
Unraveling the Complex Genomic Interplay of Sickle Cell Disease Among the Saudi Population: A Case-Control GWAS Analysis
 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Study Participant Characteristics
2.2. Top Identified Variants with Functional Consequences
3. Discussion
4. Materials and Methods
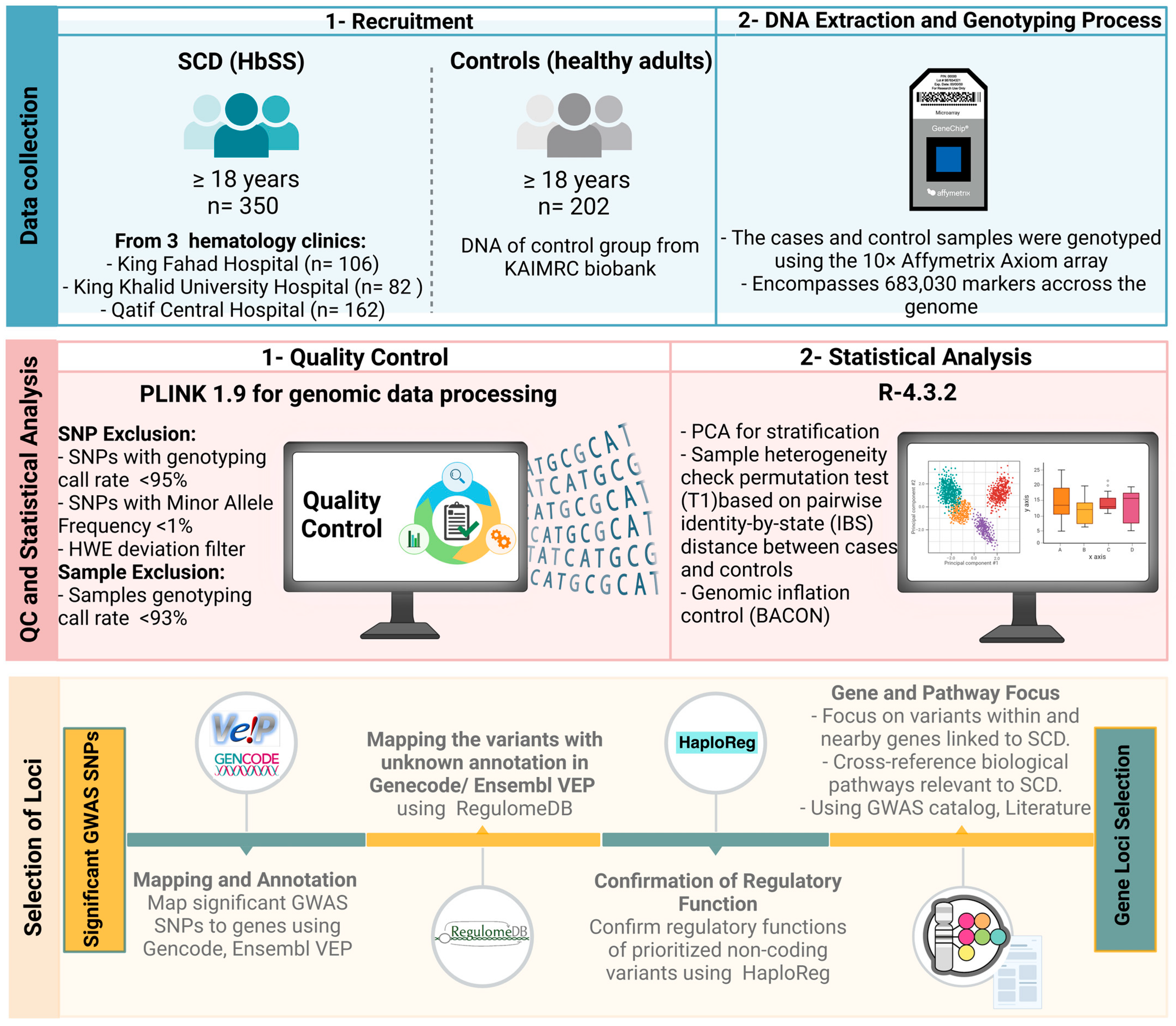
4.1. Sample Recruitment
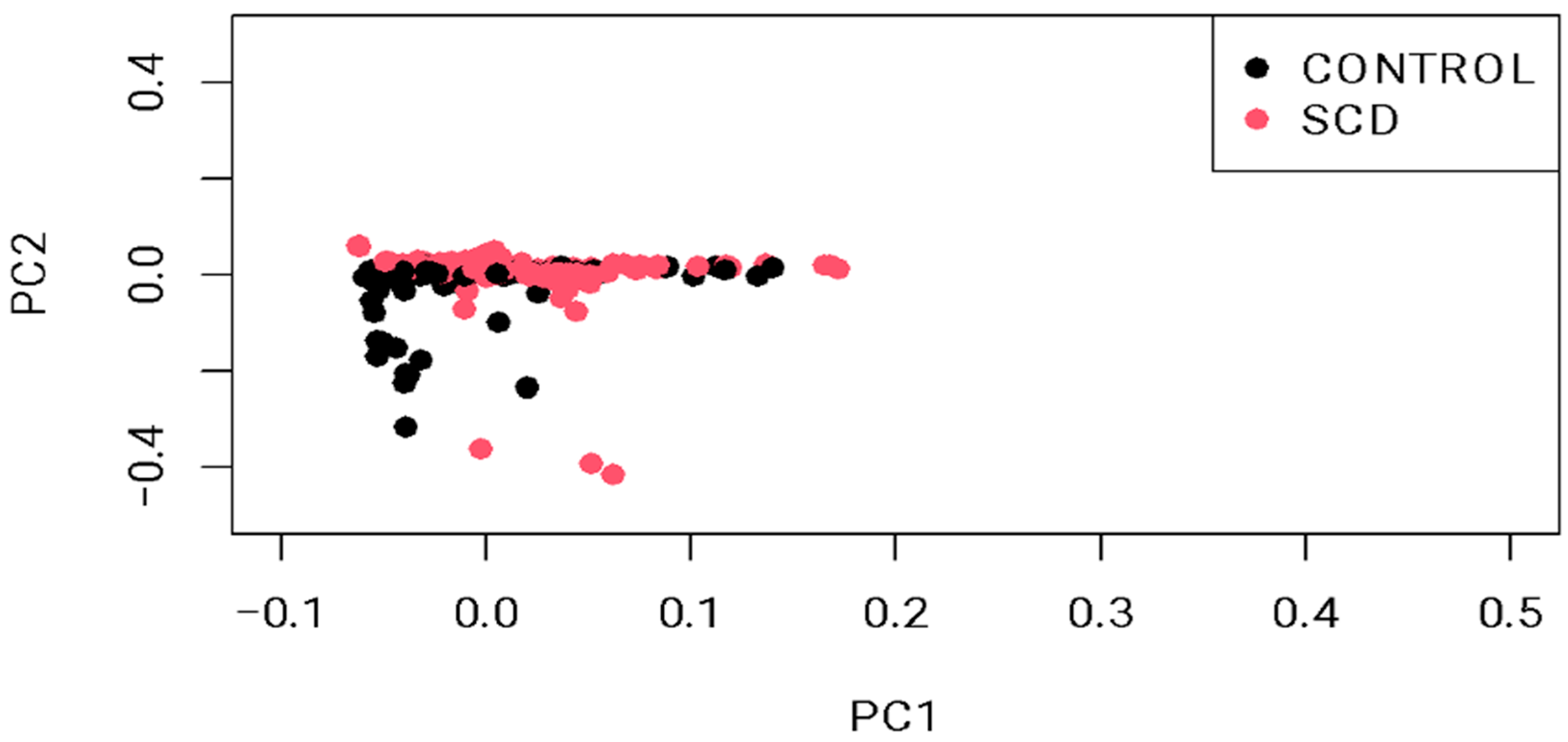
4.2. Genomic Analysis and Quality Control (QC)
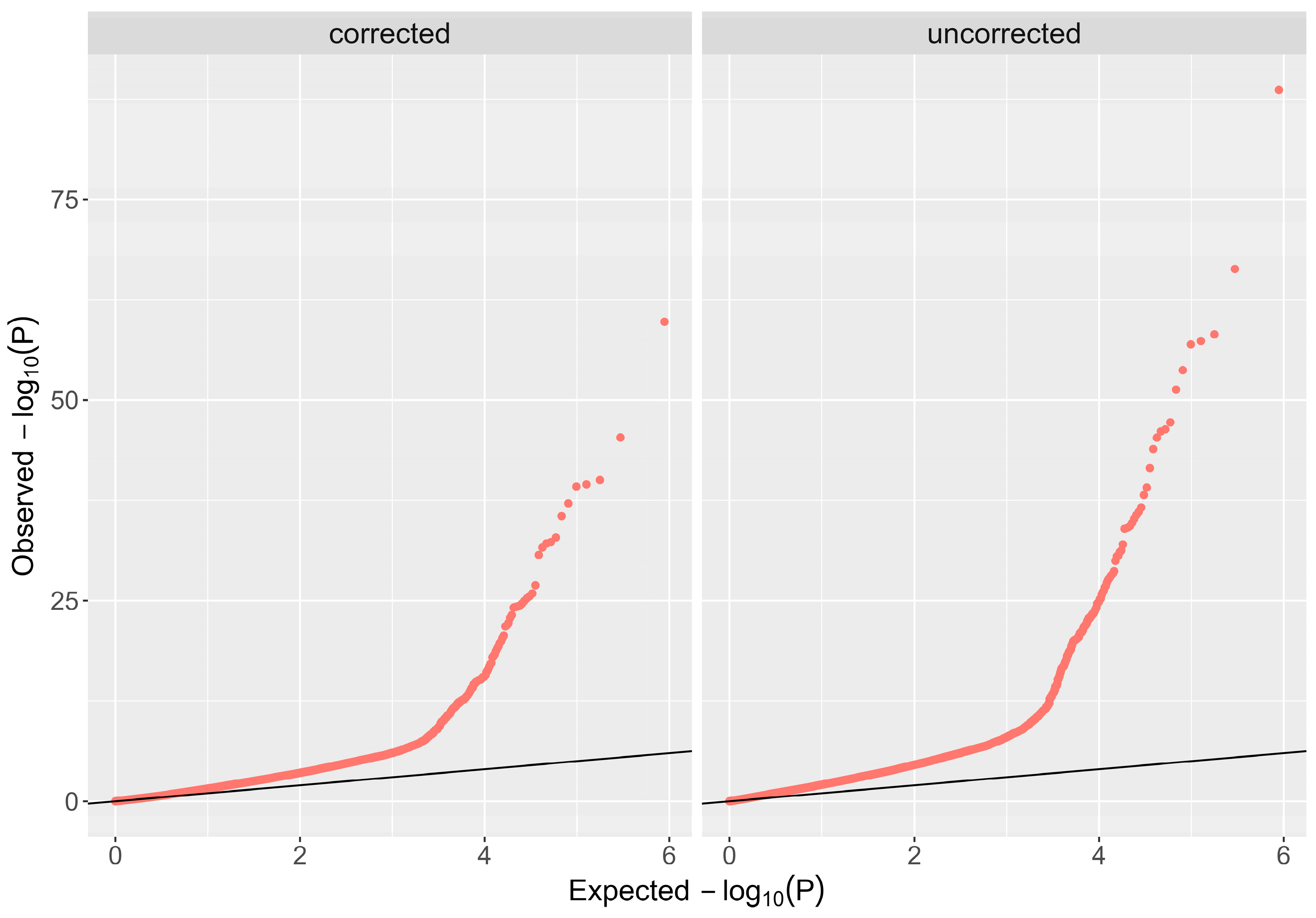
4.3. Statistical Analysis
4.4. Selection of Loci
5. Conclusions
Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: A systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023, 10, e585–e599. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global burden of sickle cell anaemia in children under five, 2010–2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [PubMed]
- El-Hazmi, M.A.F.; Al-Hazmi, A.M.; Warsy, A.S. Sickle cell disease in Middle East Arab countries. Indian J. Med. Res. 2011, 134, 597–610. [Google Scholar] [CrossRef]
- Grosse, S.D.; Odame, I.; Atrash, H.K.; Amendah, D.D.; Piel, F.B.; Williams, T.N. Sickle cell disease in Africa: A neglected cause of early childhood mortality. Am. J. Prev. Med. 2011, 41 (Suppl. S4), S398–S405. [Google Scholar] [CrossRef]
- Bin Zuair, A.; Aldossari, S.; Alhumaidi, R.; Alrabiah, M.; Alshabanat, A. The Burden of Sickle Cell Disease in Saudi Arabia: A Single-Institution Large Retrospective Study. Int. J. Gen. Med. 2023, 16, 161–171. [Google Scholar] [CrossRef]
- Alotaibi, M.M. Sickle cell disease in Saudi Arabia: A challenge or not. J. Epidemiol. Glob. Health 2017, 7, 99–101. [Google Scholar] [CrossRef]
- Al-Ali, A.K.; Alsulaiman, A.; Alfarhan, M.; Safaya, S.; Vatte, C.B.; Albuali, W.M.; Qutub, H.O.; Alzahrani, A.J.; Milton, J.N.; Steinberg, M.H. Sickle cell disease in the Eastern Province of Saudi Arabia: Clinical and laboratory features. Am. J. Hematol. 2021, 96, E117–E121. [Google Scholar] [CrossRef]
- Jastaniah, W. Epidemiology of sickle cell disease in Saudi Arabia. Ann. Saudi Med. 2011, 31, 289–293. [Google Scholar] [CrossRef]
- Al-Suliman, A.; Elsarraf, N.A.; Baqishi, M.; Homrany, H.; Bousbiah, J.; Farouk, E. Patterns of mortality in adult sickle cell disease in the Al-Hasa region of Saudi Arabia. Ann. Saudi Med. 2006, 26, 487–488. [Google Scholar] [CrossRef]
- Inusa, B.P.D.; Hsu, L.L.; Kohli, N.; Patel, A.; Ominu-Evbota, K.; Anie, K.A.; Atoyebi, W. Sickle Cell Disease-Genetics, Pathophysiology, Clinical Presentation and Treatment. Int. J. Neonatal Screen 2019, 5, 20. [Google Scholar] [CrossRef]
- Kavanagh, P.L.; Fasipe, T.A.; Wun, T. Sickle Cell Disease: A Review. JAMA 2022, 328, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, J.K.; Estepp, J.H.; Weiss, M.J.; Rashkin, S.R. Genetic Variation and Sickle Cell Disease Severity: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2023, 6, e2337484. [Google Scholar] [CrossRef] [PubMed]
- Driss, A.; Asare, K.O.; Hibbert, J.M.; Gee, B.E.; Adamkiewicz, T.V.; Stiles, J.K. Sickle Cell Disease in the Post Genomic Era: A Monogenic Disease with a Polygenic Phenotype. Genom. Insights 2009, 2009, 23–48. [Google Scholar]
- Chaturvedi, S.; Bhatnagar, P.; Bean, C.J.; Steinberg, M.H.; Milton, J.N.; Casella, J.F.; Barron-Casella, E.; Arking, D.E.; DeBaun, M.R. Genome-wide association study to identify variants associated with acute severe vaso-occlusive pain in sickle cell anemia. Blood 2017, 130, 686–688. [Google Scholar] [CrossRef]
- Griffin, P.J.; Sebastiani, P.; Edward, H.; Baldwin, C.T.; Gladwin, M.T.; Gordeuk, V.R.; Chui, D.H.; Steinberg, M.H. The genetics of hemoglobin A 2 regulation in sickle cell anemia. Am. J. Hematol. 2014, 89, 1019–1023. [Google Scholar] [CrossRef]
- Mtatiro, S.N.; Singh, T.; Rooks, H.; Mgaya, J.; Mariki, H.; Soka, D.; Mmbando, B.; Msaki, E.; Kolder, I.; Thein, S.L.; et al. Genome Wide Association Study of Fetal Hemoglobin in Sickle Cell Anemia in Tanzania. PLoS ONE 2014, 9, e111464. [Google Scholar] [CrossRef]
- Milton, J.N.; Sebastiani, P.; Solovieff, N.; Hartley, S.W.; Bhatnagar, P.; Arking, D.E.; Dworkis, D.A.; Casella, J.F.; Barron-Casella, E.; Bean, C.J.; et al. A Genome-Wide Association Study of Total Bilirubin and Cholelithiasis Risk in Sickle Cell Anemia. PLoS ONE 2012, 7, e34741. [Google Scholar] [CrossRef]
- Solovieff, N.; Milton, J.N.; Hartley, S.W.; Sherva, R.; Sebastiani, P.; Dworkis, D.A.; Klings, E.S.; Farrer, L.A.; Garrett, M.E.; Ashley-Koch, A.; et al. Fetal hemoglobin in sickle cell anemia: Genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood 2010, 115, 1815–1822. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Frohlich, D.M.; Howard, T.A.; Schultz, W.H.; Driscoll, C.; Nagasubramanian, R.; Mortier, N.A.; Kimble, A.C.; Aygun, B.; Adams, R.J.; et al. Genetic predictors for stroke in children with sickle cell anemia. Blood 2011, 117, 6681–6684. [Google Scholar] [CrossRef]
- Ashley-Koch, A.E.; Okocha, E.C.; Garrett, M.E.; Soldano, K.; De Castro, L.M.; Jonassaint, J.C.; Orringer, E.P.; Eckman, J.R.; Telen, M.J. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br. J. Haematol. 2011, 155, 386–394. [Google Scholar] [CrossRef]
- Saraf, S.L.; Zhang, X.; Shah, B.; Kanias, T.; Gudehithlu, K.P.; Kittles, R.; Machado, R.F.; Arruda, J.A.; Gladwin, M.T.; Singh, A.K.; et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica 2015, 100, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Alsultan, A.; Aleem, A.; Ghabbour, H.; AlGahtani, F.H.; Al-Shehri, A.; Osman, M.E.; Kurban, K.; Alsultan, M.S.; Bahakim, H.; Al-Momen, A.M. Sickle Cell Disease Subphenotypes in Patients from Southwestern Province of Saudi Arabia. J. Pediatr. Hematol./Oncol. 2012, 34, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Pincez, T.; Ashley-Koch, A.E.; Lettre, G.; Telen, M.J. Genetic Modifiers of Sickle Cell Disease. Hematol. Oncol. Clin. N. Am. 2022, 36, 1097–1124. [Google Scholar] [CrossRef]
- Chang, A.K.; Ginter Summarell, C.C.; Birdie, P.T.; Sheehan, V.A. Genetic modifiers of severity in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 147–164. [Google Scholar] [CrossRef]
- Alshabeeb, M.A.; Alwadaani, D.; Al Qahtani, F.H.; Abohelaika, S.; Alzahrani, M.; Al Zayed, A.; Al Saeed, H.H.; Al Ajmi, H.; Alsomaie, B.; Rashid, M.; et al. Impact of Genetic Variations on Thromboembolic Risk in Saudis with Sickle Cell Disease. Genes 2023, 14, 1919. [Google Scholar] [CrossRef]
- Witte, J.S. Genome-wide association studies and beyond. Annu. Rev. Public Health 2010, 31, 9–20. [Google Scholar] [CrossRef]
- Bhatnagar, P.; Purvis, S.; Barron-Casella, E.; DeBaun, M.R.; Casella, J.F.; Arking, D.E.; Keefer, J.R. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J. Hum. Genet. 2011, 56, 316–323. [Google Scholar] [CrossRef]
- Olender, T.; Jones, T.E.M.; Bruford, E.; Lancet, D. A unified nomenclature for vertebrate olfactory receptors. BMC Evol. Biol. 2020, 20, 42. [Google Scholar] [CrossRef]
- Roberts, T.C.; Morris, K.V. Not so pseudo anymore: Pseudogenes as therapeutic targets. Pharmacogenomics 2013, 14, 2023–2034. [Google Scholar] [CrossRef]
- Bulger, M.; Bender, M.A.; van Doorninck, J.H.; Wertman, B.; Farrell, C.M.; Felsenfeld, G.; Groudine, M.; Hardison, R. Comparative structural and functional analysis of the olfactory receptor genes flanking the human and mouse beta-globin gene clusters. Proc. Natl. Acad. Sci. USA 2000, 97, 14560–14565. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Kathiresan, S.; Lin, J.P.; Tofler, G.H.; O’Donnell, C.J. Genome-wide association and linkage analyses of hemostatic factors and hematological phenotypes in the Framingham Heart Study. BMC Med. Genet. 2007, 8 (Suppl. S1), S12. [Google Scholar] [CrossRef]
- Feingold, E.A.; Penny, L.A.; Nienhuis, A.W.; Forget, B.G. An olfactory receptor gene is located in the extended human beta-globin gene cluster and is expressed in erythroid cells. Genomics 1999, 61, 15–23. [Google Scholar] [CrossRef]
- Hou, C.; Dale, R.; Dean, A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. USA 2010, 107, 3651–3656. [Google Scholar] [CrossRef] [PubMed]
- Hanchard, N.; Elzein, A.; Trafford, C.; Rockett, K.; Pinder, M.; Jallow, M.; Harding, R.; Kwiatkowski, D.; McKenzie, C. Classical sickle beta-globin haplotypes exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC Genet. 2007, 8, 52. [Google Scholar] [CrossRef]
- Menzel, S.; Garner, C.; Rooks, H.; Spector, T.D.; Thein, S.L. HbA2 levels in normal adults are influenced by two distinct genetic mechanisms. Br. J. Haematol. 2013, 160, 101–105. [Google Scholar] [CrossRef]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272. [Google Scholar] [CrossRef]
- Kanai, M.; Akiyama, M.; Takahashi, A.; Matoba, N.; Momozawa, Y.; Ikeda, M.; Iwata, N.; Ikegawa, S.; Hirata, M.; Matsuda, K.; et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018, 50, 390–400. [Google Scholar] [CrossRef]
- Van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L.; et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [CrossRef]
- Li, J.; Glessner, J.T.; Zhang, H.; Hou, C.; Wei, Z.; Bradfield, J.P.; Mentch, F.D.; Guo, Y.; Kim, C.; Xia, Q.; et al. GWAS of blood cell traits identifies novel associated loci and epistatic interactions in Caucasian and African-American children. Hum. Mol. Genet. 2013, 22, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Sales, R.R.; Nogueira, B.L.; Tosatti, J.A.G.; Gomes, K.B.; Luizon, M.R. Do Genetic Polymorphisms Affect Fetal Hemoglobin (HbF) Levels in Patients with Sickle Cell Anemia Treated with Hydroxyurea? A Systematic Review and Pathway Analysis. Front. Pharmacol. 2021, 12, 779497. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; Despotovic, J.M.; Mortier, N.A.; Flanagan, J.M.; He, J.; Smeltzer, M.P.; Kimble, A.C.; Aygun, B.; Wu, S.; Howard, T.; et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood 2011, 118, 4985–4991. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31 (Suppl. S1), 177–183. [Google Scholar] [CrossRef]
- Ford, A.L.; Ragan, D.K.; Fellah, S.; Binkley, M.M.; Fields, M.E.; Guilliams, K.P.; An, H.; Jordan, L.C.; McKinstry, R.C.; Lee, J.M.; et al. Silent infarcts in sickle cell disease occur in the border zone region and are associated with low cerebral blood flow. Blood 2018, 132, 1714–1723. [Google Scholar] [CrossRef]
- Young, R.C.; Castro, O.; Baxter, R.P.; Dunn, R.; Armstrong, E.M.; Cook, F.J.; Sampson, C.C. The lung in sickle cell disease: A clinical overview of common vascular, infectious, and other problems. J. Natl. Med. Assoc. 1981, 73, 19–26. [Google Scholar]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 2011, 108, 3193–3198. [Google Scholar] [CrossRef]
- Zhan, Y.; Jiang, L.; Jin, X.; Ying, S.; Wu, Z.; Wang, L.; Yu, W.; Tong, J.; Zhang, L.; Lou, Y.; et al. Inhibiting RRM2 to enhance the anticancer activity of chemotherapy. Biomed. Pharmacother. 2021, 133, 110996. [Google Scholar] [CrossRef]
- McClarty, G.A.; Chan, A.K.; Engstrom, Y.; Wright, J.A.; Thelander, L. Elevated expression of M1 and M2 components and drug-induced posttranscriptional modulation of ribonucleotide reductase in a hydroxyurea-resistant mouse cell line. Biochemistry 1987, 26, 8004–8011. [Google Scholar] [CrossRef]
- Gohal, G.A.; Gosadi, I.M.; Cittana Iqbal, B.A.; Ghazwani, Y.H.; Daghriri, A.M.; Shugairi, A.A.; Daghriri, K.A.; Zurayyir, A.J.; Nemri, A.A.; Abdulhaq, M.A. Utilization of Hydroxyurea Among Patients Diagnosed with Sickle Cell Disease in Jazan, Saudi Arabia. Patient Prefer. Adherence 2022, 16, 3059–3067. [Google Scholar] [CrossRef]
- Chand, A.R.; Xu, H.; Wells, L.G.; Clair, B.; Neunert, C.; Spellman, A.E.; Clay, L.J.; Natrajan, K.; Kutlar, A. Are There True Non-Responders to Hydroxyurea in Sickle Cell Disease? A Multiparameter Analysis. Blood 2014, 124, 4073. [Google Scholar] [CrossRef]
- Chen, M.H.; Raffield, L.M.; Mousas, A.; Sakaue, S.; Huffman, J.E.; Moscati, A.; Trivedi, B.; Jiang, T.; Akbari, P.; Vuckovic, D.; et al. Trans-ethnic and Ancestry-Specific Blood-Cell Genetics in 746,667 Individuals from 5 Global Populations. Cell 2020, 182, 1198–1213.e14. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Cong, Y.; Wang, R.; Chen, Q.; Yan, C.; Gong, D. Structural insight into the human SID1 transmembrane family member 2 reveals its lipid hydrolytic activity. Nat. Commun. 2023, 14, 3568. [Google Scholar] [CrossRef] [PubMed]
- Silva-Rojas, R.; Laporte, J.; Böhm, J. STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941. [Google Scholar] [CrossRef]
- Nebor, D.; Durpes, M.C.; Mougenel, D.; Mukisi-Mukaza, M.; Elion, J.; Hardy-Dessources, M.D.; Romana, M. Association between Duffy antigen receptor for chemokines expression and levels of inflammation markers in sickle cell anemia patients. Clin. Immunol. 2010, 136, 116–122. [Google Scholar] [CrossRef]
- Drasar, E.R.; Menzel, S.; Fulford, T.; Thein, S.L. The effect of Duffy antigen receptor for chemokines on severity in sickle cell disease. Haematologica 2013, 98, e87–e89. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, L.; Xu, Y.; Song, D.; He, W.; Ji, X.; Shao, J. Hypermethylation of SCAND3 and Myo1g Gene Are Potential Diagnostic Biomarkers for Hepatocellular Carcinoma. Cancers 2020, 12, 2332. [Google Scholar] [CrossRef]
- Timoteo, V.J.; Chiang, K.M.; Yang, H.C.; Pan, W.H. Common and ethnic-specific genetic determinants of hemoglobin concentration between Taiwanese Han Chinese and European Whites: Findings from comparative two-stage genome-wide association studies. J. Nutr. Biochem. 2023, 111, 109126. [Google Scholar] [CrossRef]
- Naitza, S.; Porcu, E.; Steri, M.; Taub, D.D.; Mulas, A.; Xiao, X.; Strait, J.; Dei, M.; Lai, S.; Busonero, F.; et al. A genome-wide association scan on the levels of markers of inflammation in Sardinians reveals associations that underpin its complex regulation. PLoS Genet. 2012, 8, e1002480. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Dupuis, J.; Larson, M.G.; Lunetta, K.L.; Booth, S.L.; Govindaraju, D.R.; Kathiresan, S.; Keaney, J.F.; Keyes, M.J.; Lin, J.P.; et al. Genome-wide association with select biomarker traits in the Framingham Heart Study. BMC Med. Genet. 2007, 8 (Suppl. S1), S11. [Google Scholar] [CrossRef]
- Lin, Z.; Shi, J.L.; Chen, M.; Zheng, Z.M.; Li, M.Q.; Shao, J. CCL2: An important cytokine in normal and pathological pregnancies: A review. Front. Immunol. 2022, 13, 1053457. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Escobar, R.; Dona, I.; Montes, A.T.; Bartra, J.; Sanchez, N.P.; Laguna, J.; Cruz-Amaya, A.; de Santamaria, R.S.; Nuñez, R.; Salas, M.; et al. FCERIA Single Nucleotide Polymorphisms Associated with Nonsteroidal Anti-inflammatory Drug-induced Acute Urticaria/Angioedema. J. Allergy Clin. Immunol. 2022, 149 (Suppl. S2), AB61. [Google Scholar] [CrossRef]
- Doña, I.; Blanca-López, N.; Torres, M.J.; Gómez, F.; Fernández, J.; Zambonino, M.A.; Monteseirín, F.J.; Canto, G.; Blanca, M.; Cornejo-García, J.A. NSAID-induced urticaria/angioedema does not evolve into chronic urticaria: A 12-year follow-up study. Allergy 2014, 69, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Scheunemann, L.P.; Ataga, K.I. Delayed hemolytic transfusion reaction in sickle cell disease. Am. J. Med. Sci. 2010, 339, 266–269. [Google Scholar] [CrossRef]
- Kuriri, F.A.; Ahmed, A.; Alanazi, F.; Alhumud, F.; Ageeli Hakami, M.; Atiatalla Babiker Ahmed, O. Red Blood Cell Alloimmunization and Autoimmunization in Blood Transfusion-Dependent Sickle Cell Disease and β-Thalassemia Patients in Al-Ahsa Region, Saudi Arabia. Anemia 2023, 2023, 3239960. [Google Scholar] [CrossRef]
- Suhre, K. Genetic associations with ratios between protein levels detect new pQTLs and reveal protein-protein interactions. Cell Genom. 2024, 4, 100506. [Google Scholar] [CrossRef]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429.e19. [Google Scholar] [CrossRef]
- López-López, S.; Romero de Ávila, M.J.; Hernández de León, N.C.; Ruiz-Marcos, F.; Baladrón, V.; Nueda, M.L.; Laborda, J.; García-Ramírez, J.J.; Monsalve, E.M.; Díaz-Guerra, M.J. NOTCH4 Exhibits Anti-Inflammatory Activity in Activated Macrophages by Interfering with Interferon-γ and TLR4 Signaling. Front. Immunol. 2021, 12, 734966. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Rivera, N.V.; Ronninger, M.; Shchetynsky, K.; Franke, A.; Nöthen, M.M.; Müller-Quernheim, J.; Schreiber, S.; Adrianto, I.; Karakaya, B.; van Moorsel, C.H.; et al. High-Density Genetic Mapping Identifies New Susceptibility Variants in Sarcoidosis Phenotypes and Shows Genomic-driven Phenotypic Differences. Am. J. Respir. Crit. Care Med. 2016, 193, 1008–1022. [Google Scholar] [CrossRef]
- El Sharu, H.; Zweigle, J.; Jamil, M.; Singh, S.; Cowles, S.; Liles, D.K. Outcomes of Sarcoidosis on Patients with Sickle Cell Disease—A Review of the National Inpatient Database 2016–2020. Blood 2023, 142 (Suppl. S1), 1135. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- RegulomeDB Search—RegulomeDB. Available online: https://regulomedb.org/regulome-search (accessed on 2 September 2024).
- HaploReg v4.2. Available online: https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php (accessed on 2 September 2024).
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- Wong, K.; Lai, W.K.; Jackson, D.E. HLA Class II regulation of immune response in sickle cell disease patients: Susceptibility to red blood cell alloimmunization (systematic review and meta-analysis). Vox Sang. 2022, 117, 1251–1261. [Google Scholar] [CrossRef]
- Tamouza, R.; Neonato, M.G.; Busson, M.; Marzais, F.; Girot, R.; Labie, D.; Elion, J.; Charron, D. Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum. Immunol. 2002, 63, 194–199. [Google Scholar] [CrossRef]
- Mahdi, N.; Al-Ola, K.; Al-Subaie, A.M.; Ali, M.E.; Al-Irhayim, Z.; Al-Irhayim, A.Q.; Almawi, W.Y. HLA class II haplotypes distinctly associated with vaso-occlusion in children with sickle cell disease. Clin. Vaccine Immunol. 2008, 15, 729–731. [Google Scholar] [CrossRef]
- Martins, J.O.; Pagani, F.; Dezan, M.R.; Oliveira, V.B.; Conrado, M.; Ziza, K.C.; Gualandro, S.F.; Langui, D.M.; Bordin, J.O.; Rocha, V.; et al. Impact of HLA-G +3142C>G on the development of antibodies to blood group systems other than the Rh and Kell among sensitized patients with sickle cell disease. Transfus. Apher. Sci. 2022, 61, 103447. [Google Scholar] [CrossRef]
- Nagata, Y.; Suzuki, R. FcεRI: A Master Regulator of Mast Cell Functions. Cells 2022, 11, 622. [Google Scholar] [CrossRef]
- Himadewi, P.; Wang, X.Q.D.; Feng, F.; Gore, H.; Liu, Y.; Yu, L.; Kurita, R.; Nakamura, Y.; Pfeifer, G.P.; Liu, J.; et al. 3′HS1 CTCF binding site in human β-globin locus regulates fetal hemoglobin expression. Elife 2021, 10, e70557. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Bagu, E.T.; Patten, S.A.; Molidperee, S.; Parent, S.; Barchi, S.; Villemure, I.; Tremblay, A.; Moldovan, F. Differential Regulation of POC5 by ERα in Human Normal and Scoliotic Cells. Genes 2023, 14, 1111. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.Y.; Wang, L.; Song, Y.Y.; Gu, J.; Hu, X.; Yuan, C.; Yang, M.; Pei, W.J.; Zhang, Y.; Gao, J.L. Sidt2 is a key protein in the autophagy-lysosomal degradation pathway and is essential for the maintenance of kidney structure and filtration function. Cell Death Dis. 2021, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Donkor, E.S.; Sey, F.; Dzudzor, B.; Dankwah, G.B.; Otu, K.H.; Doku, A.; Dale, C.A.; Ekem, I. Levels of Soluble Endothelium Adhesion Molecules and Complications among Sickle Cell Disease Patients in Ghana. Diseases 2018, 6, 29. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Cases (n = 350) | Controls (n = 202) |
|---|---|---|
| Age (mean ± SD) | 32.7 ± 10.2 | 29.4 ± 8.4 |
| Male/female | 195 (56%)/155 (44%) | 103 (51%)/99 (49%) |
| CHR | Gene | SNP | Variant Type | MAF (Cases) | MAF (Controls) | p-Value |
|---|---|---|---|---|---|---|
| 1 | ACKR1 | rs12075 (A>G) | Missense | 0.010 | 0.217 | 9.35 × 10−13 |
| rs12074934 (T>G) | Intergenic | 0.487 | 0.218 | 1.71 × 10−9 | ||
| rs863002 (C>T) | Regulatory (CTCF site) | 0.010 | 0.176 | 4.45 × 10−11 | ||
| 6 | AGER | rs1800684 (T>A) | Upstream | 0.017 | 0.122 | 2.44 × 10−8 |
| 1 | FCER1A | rs2494250 (C>G) | Intron (500 B downstream) | 0.011 | 0.174 | 3.31 × 10−11 |
| 11 | HBBP1, HBD | rs2071348 (T>G) | Intron | 0.467 | 0.164 | 6.77 × 10−12 |
| 11 | HBE1, HBG2 | rs2213170 (G>A) | Intron | 0.440 | 0.015 | 1.78 × 10−11 |
| rs7130110 (C>G) | Regulatory | 0.466 | 0.179 | 9.21 × 10−11 | ||
| rs2213169 (G>A) | Intron | 0.437 | 0.015 | 2.17 × 10−11 | ||
| 11 | HBG2 | rs2236794 (C>T) | Downstream | 0.070 | 0.639 | 4.68 × 10−46 |
| 6 | HLA-A | rs2844806 (T>C) | Intergenic | 0.330 | 0.536 | 5.92 × 10−9 |
| HLA-G | rs2524035 (G>A) | Intron | 0.095 | 0.239 | 2.85 × 10−8 | |
| HLA-DRB1/HLA-DQB1 | rs3135006 (C>T) | Intergenic | 0.082 | 0.294 | 8.82 × 10−14 | |
| rs2395522 (T>A) | Intergenic | 0.409 | 0.617 | 8.00 × 10−9 | ||
| 11 | MMP26 | rs6578521 (G>A) | Intron | 0.069 | 0.510 | 1.38 × 10−33 |
| rs11822851(A>G) | Intron | 0.466 | 0.141 | 2.06 × 10−13 | ||
| 1 | MPTX1/CADM3-AS1 | rs3845624 (C>A) | Intergenic | 0.018 | 0.233 | 8.35 × 10−15 |
| 6 | NOTCH4 | rs3132946 (G>A) | Intron | 0.016 | 0.143 | 1.02 × 10−9 |
| rs3132940 (G>T) | Intron | 0.016 | 0.140 | 1.45 × 10−9 | ||
| rs3096702 (G>A) | Upstream | 0.077 | 0.222 | 7.92 × 10−9 | ||
| rs9267898 (C>T) | Intergenic | 0.063 | 0.209 | 1.65 × 10−9 | ||
| 1 | OR10J8P/OR10J9P | rs6687840 (T>C) | Intergenic | 0.182 | 0.453 | 2.31 × 10−15 |
| rs4446959 (C>T) | Intergenic | 0.180 | 0.459 | 3.17 × 10−16 | ||
| 11 | OR51B5 | rs147062602 (CAGCCCCAG9GTCTGTGG>ins) | Frameshift | 0.351 | 0.018 | 5.75 × 10−10 |
| rs10838058 (A>G) | Intron | 0.097 | 0.374 | 1.66 x10−18 | ||
| rs10837853 (G>A) | Intron | 0.306 | 0.611 | 3.82 × 10−16 | ||
| rs78253695 (GTC>del) | Intron | 0.114 | 0.342 | 6.78 × 10−14 | ||
| rs180750244 (G>A) | Intron | 0.017 | 0.153 | 3.02 × 10−10 | ||
| 11 | OR51S1 | rs12361955 (A>G) | Missense | 0.077 | 0.425 | 2.09 × 10−25 |
| 11 | OR51V1 | rs7933549 (G>A) | Missense | 0.484 | 0.015 | 1.95 × 10−12 |
| 11 | OR52A1 | rs112098990 (C>del) | Frameshift | 0.029 | 0.308 | 9.87 × 10−20 |
| 11 | OR52A5 | rs2472530 (A>G) | Missense | 0.453 | 0.150 | 6.73 × 10−12 |
| 5 | POC5 | rs2307111 (C>T) | Missense | 0.397 | 0.592 | 4.47 × 10−8 |
| 11 | RRM1 | rs55945048 (T>C) | Downstream | 0.355 | 0.101 | 1.61 × 10−9 |
| 6 | SCAND3 | rs450630 (A>G) | Missense | 0.357 | 0.552 | 4.71 × 10−8 |
| 11 | SIDT2 | rs10535646 (TGC>del) | Upstream | 0.296 | 0.497 | 9.81 × 10−9 |
| 11 | STIM1 | rs10767695(A>G) | Intron-NMD | 0.597 | 0.300 | 4.23 × 10−11 |
| rs7120828 (C>T) | Intron-NMD | 0.401 | 0.161 | 2.47 × 10−8 | ||
| rs4243966 (T>C) | Intron-NMD | 0.044 | 0.223 | 1.04 × 10−12 | ||
| 11 | TRIM5 | rs10838525 (C>T) | Missense | 0.050 | 0.267 | 2.26 × 10−15 |
| rs11038628 (C>T) | Missense | 0.330 | 0.074 | 7.03 × 10−10 | ||
| rs12786650 (T>C) | Intron | 0.078 | 0.337 | 8.82 × 10−18 | ||
| rs57956987 (T>C) | Intron | 0.030 | 0.165 | 4.84 × 10−10 | ||
| 11 | TRIM6 | rs3740999 (A>C) | Splice donor | 0.075 | 0.283 | 1.45 × 10−13 |
| rs11038294 (C>T) | Intron | 0.107 | 0.282 | 4.55 × 10−10 | ||
| rs12272467 (A>G) | Regulatory (TF site) | 0.600 | 0.255 | 1.76 × 10−14 | ||
| 11 | TRIM22 | rs67573252 (T>G) | Intron-NMD | 0.050 | 0.228 | 2.54 × 10−12 |
| 11 | TRIM 34 | rs2342380 (A>G) | Intron | 0.136 | 0.345 | 1 × 10−11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghubayshi, A.; Wijesinghe, D.; Alwadaani, D.; Algahtani, F.H.; Abohelaika, S.; Alzahrani, M.; Al Saeed, H.H.; Al Zayed, A.; Alshammari, S.; Alhendi, Y.; et al. Unraveling the Complex Genomic Interplay of Sickle Cell Disease Among the Saudi Population: A Case-Control GWAS Analysis. Int. J. Mol. Sci. 2025, 26, 2817. https://doi.org/10.3390/ijms26062817
Alghubayshi A, Wijesinghe D, Alwadaani D, Algahtani FH, Abohelaika S, Alzahrani M, Al Saeed HH, Al Zayed A, Alshammari S, Alhendi Y, et al. Unraveling the Complex Genomic Interplay of Sickle Cell Disease Among the Saudi Population: A Case-Control GWAS Analysis. International Journal of Molecular Sciences. 2025; 26(6):2817. https://doi.org/10.3390/ijms26062817
Chicago/Turabian StyleAlghubayshi, Ali, Dayanjan Wijesinghe, Deemah Alwadaani, Farjah H. Algahtani, Salah Abohelaika, Mohsen Alzahrani, Hussain H. Al Saeed, Abdullah Al Zayed, Suad Alshammari, Yaseen Alhendi, and et al. 2025. "Unraveling the Complex Genomic Interplay of Sickle Cell Disease Among the Saudi Population: A Case-Control GWAS Analysis" International Journal of Molecular Sciences 26, no. 6: 2817. https://doi.org/10.3390/ijms26062817
APA StyleAlghubayshi, A., Wijesinghe, D., Alwadaani, D., Algahtani, F. H., Abohelaika, S., Alzahrani, M., Al Saeed, H. H., Al Zayed, A., Alshammari, S., Alhendi, Y., Alsomaie, B., Alsaleh, A., & Alshabeeb, M. A. (2025). Unraveling the Complex Genomic Interplay of Sickle Cell Disease Among the Saudi Population: A Case-Control GWAS Analysis. International Journal of Molecular Sciences, 26(6), 2817. https://doi.org/10.3390/ijms26062817