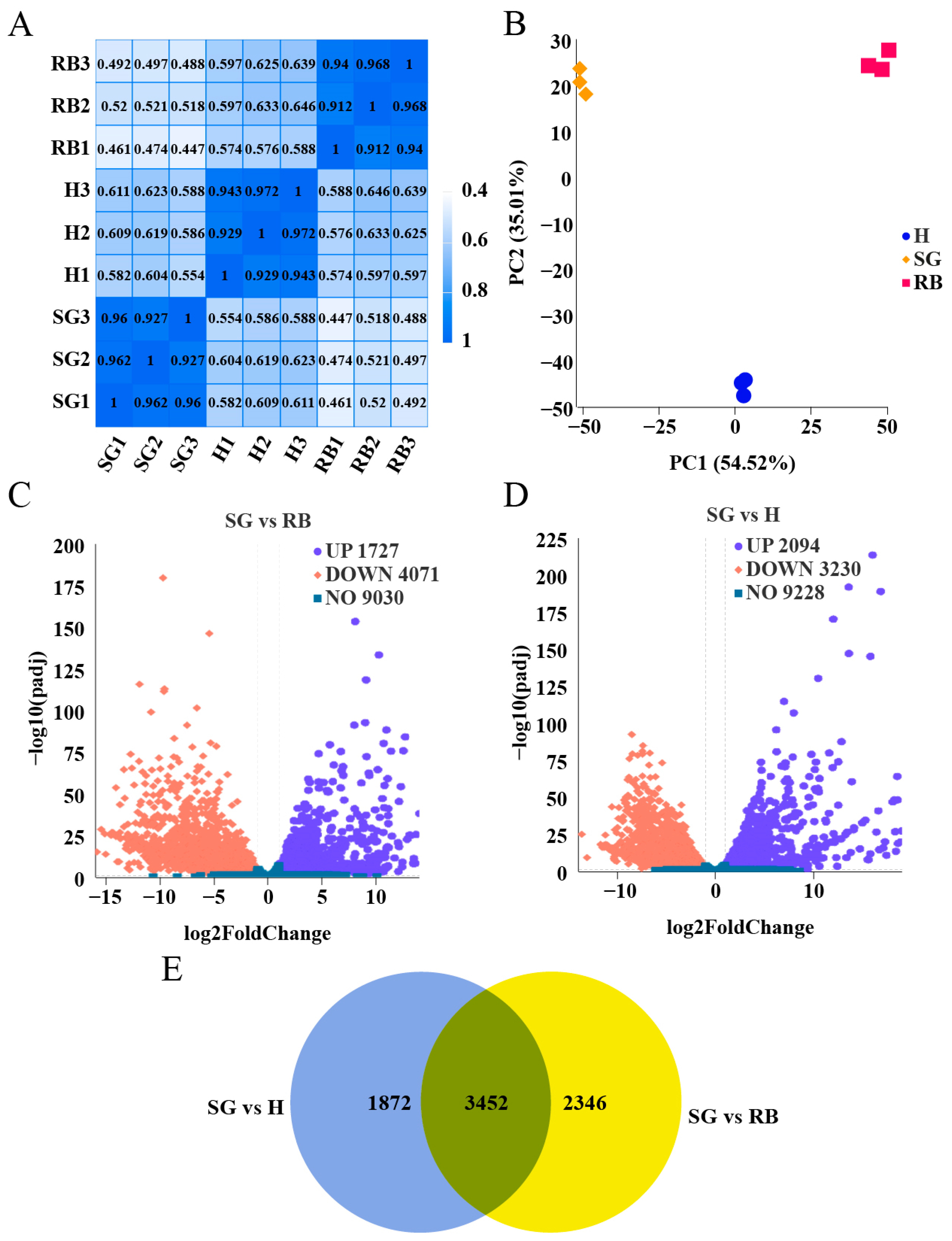
2.1. Overview of RNA-Seq Data
2.2. Transcriptome Sequencing Analysis
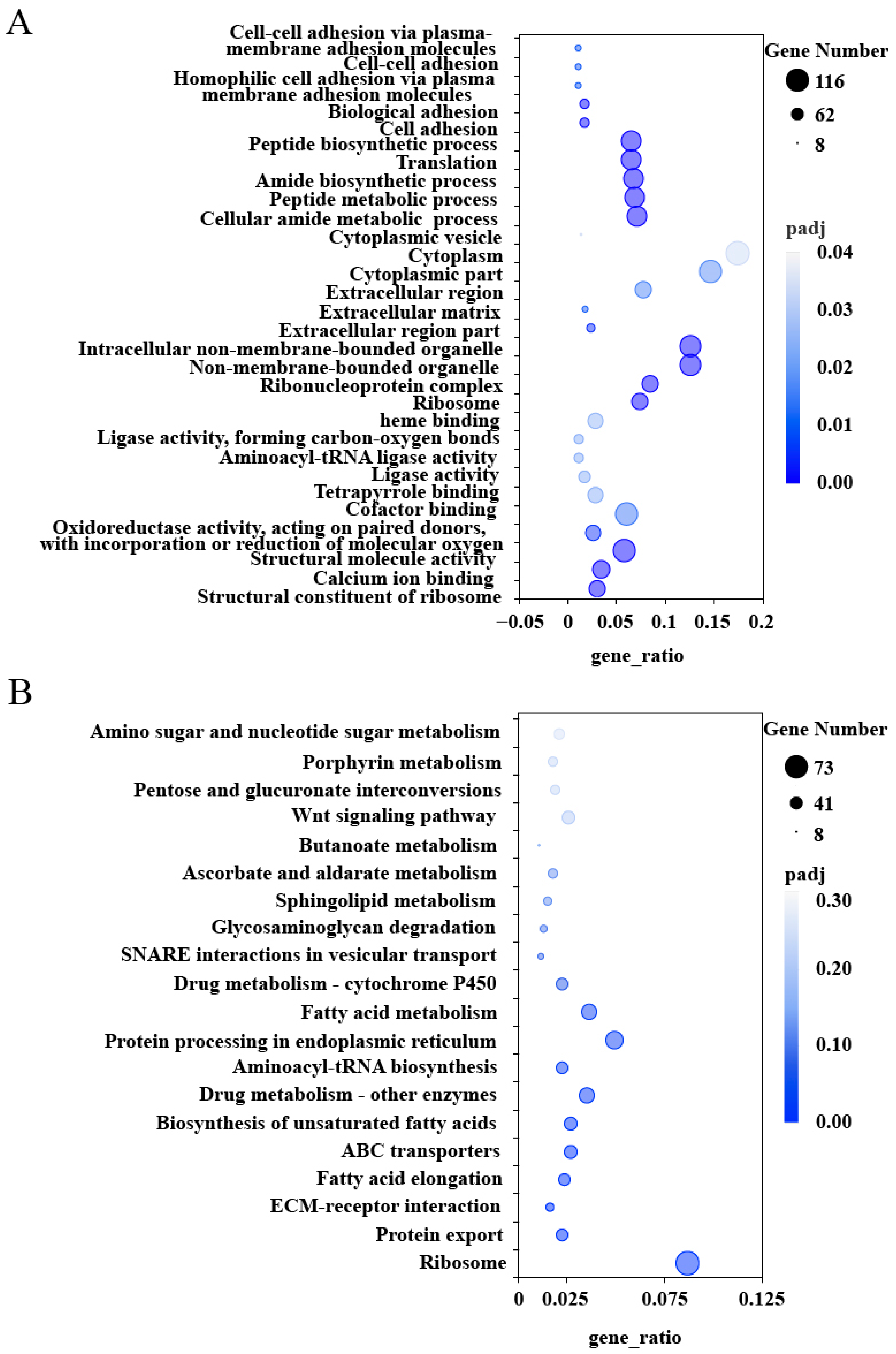
2.3. GO and KEGG Enrichment Analysis
2.4. RT-qPCR Validation
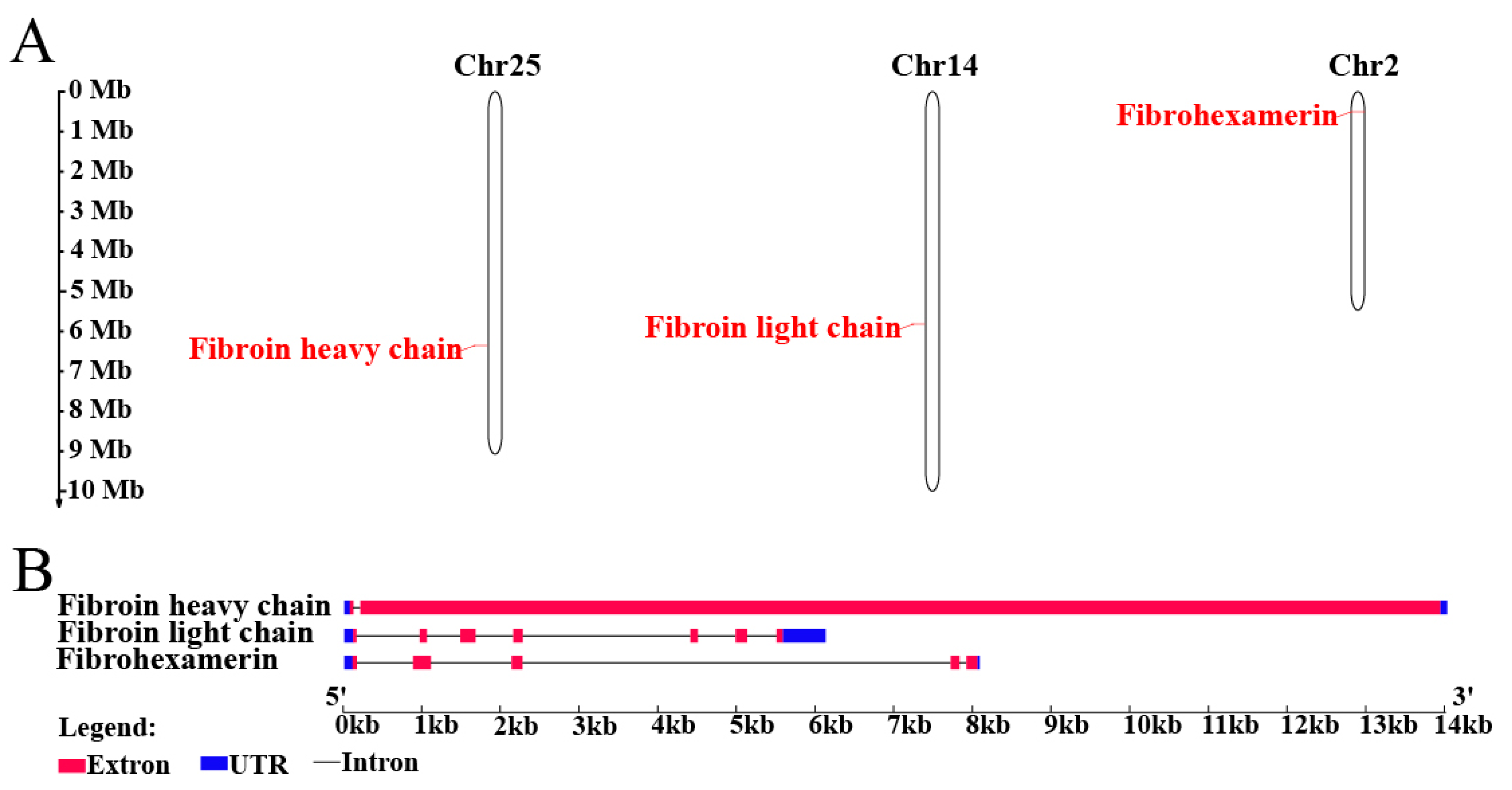
2.5. Silk Fibroins Analysis and Phylogenetic Tree Construction
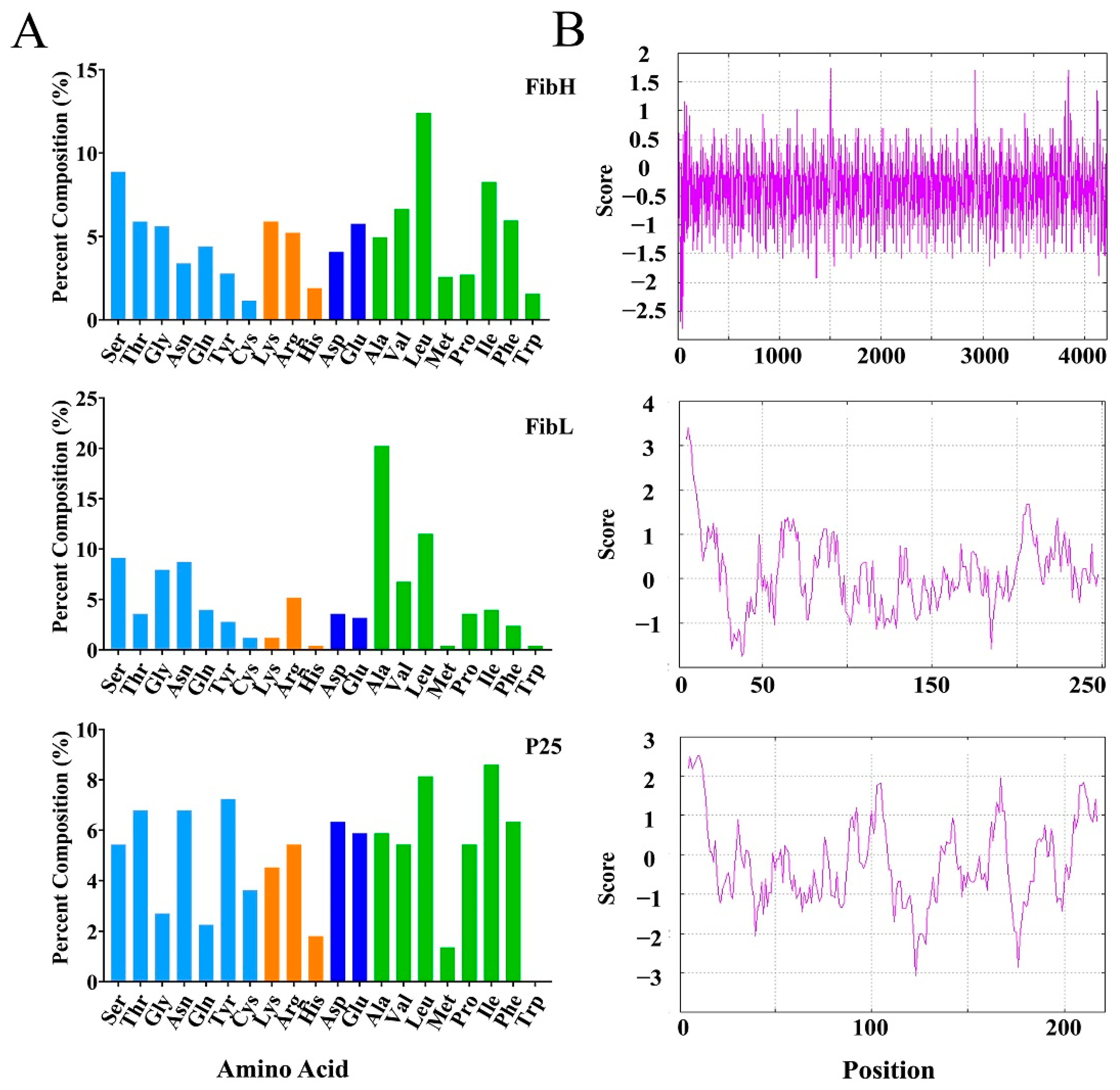
2.6. Amino Acid Composition and Hydrophilicity
2.7. Expression Profile of Silk Fibroin Genes
3. Discussion
4. Materials and Methods
4.1. Insect Culture
4.2. RNA Sequencing and Analysis
4.3. Bioinformatic Analysis
4.4. Phylogenetic Analysis
4.5. RNA Preparation and RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Furlong, M.J.; Wright, D.J.; Dosdall, L.M. Diamondback moth ecology and management: Problems, progress, and prospects. Annu. Rev. Entomol. 2013, 58, 517–541. [Google Scholar] [PubMed]
- Gangurde, S.M.; Wankhede, S. Biology of diamond back moth, Plutella xylostella Linn. Int. J. Plant Prot. 2009, 2, 165–166. [Google Scholar]
- Zalucki, M.P.; Asad, S.; Rehan, S.; David, A.; Liu, S.S.; Furlong, M.J. Estimating the economic cost of one of the world’s major insect pests, Plutella xylostella (Lepidoptera: Plutellidae): Just how long is a piece of string? J. Econ. Entomol. 2012, 105, 1125–1129. [Google Scholar]
- Dunn, T.P.S.; Champagne, D.E.; Riley, D.G.; Smith, H.; Bennett, J.E. A target site mutation associated with diamide insecticide resistance in the diamondback moth Plutella xylostella (lepidoptera: Plutellidae) is widespread in south georgia and florida populations. J. Econ. Entomol. 2022, 115, 289–296. [Google Scholar]
- Wang, J.; Zheng, X.B.; Yuan, J.J.; Wang, S.Y.; Xu, B.Y.; Wang, S.L.; Zhang, Y.J.; Wu, Q.J. Insecticide resistance monitoring of the diamondback moth (Lepidoptera: Plutellidae) populations in China. J. Econ. Entomol. 2021, 114, 1282–1290. [Google Scholar]
- Kirshboim, S.; Ishay, J.S. Silk produced by hornets: Thermophotovoltaic properties—A review. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2000, 127, 1–20. [Google Scholar]
- Wolff, J.O.; Lovtsova, J.; Gorb, E.; Dai, Z.D.; Ji, A.H.; Zhao, Z.H.; Jiang, N.; Gorb, S.N. Strength of silk attachment to Ilex chinensis leaves in the tea bagworm Eumeta minuscula (Lepidoptera, Psychidae). J. R. Soc. Interface 2017, 14, 20170007. [Google Scholar]
- Okada, S.; Weisman, S.; Trueman, H.E.; Mudie, S.T.; Haritos, V.S.; Sutherland, T.D. An Australian webspinner species makes the finest known insect silk fibers. Int. J. Biol. Macromol. 2008, 43, 271–275. [Google Scholar]
- Kaur, J.; Rajkhowa, R.; Tsuzuki, T.; Millington, K.; Zhang, J.; Wang, X. Photoprotection by silk cocoons. Biomacromolecules 2013, 14, 3660–3667. [Google Scholar]
- Talekar, N.S.; Shelton, A.M. Biology, ecology, and management of the diamondback moth. Annu. Rev. Entomol. 2003, 38, 275–301. [Google Scholar]
- Harcourt, D.G. Biology of the diamondback moth, Plutella maculipennis (Curt.) (Lepidoptera: Plutellidae), in Eastern Ontario. II. life-history, behaviour, and host relationships. Can. Entomol. 1957, 89, 554–564. [Google Scholar]
- Zhu, J.Y.; Xiang, Z.W.; Zhang, S.Z.; Wu, L.N.; Liu, T.X. Adaptations of Plutella xylostella adult females and larvae to waxy host plants. J. Pest Sci. 2022, 95, 203–214. [Google Scholar]
- Zhu, J.Y.; Xiang, Z.W.; Yu, W.Y.; Wang, W.W.; Liu, T.X.; Zhang, S.Z. Pieris rapae larvae facilitate the oviposition of Plutella xylostella by spinning silk on waxy host plants. Entomol. Gen. 2022, 42, 641–648. [Google Scholar]
- Sun, L.; Sun, B.; Chen, L.; Ge, Q.; Chen, K. Identification of genes associated with the silk gland size using multi-omics in silkworm (Bombyx mori ). Insect Mol. Biol. 2024, 33, 1–16. [Google Scholar] [PubMed]
- Liu, J.; Ding, Y.; Wang, Y.; Jiang, Y.; Wu, J.; Zhang, Y.; Zhang, J.; Miao, X.; Sun, Y.; Xue, X.; et al. Enhanced specific surface area and mechanical property of silk nanofibers aerogel for potential hemostasis applications. Int. J. Biol. Macromol. 2024, 277, 134345. [Google Scholar]
- Wang, W.; Ji, L.; Jing, X.; Zhao, P.; Xia, Q. MicroRNA let-7 targets BmCDK1 to regulate cell proliferation and endomitosis of silk gland in the silkworm, Bombyx mori. Insect Sci. 2024, 31, 1026–1040. [Google Scholar]
- Shi, Y.; Lin, G.; Fu, X.; Keller, M.; Smagghe, G.; Liu, T. Cocoon-spinning behavior and 20-hydroxyecdysone regulation of fibroin genes in Plutella xylostella. Front. Physiol. 2020, 11, 574800. [Google Scholar]
- Gamo, T.; Lauferc, T.I.A.H. Polypeptides of fibroin and sericin secreted from the different sections of the silk gland in Bombyx mori. Insect Biochem. 1977, 7, 285–295. [Google Scholar]
- Okamoto, H.; Ishikawa, E.; Suzuki, Y. Structural analysis of sericin genes. Homologies with fibroin gene in the 5′ flanking nucleotide sequences. J. Biol. Chem. 1982, 257, 15192–15199. [Google Scholar]
- Ishikawa, E.; Suzuki, Y. Tissue- and stage-specific expression of sericin genes in the middle silk gland of Bombyx mori: (sericin mRNA/northern blotting/gene regulation). Dev. Growth. Diff. 1985, 27, 73–82. [Google Scholar]
- Inoue, S.; Tanaka, K.; Arisaka, F.; Kimura, S.; Ohtomo, K.; Mizuno, S. Silk fibroin of Bombyx mori is secreted, assembling a high molecular mass elementary unit consisting of H-chain, L-chain, and P25, with a 6:6:1 molar ratio. J. Biol. Chem. 2000, 275, 40517–40528. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Mori, K.; Mizuno, S. Immunological identification of the major disulfide-linked light component of silk fibroin. J. Biochem. 1993, 114, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Inoue, S.; Mizuno, S. Hydrophobic interaction of P25, containing Asn-linked oligosaccharide chains, with the H-L complex of silk fibroin produced by Bombyx mori. Insect Biochem. Mol. Biol. 1999, 29, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Peng, Z.; Lv, J.; Zhang, Q.; Wang, X.; Xia, Q. Developmental and nuclear proteomic signatures characterize the temporal regulation of fibroin synthesis during the last molting-feeding transition of silkworm. Int. J. Biol. Macromol. 2024, 274, 133028. [Google Scholar] [CrossRef]
- Ma, Y.; Li, Q.; Tang, Y.; Zhang, Z.; Liu, R.; Luo, Q.; Wang, Y.; Hu, J.; Chen, Y.; Li, Z.; et al. The architecture of silk-secreting organs during the final larval stage of silkworms revealed by single-nucleus and spatial transcriptomics. Cell. Rep. 2024, 43, 114460. [Google Scholar] [CrossRef]
- Singh, D.; Mosahari, P.V.; Sharma, P.; Neog, K.; Bora, U. Comparative genomic and phylogenetic analysis of the complete mitochondrial genome of Cricula trifenestrata (Helfer) among lepidopteran insects. Genome 2024, 67, 424–439. [Google Scholar] [CrossRef]
- Heckenhauer, J.; Plotkin, D.; Martinez, J.I.; Bethin, J.; Pauls, S.U.; Frandsen, P.B.; Kawahara, A.Y. Genomic resources of aquatic Lepidoptera, Elophila obliteralis and Hyposmocoma kahamanoa, reveal similarities with Trichoptera in amino acid composition of major silk genes. G3-Genes. Genom. Genet. 2024, 14, jkae93. [Google Scholar] [CrossRef]
- Rindos, M.; Kucerova, L.; Rouhova, L.; Sehadova, H.; Sery, M.; Hradilova, M.; Konik, P.; Zurovec, M. Comparison of silks from Pseudoips prasinana and Bombyx mori shows molecular convergence in fibroin heavy chains but large differences in other silk components. Int. J. Biol. Macromol. 2021, 22, 8246. [Google Scholar] [CrossRef]
- Wu, N.; Zhang, S.; Li, X.; Cao, Y.; Liu, X.; Wang, Q.; Liu, Q.; Liu, H.; Hu, X.; Zhou, X.J.; et al. Fall webworm genomes yield insights into rapid adaptation of invasive species. Nat. Ecol. Evol. 2019, 3, 105–115. [Google Scholar] [CrossRef]
- Kmet, P.; Kucerova, L.; Sehadova, H.; Chia-Hsiang, W.B.; Wu, Y.L.; Zurovec, M. Identification of silk components in the bombycoid moth Andraca theae (Endromidae) reveals three fibroin subunits resembling those of Bombycidae and Sphingidae. J. Insect Physiol. 2023, 147, 104523. [Google Scholar] [CrossRef]
- Shi, R.; Ma, S.Y.; He, T.; Peng, J.; Zhang, T.; Chen, X.X.; Wang, X.G.; Chang, J.S.; Xia, Q.Y.; Zhao, P. Deep insight into the transcriptome of the single silk gland of Bombyx mori. Int. J. Mol. Sci. 2019, 20, 2491. [Google Scholar] [CrossRef] [PubMed]
- Mita, K.; Ichimura, S.; James, T.C. Highly repetitive structure and its organization of the silk fibroin gene. J. Mol. Evol. 1994, 38, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Yokoi, K.; Yamamoto, K.; Jouraku, A. An update of KAIKObase, the silkworm genome database. Database 2021, 2021, baaa099. [Google Scholar] [CrossRef]
- Yonemura, N.; Sehnal, F. The design of silk fiber composition in moths has been conserved for more than 150 million years. J. Mol. Evol. 2006, 63, 42–53. [Google Scholar] [CrossRef]
- Tamura, T.; Inoue, H.; Suzuki, Y. The fibroin genes of Antheraea yamamai and Bombyx mori are different in their core regions but reveal a striking sequence similarity in their 5′ ends and 5′ flanking regions. Mol. Gen. Genet. MGG 1987, 207, 189–195. [Google Scholar] [CrossRef]
- Cui, Y.; Zhu, Y.; Lin, Y.; Chen, L.; Feng, Q.; Wang, W.; Xiang, H. New insight into the mechanism underlying the silk gland biological process by knocking out fibroin heavy chain in the silkworm. BMC Genom. 2018, 19, 215. [Google Scholar]
- Ye, X.; Dai, X.; Wang, X.; Yu, S.; Wu, M.; Zhao, S.; Ruan, J.; Zhong, B. Mechanism of silk secretion revealed by proteomic analysis of silkworm cocoons with fibroin light chain mutations. J. Proteom. 2022, 265, 104649. [Google Scholar] [CrossRef]
- Zafar, M.S.; Belton, D.J.; Hanby, B.; Kaplan, D.L.; Perry, C.C. Functional material features of Bombyx mori silk light versus heavy chain proteins. Biomacromolecules 2015, 16, 606–614. [Google Scholar] [CrossRef]
- Adhav, A.; Harne, S.; Bhide, A.; Giri, A.; Gayathri, P.; Joshi, R. Mechanistic insights into enzymatic catalysis by trehalase from the insect gut endosymbiont Enterobacter cloacae. FEBS J. 2019, 286, 1700–1716. [Google Scholar]
- Lin, C.; Top, D.; Manahan, C.C.; Young, M.W.; Crane, B.R. Circadian clock activity of cryptochrome relies on tryptophan-mediated photoreduction. Proc. Natl. Acad. Sci. USA 2018, 115, 3822–3827. [Google Scholar] [PubMed]
- Oxenkrug, G.; Navrotska, V. Extension of life span by down-regulation of enzymes catalyzing tryptophan conversion into kynurenine: Possible implications for mechanisms of aging. Exp. Biol. Med. 2023, 248, 573–577. [Google Scholar]
- Zabelina, V.; Takasu, Y.; Sehadova, H.; Yonemura, N.; Nakajima, K.; Sezutsu, H.; Sery, M.; Zurovec, M.; Sehnal, F.; Tamura, T. Mutation in Bombyx mori fibrohexamerin (P25) gene causes reorganization of rough endoplasmic reticulum in posterior silk gland cells and alters morphology of fibroin secretory globules in the silk gland lumen. Insect Biochem. Mol. Biol. 2021, 135, 103607. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.Y.; Zhang, X.L.; Zhao, D.C.; Qin, L.X.; Jiang, W.C.; Hu, W.B.; Liu, X.; Xia, Q.Y.; Dong, Z.M.; Zhao, P. Identification and characterization of sericin5 reveals non-cocoon silk sericin components with high β-sheet content and adhesive strength. Acta Biomater. 2022, 150, 96–110. [Google Scholar]
- Chaitanya, R.K.; Sridevi, P.; Senthilkumaran, B.; Dutta, G.A. Effect of juvenile hormone analog, methoprene on H-fibroin regulation during the last instar larval development of Corcyra cephalonica. Gen. Comp. Endocrinol. 2013, 181, 10–17. [Google Scholar]
- Kimoto, M.; Tsubota, T.; Uchino, K.; Sezutsu, H.; Takiya, S. LIM-homeodomain transcription factor Awh is a key component activating all three fibroin genes, fibH, fibL and fhx, in the silk gland of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2015, 56, 29–35. [Google Scholar]
- Masuoka, Y.; Cao, W.; Jouraku, A.; Sakai, H.; Sezutsu, H.; Yokoi, K. Co-expression network and time-course expression analyses to identify silk protein regulatory factors in Bombyx mori. Insects 2022, 13, 131. [Google Scholar] [CrossRef]
- Chaitanya, R.K.; Dutta-Gupta, A. Light chain fibroin and P25 genes of Corcyra cephalonica: Molecular cloning, characterization, tissue-specific expression, synchronous developmental and 20-hydroxyecdysone regulation during the last instar larval development. Gen. Comp. Endocrinol. 2010, 167, 113–121. [Google Scholar]
- Jiang, D.; Qian, C.; Wang, D.H.; Wang, F.L.; Zhao, S.; Yang, Y.H.; Baxter, S.W.; Wang, X.L.; Wu, Y.D. Varying contributions of three ryanodine receptor point mutations to diamide insecticide resistance in Plutella xylostella. Pest Manag. Sci. 2021, 77, 4874–4883. [Google Scholar]
- Zhu, J.Y.; Xiang, Z.W.; Zhang, S.Z.; Kang, Z.W.; Fan, Y.L.; Liu, T.X. A new pest management strategy: Transforming a non-host plant into a dead-end trap crop for the diamondback moth Plutella xylostella L. Pest Manag. Sci. 2021, 77, 1094–1101. [Google Scholar]
- Zhang, H.; Li, H.C.; Miao, X.X. Feasibility, limitation and possible solutions of RNAi-based technology for insect pest control. Insect Sci. 2013, 20, 15–30. [Google Scholar] [PubMed]
- Chao, J.; Li, Z.; Sun, Y.; Aluko, O.O.; Wu, X.; Wang, Q.; Liu, G. MG2C: A user-friendly online tool for drawing genetic maps. Mol. Hortic. 2021, 1, 16. [Google Scholar] [PubMed]
- Almagro, A.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree visualization by one table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Q20 | Q30 | GC (%) |
|---|---|---|---|---|---|---|---|
| SG1 | 41,191,356 | 6.18 G | 39,767,162 | 5.97 G | 97.41 | 93.26 | 51.49 |
| SG2 | 48,942,932 | 7.34 G | 47,060,438 | 7.06 G | 96.99 | 92.46 | 51.10 |
| SG3 | 41,824,838 | 6.27 G | 40,723,700 | 6.11 G | 96.71 | 91.89 | 50.45 |
| H1 | 42,368,122 | 6.36 G | 41,705,096 | 6.26 G | 97.47 | 93.28 | 48.56 |
| H2 | 46,174,644 | 6.93 G | 45,491,500 | 6.82 G | 97.32 | 92.87 | 47.77 |
| H3 | 42,519,786 | 6.38 G | 41,719,820 | 6.26 G | 97.53 | 93.38 | 48.42 |
| RB1 | 46,530,060 | 6.98 G | 45,525,936 | 6.83 G | 97.28 | 92.97 | 52.93 |
| RB2 | 44,564,120 | 6.68 G | 43,548,404 | 6.53 G | 97.54 | 93.48 | 51.42 |
| RB3 | 43,085,530 | 6.46 G | 41,825,324 | 6.27 G | 97.48 | 93.48 | 51.76 |
| Primers | Direction | Primer Sequence (5′-3′) |
|---|---|---|
| FibH | Forward | GCTCAAGTTCCGCAGCAACCA |
| Reverse | CCCGCAGCAGCAGTGTTTGAA | |
| FibL | Forward | CACACTAACCGCCGTGAACGT |
| Reverse | GCCAGCAGGTTGATGGTCTGTC | |
| P25 | Forward | CCCGCCGAAATCTGCAATCCA |
| Reverse | CGTTGGCGTTGCATCTGGAGTT | |
| Udp | Forward | TGGCAACAGTGGACCGAGATCA |
| Reverse | GCAGCAACTTGACACAGTGGCA | |
| Fer3 | Forward | TGAGGTGGCGGATCACAGCAA |
| Reverse | CCTCCACTCCAGAGCAGCATCA | |
| Ser | Forward | GCTGGTGTCGTGCAGCAACT |
| Reverse | CGTCGCCGCGTTCATCTTCTT | |
| Sec62 | Forward | TCGGCTTTGTGGCTTCTTTCTGG |
| Reverse | CTCCTCCTCGTCGTCGGAATGT | |
| Pcp36a1 | Forward | TCGTCATTCTCGTCGTCCGTCA |
| Reverse | GGTTCCTCGTCTCCGTTCTGGT | |
| RPL32 | Forward | CAATCAGGCCAATTTACCGC |
| Reverse | CTGCGTTTACGCCAGTTACG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, R.-C.; Meng, F.-X.; Zeng, Q.-H.; Wang, Y.-J.; Liu, T.-X.; Chu, D.; Zhang, S.-Z. Comprehensive Transcriptomic Analyses of Silk-Associated Genes and Functional Characterization of Key Silk Fibroins in Plutella xylostella. Int. J. Mol. Sci. 2025, 26, 2842. https://doi.org/10.3390/ijms26072842
Niu R-C, Meng F-X, Zeng Q-H, Wang Y-J, Liu T-X, Chu D, Zhang S-Z. Comprehensive Transcriptomic Analyses of Silk-Associated Genes and Functional Characterization of Key Silk Fibroins in Plutella xylostella. International Journal of Molecular Sciences. 2025; 26(7):2842. https://doi.org/10.3390/ijms26072842
Chicago/Turabian StyleNiu, Rui-Chang, Fan-Xin Meng, Qing-Hui Zeng, Yi-Jing Wang, Tong-Xian Liu, Dong Chu, and Shi-Ze Zhang. 2025. "Comprehensive Transcriptomic Analyses of Silk-Associated Genes and Functional Characterization of Key Silk Fibroins in Plutella xylostella" International Journal of Molecular Sciences 26, no. 7: 2842. https://doi.org/10.3390/ijms26072842
APA StyleNiu, R.-C., Meng, F.-X., Zeng, Q.-H., Wang, Y.-J., Liu, T.-X., Chu, D., & Zhang, S.-Z. (2025). Comprehensive Transcriptomic Analyses of Silk-Associated Genes and Functional Characterization of Key Silk Fibroins in Plutella xylostella. International Journal of Molecular Sciences, 26(7), 2842. https://doi.org/10.3390/ijms26072842