
Unraveling the Mystery of Insulin Resistance: From Principle Mechanistic Insights and Consequences to Therapeutic Interventions

 ,
,  ,
,  , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Definition and Clinical Relevance
3. Risk and Contributing Factors
4. Global Epidemiology of IR
5. Prevalence of IR in Specific Populations
6. Molecular Mechanisms of Insulin Signaling
6.1. Insulin Receptor: Structure and Function
6.2. Intracellular Signaling Pathways
6.3. Role of Kinases and Phosphatases
6.4. AKT Pathway of Insulin Action
6.5. Interplay of PKC Isoforms
6.6. Alternate Insulin Signaling: GRB2-SOS-RAS-MAPK Cascade
6.7. Modulation of Insulin Action
6.8. Role of Lipid Phosphatases
6.9. Regulatory Roles of Grb, SOCS, Trb3 and IP7
6.10. Role of Phosphorylation Cascade Induced Activated Serine—Threonine Kinases
7. Cellular and Tissue Specificity of Insulin Action
7.1. Adipose Tissue
7.2. Skeletal Muscle
7.3. Hepatic Insulin Action
8. Insulin Resistance
8.1. Factors Contributing to Insulin Resistance
8.1.1. Obesity and Adipose Tissue Dysfunction
8.1.2. Inflammatory Mechanisms in Insulin Resistance
8.1.3. Role of Oxidative Stress
8.1.4. Mitochondrial Distress
8.1.5. Lysosomal Distress
8.1.6. Dysfunction of Endoplasmic Reticulum
8.1.7. Genetic Factors in Insulin Resistance
8.1.8. Lifestyle and Nutritional Factors in Insulin Resistance Risk
8.1.9. Relationship Between Age and Insulin Resistance
9. Tissue Specific Insulin Resistance
9.1. Role of Skeletal Muscle in Insulin Resistance
9.2. Role of Liver in Insulin Resistance
9.3. Role of Adipose Tissue in Insulin Resistance
9.4. Role of Myocardial Tissue in Insulin Resistance
9.5. Role of Other Cell Types and Tissues in Insulin Resistance
9.5.1. Hypothalamic Neurons
9.5.2. Pancreatic β Cells
9.5.3. Vascular Endothelial Cells
9.5.4. Macrophages
10. Consequences of Insulin Resistance
11. Therapeutic Modalities Targeting Insulin Resistance
11.1. Lifestyle Modifications
11.2. Pharmacologic Interventions
11.2.1. Currently Used Medications
11.2.2. Recent Drug Targets for Insulin Resistance
11.2.3. Future Insulin Resistance Drug Targets
11β—Hydroxysteroid Dehydrogenase (11β-HSD)
ACRP-30 (Adiponectin)
Fetuin-A
Visfatin/NAMPT (Nicotinamide Phosphoribosyl Transferase)
Metrnl
PEDF (Pigment Epithelium-Derived Factor)
Vaspin (Serpin A12)
G Protein-Coupled Estrogen Receptor (GPER)
Gene Therapy
12. Personalized Therapies for Insulin Resistance
13. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| T1DM | Type-1 diabetes mellitus |
| T2DM | Type-2 diabetes mellitus |
| IR | Insulin resistance |
| HOMA-IR | Homeostatic model assessment for insulin resistance |
| IGF | Insulin like growth factor |
| GRB | Growth factor receptor-bound protein |
| SHC | Src homology 2 domain-containing adapter protein |
| CETP | Cholesteryl ester transfer protein |
| PH | Pleckstrin homology |
| SH2 | Src homology -2 |
| IRS | Insulin receptor substrate |
| APS | Adapter protein with a PH and SH2 domain |
| Ras | Rat sarcoma virus oncogene |
| MAPK | Mitogen activated protein kinase |
| IRR | Insulin related receptor |
| IGF-1R | IGF-1 receptor |
| mMRA | messenger RNA |
| IR-A | Insulin receptor-A |
| KO | knockout |
| ERK | Extracellular signal-regulated kinase |
| DOK4 | Docking protein4 |
| GTP | Guanosine triphosphate |
| GAB | Grb2-associated binder |
| Cbl gene | Casitas B-lineage Lymphoma gene |
| CAP | catabolite activator protein |
| DOK | Docking protein |
| PI3K | Phosphatidylinositol 3-kinase |
| AKT | serine/threonine-protein kinase also known as protein kinase B |
| Pik3r1 | Phosphoinositide-3-Kinase Regulatory Subunit 1 |
| Src | Steroid receptor coactivator |
| Csk | C-Terminal Src Kinase |
| DOCK | Dedicator of cytokinesis protein |
| Crk | Proto-oncogene c-Crk protein |
| PKB | Protein kinase B |
| mTORC | Mammalian target of rapamycin complex 1 |
| DNAPK | DNA-dependent protein kinase |
| FOXO1 | Forkhead box protein O1 |
| TBC1D4 | TBC1D4 (TBC1 Domain Family Member 4) |
| PGC | Peroxisome proliferator-activated receptor-gamma coactivator |
| PDE3B | PDE3B phosphodiesterase 3B |
| c-AMP | Cyclic adenosine monophosphate |
| Cip 1 | Cdk-interacting protein-1 |
| WAF 1 | wildtype p53-activated fragment 1 |
| p27Kip1 | Cyclin-dependent kinase inhibitor 1B |
| IKK | IκB kinase |
| PKC | Protein Kinase C |
| nPKCs | Novel protein kinases |
| aPKCs | atypical protein kinases |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| SOS | Son of Sevenless (a set of genes) |
| MEK | Mitogen-activated protein kinase kinas |
| PTP1B | Protein tyrosine phosphatase 1B |
| LAR | leukocyte common antigen-related protein |
| PP2A | Protein Phosphatase 2A |
| PP2B | Protein Phosphatase 2B |
| S6K | S6 kinase p70 |
| PHLPP-1 | PH domain leucine-rich repeat protein phosphatase 1 |
| PTEN | Phosphatase and tensin homolog |
| SHIP | SH2 domain-containing inositol 5-phosphatases |
| SOCS | Suppressor of Cytokine Signaling |
| IP7 | Inositol pyrophosphate |
| IP6K1 | Inositol hexakisphosphate kinase 1 |
| Trb3 | Tribbles homolog 3 |
| JNK | c-Jun N-terminal kinase |
| Ser-307 | Serine residue at position 307 |
| DAG | Diacylglycerol |
| PKA | Protein kinase A |
| PPARγ | Peroxisome proliferator-activated receptor-γ |
| GLUT | Glucose transporter |
| GAP | GTPase-activating protein |
| RAC-1 | Ras-related C3 botulinum toxin substrate 1 |
| GYS | Glycogen synthase |
| GSK | Glycogen synthase kinase |
| IRTK | Insulin-Induced Receptor Tyrosine Kinase |
| HGP | Hepatic glucose production |
| G6PC1 | Glucose-6-phosphatase catalytic subunit 1 |
| PEPCK | Phosphoenolpyruvate carboxylase |
| SREBP-1 | Sterol regulatory element-binding protein |
| ACC1 | Acetyl-CoA carboxylase 1 |
| GPAT1 | Glycerol-3-phosphate acyltransferase |
| NAFLD | Non-alcoholic fatty liver disease |
| ROS | Reactive oxygen species |
| ER | Endoplasmic reticulum |
| NFκB | Nuclear factor kappa B |
| TLR4 | Toll-like receptor 4 |
| CerS | Ceramide synthase |
| FFA | Free fatty acids |
| MCP-1 | Monocyte chemoattractant protein-1 |
| TNF- α | Tumor necrosis factor alpha |
| IL | Interleukin |
| CLS | Crown like structure |
| JAK-STAT | Janus kinase signal transducer and activator of transcription |
| NOX | NADPH oxidase |
| GPX | Glutathione peroxidase |
| Mfn1 | Mitofusin1 |
| Drp1 | Dynamin-related protein 1 |
| VLDL | Very low-density lipoprotein |
| DAMP | Damage-associated molecular patterns |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| SERCA | Sarcoendoplasmic reticulum calcium transport ATPase |
| TFEB | Transcription factor EB |
| PC | Phosphatidylcholine |
| PERK | Protein kinase R like protein kinase |
| ATF | Activating transcription factor |
| IRE-1 | Inositol-requiring enzyme type 1 |
| XBP1 | X-box binding protein 1 |
| F25BS | Los Angeles insulin |
| F25BL | Chicago insulin |
| PTPN1 | Protein tyrosine phosphatase N1 |
| LDLR | Low density lipoprotein receptor |
| IGF1R | Insulin-like growth factor receptor-1 |
| AgRP | Agouti-related protein |
| POMC | Pro-opiomelanocortin |
| Ins1 | Insulin 1 gene |
| VEC | Vascular endothelial cell |
| VCAM | Vascular cell adhesion molecule |
| eNOS | endothelial nitric-oxide synthase |
| iNOS | Inducible nitric-oxide synthase |
| CVD | Cardiovascular disease |
| SLI | silent lacunar infarction |
| Ty-G | Triglyceride-glucose index |
| END | Early neurological degeneration |
| PCOS | Polycystic ovarian syndrome |
| INSR | Insulin receptor |
| NALP3 | Nucleotide-binding domain, leucine-rich repeat/pyrin domain-containing-3 |
References
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Freeman, A.M.; Acevedo, L.A.; Pennings, N. Insulin Resistance. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507839/ (accessed on 13 March 2025).
- Videira-Silva, A.; Albuquerque, C.; Fonseca, H. Acanthosis nigricans as a clinical marker of insulin resistance among overweight adolescents. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 99–103. [Google Scholar] [CrossRef]
- Buscemi, C.; Randazzo, C.; Barile, A.M.; Bo, S.; Ponzo, V.; Caldarella, R.; Malavazos, A.E.; Caruso, R.; Colombrita, P.; Lombardo, M.; et al. Factors associated with body weight gain and insulin-resistance: A longitudinal study. Nutr. Diabetes 2024, 14, 21. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Bousvarou, M.D.; Kostara, C.E.; Papakonstantinou, E.J.; Salamou, E.; Guzman, E. Insulin resistance and cardiovascular disease. J. Int. Med. Res. 2023, 51, 3000605231164548. [Google Scholar] [CrossRef]
- Taylor, R. Insulin resistance and type 2 diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Janssen, J.A.M.J.L. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef]
- Zhao, X.; An, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. The crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef]
- Heindel, J.J.; Lustig, R.H.; Howard, S.; Corkey, B.E. Obesogens: A unifying theory for the global rise in obesity. Int. J. Obes. 2024, 48, 449–460. [Google Scholar] [CrossRef]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Renal Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef]
- Lee, Y.S.; Olefsky, J. Chronic tissue inflammation and metabolic disease. Genes Dev. 2021, 35, 307–328. [Google Scholar] [CrossRef]
- Tyagi, A.; Pugazhenthi, S. Targeting Insulin Resistance to Treat Cognitive Dysfunction. Mol. Neurobiol. 2021, 58, 2672–2691. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Chathoth, S.; Ismail, M.H.; Alghamdi, H.M.; Zakaria, H.M.; Hassan, K.A.; Alshomimi, S.; Vatte, C.; Cyrus, C.; Alsaif, H.S.; Mostafa, A.; et al. Insulin resistance induced by de novo pathway-generated C16-ceramide is associated with type 2 diabetes in an obese population. Lipids Health Dis. 2022, 21, 24. [Google Scholar] [CrossRef]
- Le, T.K.C.; Dao, X.D.; Nguyen, D.V.; Luu, D.H.; Bui, T.M.H.; Le, T.H.; Nguyen, H.T.; Le, T.N.; Hosaka, T.; Nguyen, T.T.T. Insulin signaling and its application. Front. Endocrinol. 2023, 14, 1226655. [Google Scholar] [CrossRef]
- Kumar, S.; Senapati, S.; Bhattacharya, N.; Bhattacharya, A.; Maurya, S.K.; Husain, H.; Bhatti, J.S.; Pandey, A.K. Mechanism and recent updates on insulin-related disorders. World J. Clin. Cases 2023, 11, 5840–5856. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef] [PubMed]
- Caricilli, A.M.; Saad, M.J. The role of gut microbiota on insulin resistance. Nutrients 2013, 5, 829–851. [Google Scholar] [CrossRef]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Maleki, M.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. Pathophysiology of Physical Inactivity-Dependent Insulin Resistance: A Theoretical Mechanistic Review Emphasizing Clinical Evidence. J. Diabetes Res. 2021, 2021, 7796727. [Google Scholar] [CrossRef]
- Parker, K.M.; Tucker, L.A.; Bailey, B.W.; Davidson, L.E. Relationship between Sitting Time and Insulin Resistance in 6931 U.S. Adults: The Mediating Role of Abdominal Adiposity. J. Diabetes Res. 2023, 2023, 5015572. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.O.; Conde, S.V. Impact of Diet Composition on Insulin Resistance. Nutrients 2022, 14, 3716. [Google Scholar] [CrossRef]
- Zyoud, S.H. Mapping the landscape of research on insulin resistance: A visualization analysis of randomized clinical trials. J. Health Popul. Nutr. 2024, 43, 6. [Google Scholar] [CrossRef]
- Abildinova, G.Z.; Benberin, V.V.; Vochshenkova, T.A.; Afshar, A.; Mussin, N.M.; Kaliyev, A.A.; Zhussupova, Z.; Tamadon, A. Global trends and collaborative networks in gut microbiota-insulin resistance research: A comprehensive bibliometric analysis (2000–2024). Front. Med. 2024, 11, 1452227. [Google Scholar] [CrossRef]
- Magliano, D.J.; Boyko, E.J. IDF Diabetes Atlas 10th Edition Scientific Committee IDFDIABETESATLAS [Internet], 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK581934/ (accessed on 14 February 2025).
- Goh, L.P.W.; Sani, S.A.; Sabullah, M.K.; Gansau, J.A. The Prevalence of Insulin Resistance in Malaysia and Indonesia: An Updated Systematic Review and Meta-Analysis. Medicina 2022, 58, 826. [Google Scholar] [CrossRef]
- Ferreira, J.R.S.; Zandonade, E.; de Paula Alves Bezerra, O.M.; Salaroli, L.B. Insulin resistance by the triglyceride-glucose index in a rural Brazilian population. Arch. Endocrinol. Metab. 2022, 66, 848–855. [Google Scholar] [CrossRef]
- Parcha, V.; Heindl, B.; Kalra, R.; Li, P.; Gower, B.; Arora, G.; Arora, P. Insulin Resistance and Cardiometabolic Risk Profile Among Nondiabetic American Young Adults: Insights From NHANES. J. Clin. Endocrinol. Metab. 2022, 107, e25–e37. [Google Scholar] [CrossRef]
- El-Kebbi, I.M.; Bidikian, N.H.; Hneiny, L.; Nasrallah, M.P. Epidemiology of type 2 diabetes in the Middle East and North Africa: Challenges and call for action. World J. Diabetes 2021, 12, 1401–1425. [Google Scholar] [CrossRef] [PubMed]
- Elshebiny, A.; Alrashed, A.; Albuwaydi, Z.; Aljassim, S.; Alhammad, F.; Alhajji, R. An Assessment of the 10-Year Risk of Developing Type 2 Diabetes Among Saudi Adults Based on the Finnish Diabetes Risk Score. Cureus 2022, 14, e32034. [Google Scholar] [CrossRef]
- Bukhari, H.M. Sociodemographic Variables Associated with the Prevalence of Insulin Resistance Using a Non-Invasive Score System Among Adults in the Makkah Region of Saudi Arabia. Curr. Res. Nutr. Food Sci. 2023, 11, 685–695. [Google Scholar] [CrossRef]
- Fahed, M.; Abou Jaoudeh, M.G.; Merhi, S.; Mosleh, J.M.B.; Ghadieh, R.; Al Hayek, S.; El Hayek Fares, J.E. Evaluation of risk factors for insulin resistance: A cross sectional study among employees at a private university in Lebanon. BMC Endocr. Disord. 2020, 20, 85. [Google Scholar] [CrossRef]
- Elrayess, M.A.; Rizk, N.M.; Fadel, A.S.; Kerkadi, A. Prevalence and Predictors of Insulin Resistance in Non-Obese Healthy Young Females in Qatar. Int. J. Environ. Res. Public Health 2020, 17, 5088. [Google Scholar] [CrossRef]
- Raygor, V.; Abbasi, F.; Lazzeroni, L.C.; Kim, S.; Ingelsson, E.; Reaven, G.M.; Knowles, J.W. Impact of race/ethnicity on insulin resistance and hypertriglyceridaemia. Diabetes Vasc. Dis. Res. 2019, 16, 153–159. [Google Scholar] [CrossRef]
- Kanaya, A.M.; Herrington, D.; Vittinghoff, E.; Ewing, S.K.; Liu, K.; Blaha, M.J.; Dave, S.S.; Qureshi, F.; Kandula, N.R. Understanding the high prevalence of diabetes in U.S. south Asians compared with four racial/ethnic groups: The MASALA and MESA studies. Diabetes Care 2014, 37, 1621–1628. [Google Scholar] [CrossRef]
- Williams, D.R.; Mohammed, S.A.; Leavell, J.; Collins, C. Race, socioeconomic status, and health: Complexities, ongoing challenges, and research opportunities. Ann. N. Y. Acad. Sci. 2010, 1186, 69–101. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef]
- Wang, D.; Liu, G.; Meng, Y.; Chen, H.; Ye, Z.; Jing, J. The Configuration of GRB2 in Protein Interaction and Signal Transduction. Biomolecules 2024, 14, 259. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.C. Understanding insulin and its receptor from their three-dimensional structures. Mol. Metab. 2021, 52, 101255. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M. Structure and function of the insulin receptor-a personal perspective. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: Contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Mendoza, C.; Hanegan, C.; Sperry, A.; Vargas, L.; Case, T.; Bikman, B.; Mizrachi, D. Insulin receptor-inspired soluble insulin binder. Eur. J. Cell Biol. 2023, 102, 151293. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M. The insulin receptor substrate (IRS) proteins: At the intersection of metabolism and cancer. Cell Cycle 2011, 10, 1750–1756. [Google Scholar] [CrossRef]
- Nawaratne, R.; Gray, A.; Jørgensen, C.H.; Downes, C.P.; Siddle, K.; Sethi, J.K. Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: Role of serine 24 phosphorylation. Mol. Endocrinol. 2006, 20, 1838–1852. [Google Scholar] [CrossRef]
- Wagner, M.J.; Stacey, M.M.; Liu, B.A.; Pawson, T. Molecular mechanisms of SH2- and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a008987. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nakamura, K.; Tokita, R.; Teramoto, N.; Sugihara, H.; Kato, H.; Yamanouchi, K.; Minami, S. Disruption of insulin receptor substrate-2 impairs growth but not insulin function in rats. J. Biol. Chem. 2020, 295, 11914–11927. [Google Scholar] [CrossRef] [PubMed]
- Martínez Báez, A.; Ayala, G.; Pedroza-Saavedra, A.; González-Sánchez, H.M.; Chihu Amparan, L. Phosphorylation Codes in IRS-1 and IRS-2 Are Associated with the Activation/Inhibition of Insulin Canonical Signaling Pathways. Curr. Issues Mol. Biol. 2024, 46, 634–649. [Google Scholar] [CrossRef]
- Guijarro, L.G.; Justo Bermejo, F.J.; Boaru, D.L.; De Castro-Martinez, P.; De Leon-Oliva, D.; Fraile-Martínez, O.; Garcia-Montero, C.; Alvarez-Mon, M.; Toledo-Lobo, M.D.V.; Ortega, M.A. Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes? Cancers 2023, 15, 4651. [Google Scholar] [CrossRef]
- Nagao, H.; Cai, W.; Wewer Albrechtsen, N.J.; Steger, M.; Batista, T.M.; Pan, H.; Dreyfuss, J.M.; Mann, M.; Kahn, C.R. Distinct signaling by insulin and IGF-1 receptors and their extra- and intracellular domains. Proc. Natl. Acad. Sci. USA 2021, 118, e2019474118. [Google Scholar] [CrossRef] [PubMed]
- Tsay, A.; Wang, J.C. The Role of PIK3R1 in Metabolic Function and Insulin Sensitivity. Int. J. Mol. Sci. 2023, 24, 12665. [Google Scholar] [CrossRef]
- Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef]
- Fox, M.; Mott, H.R.; Owen, D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem. Soc. Trans. 2020, 48, 1397–1417. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Selinger, E.S.; Ballou, L.M.; Lin, R.Z. Ablation of PI3K p110-α prevents high-fat diet-induced liver steatosis. Diabetes 2011, 60, 1483–1492. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Ueki, K.; Engelman, J.A.; Kahn, R.C.; Cantley, L.C. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol. Cell. Biol. 2005, 25, 1596–1607. [Google Scholar] [CrossRef]
- Ueki, K.; Fruman, D.A.; Brachmann, S.M.; Tseng, Y.H.; Cantley, L.C.; Kahn, C.R. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol. Cell. Biol. 2002, 22, 965–977. [Google Scholar] [CrossRef]
- Hanke, S.; Mann, M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol. Cell. Proteom. 2009, 8, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Saini, V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J. Diabetes 2010, 1, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef]
- Somale, D.; Di Nardo, G.; di Blasio, L.; Puliafito, A.; Vara-Messler, M.; Chiaverina, G.; Palmiero, M.; Monica, V.; Gilardi, G.; Primo, L.; et al. Activation of RSK by phosphomimetic substitution in the activation loop is prevented by structural constraints. Sci. Rep. 2020, 10, 591. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, S.; Wang, P.; Ouyang, J.; Zhou, N.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) drives chronic kidney disease progression in male mice. Nat. Commun. 2023, 14, 1334. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Huang, J.; Düvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef]
- Marchelek-Mysliwiec, M.; Nalewajska, M.; Turoń-Skrzypińska, A.; Kotrych, K.; Dziedziejko, V.; Sulikowski, T.; Pawlik, A. The Role of Forkhead Box O in Pathogenesis and Therapy of Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 11611. [Google Scholar] [CrossRef]
- Farhan, M.; Silva, M.; Xingan, X.; Huang, Y.; Zheng, W. Role of FOXO Transcription Factors in Cancer Metabolism and Angiogenesis. Cells 2020, 9, 1586. [Google Scholar] [CrossRef]
- Cartee, G.D. Roles of TBC1D1 and TBC1D4 in insulin- and exercise-stimulated glucose transport of skeletal muscle. Diabetologia 2015, 58, 19–30. [Google Scholar] [CrossRef]
- Aslam, M.; Ladilov, Y. Emerging Role of cAMP/AMPK Signaling. Cells 2022, 11, 308. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118, Erratum in Proc. Natl. Acad. Sci. USA 2021, 118, e2101572118.. [Google Scholar] [CrossRef]
- Kreis, N.N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef]
- Jeong, S.J.; Dasgupta, A.; Jung, K.J.; Um, J.H.; Burke, A.; Park, H.U.; Brady, J.N. PI3K/AKT inhibition induces caspase-dependent apoptosis in HTLV-1-transformed cells. Virology 2008, 370, 264–272. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell. 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, U.; Kishimoto, A.; Nishizuka, Y. The protein kinase C family: Heterogeneity and its implications. Annu. Rev. Biochem. 1989, 58, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Farese, R.V.; Sajan, M.P. Metabolic functions of atypical protein kinase C: “good” and “bad” as defined by nutritional status. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E385–E394. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Sajan, M.P.; Yang, H.; Li, P.; Mastorides, S.; Gower, W.R., Jr.; Nimal, S.; Choi, C.S.; Kim, S.; Shulman, G.I.; et al. Muscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J. Clin. Investig. 2007, 117, 2289–2301, Erratum in J. Clin. Investig. 2007, 117, 3141. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Hauser, C.; Kahn, B.B.; Haj, F.G.; Neel, B.G.; Tonks, N.K.; Tiganis, T. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 2005, 25, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.G.; Cho, Y.R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Arora, D.K.; Machhadieh, B.; Matti, A.; Wadzinski, B.E.; Ramanadham, S.; Kowluru, A. High glucose exposure promotes activation of protein phosphatase 2A in rodent islets and INS-1 832/13 β-cells by increasing the posttranslational carboxylmethylation of its catalytic subunit. Endocrinol. 2014, 155, 380–391. [Google Scholar] [CrossRef]
- Nygren, P.J.; Scott, J.D. Regulation of the phosphatase PP2B by protein-protein interactions. Biochem. Soc. Trans. 2016, 44, 1313–1319. [Google Scholar] [CrossRef]
- Cha, J.H.; Jeong, Y.; Oh, A.R.; Lee, S.B.; Hong, S.S.; Kim, K. Emerging roles of PHLPP phosphatases in metabolism. BMB Rep. 2021, 54, 451–457. [Google Scholar] [CrossRef]
- Baffi, T.R.; Cohen-Katsenelson, K.; Newton, A.C. PHLPPing the Script: Emerging Roles of PHLPP Phosphatases in Cell Signaling. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 723–743. [Google Scholar] [CrossRef]
- Metcalfe, L.K.; Smith, G.C.; Turner, N. Defining lipid mediators of insulin resistance-controversies and challenges. J. Mol. Endocrinol. 2018, 62, R65–R82. [Google Scholar] [CrossRef] [PubMed]
- Tariq, K.; Luikart, B.W. Striking a balance: PIP2 and PIP3 signaling in neuronal health and disease. Explor. Neuroprot. Ther. 2021, 1, 86–100. [Google Scholar] [CrossRef]
- Lee, M.F.; Trotman, L.C. PTEN: Bridging Endocytosis and Signaling. Cold Spring Harb. Perspect. Med. 2020, 10, a036103. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Barber, T.M.; Van de Bunt, M.; Rudge, S.A.; Zhang, Q.; Lachlan, K.L.; Cooper, N.S.; Linden, H.; Levy, J.C.; Wakelam, M.J.; et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. New Engl. J. Med. 2012, 367, 1002–1011. [Google Scholar] [CrossRef]
- Peyrou, M.; Bourgoin, L.; Poher, A.L.; Altirriba, J.; Maeder, C.; Caillon, A.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; Foti, M. Hepatic PTEN deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J. Hepatol. 2015, 62, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Kotzampasi, D.M.; Premeti, K.; Papafotika, A.; Syropoulou, V.; Christoforidis, S.; Cournia, Z.; Leondaritis, G. The orchestrated signaling by PI3Kα and PTEN at the membrane interface. Comput. Struct. Biotechnol. J. 2022, 20, 5607–5621. [Google Scholar] [CrossRef]
- Bradshaw, W.J.; Kennedy, E.C.; Moreira, T.; Smith, L.A.; Chalk, R.; Katis, V.L.; Benesch, J.L.P.; Brennan, P.E.; Murphy, E.J.; Gileadi, O. Regulation of inositol 5-phosphatase activity by the C2 domain of SHIP1 and SHIP2. Structure 2024, 32, 453–466.e6. [Google Scholar] [CrossRef]
- Lehtonen, S. SHIPping out diabetes-Metformin, an old friend among new SHIP2 inhibitors. Acta Physiol. 2020, 228, e13349. [Google Scholar] [CrossRef]
- Kagawa, S.; Soeda, Y.; Ishihara, H.; Oya, T.; Sasahara, M.; Yaguchi, S.; Oshita, R.; Wada, T.; Tsuneki, H.; Sasaoka, T. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology 2008, 149, 642–650. [Google Scholar] [CrossRef]
- Holt, L.J.; Siddle, K. Grb10 and Grb14: Enigmatic regulators of insulin action--and more? Biochem. J. 2005, 388 Pt 2, 393–406. [Google Scholar] [CrossRef]
- Moorwood, K.; Smith, F.M.; Garfield, A.S.; Cowley, M.; Holt, L.J.; Daly, R.J.; Ward, A. Grb7, Grb10 and Grb14, encoding the growth factor receptor-bound 7 family of signalling adaptor proteins have overlapping functions in the regulation of fetal growth and post-natal glucose metabolism. BMC Biol. 2024, 22, 221. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Smith, F.M.; Holt, L.J.; Garfield, A.S.; Charalambous, M.; Koumanov, F.; Perry, M.; Bazzani, R.; Sheardown, S.A.; Hegarty, B.D.; Lyons, R.J.; et al. Mice with a disruption of the imprinted Grb10 gene exhibit altered body composition, glucose homeostasis, and insulin signaling during postnatal life. Mol. Cell. Biol. 2007, 27, 5871–5886. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Holt, L.J.; Lyons, R.J.; Ryan, A.S.; Beale, S.M.; Ward, A.; Cooney, G.J.; Daly, R.J. Dual ablation of Grb10 and Grb14 in mice reveals their combined role in regulation of insulin signaling and glucose homeostasis. Mol. Endocrinol. 2009, 23, 1406–1414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Villanueva-Hayes, C.; Millership, S.J. Imprinted Genes Impact Upon Beta Cell Function in the Current (and Potentially Next) Generation. Front. Endocrinol. 2021, 12, 660532. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 2004, 24, 5434–5446, Erratum in Mol. Cell. Biol. 2005, 25, 8762. [Google Scholar] [CrossRef]
- Sobah, M.L.; Liongue, C.; Ward, A.C. SOCS Proteins in Immunity, Inflammatory Diseases, and Immune-Related Cancer. Front. Med. 2021, 8, 727987. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, S.; Han, S.; Jin, K.; Yu, T.; Chen, H.; Zhou, X.; Tan, Z.; Zhang, G. SOCS2 Suppresses Inflammation and Apoptosis during NASH Progression through Limiting NF-κB Activation in Macrophages. Int. J. Biol. Sci. 2021, 17, 4165–4175. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef]
- Liew, C.W.; Bochenski, J.; Kawamori, D.; Hu, J.; Leech, C.A.; Wanic, K.; Malecki, M.; Warram, J.H.; Qi, L.; Krolewski, A.S.; et al. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse beta cells. J. Clin. Investig. 2010, 120, 2876–2888. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jeong, H.W.; Choi, R.H.; Koh, H.J. Obesity-induced TRB3 negatively regulates Brown adipose tissue function in mice. Biochem. Biophys. Res. Commun. 2021, 547, 29–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shears, S.B.; Wang, H. Metabolism and Functions of Inositol Pyrophosphates: Insights Gained from the Application of Synthetic Analogues. Molecules 2020, 25, 4515. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, S.; Kim, M.G.; Ahn, H.; Kim, S. Inositol Pyrophosphates: Signaling Molecules with Pleiotropic Actions in Mammals. Molecules 2020, 25, 2208. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdul-Hay, S.O.; Kang, D.; McBride, M.; Li, L.; Zhao, J.; Leissring, M.A. Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. PLoS ONE 2011, 6, e20818. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rajan, M.R.; Fagerholm, S.; Jönsson, C.; Kjølhede, P.; Turkina, M.V.; Strålfors, P. Phosphorylation of IRS1 at serine 307 in response to insulin in human adipocytes is not likely to be catalyzed by p70 ribosomal S6 kinase. PLoS ONE 2013, 8, e59725. [Google Scholar] [CrossRef]
- Copps, K.D.; Hancer, N.J.; Opare-Ado, L.; Qiu, W.; Walsh, C.; White, M.F. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010, 11, 84–92. [Google Scholar] [CrossRef]
- Edick, A.M.; Auclair, O.; Burgos, S.A. Role of Grb10 in mTORC1-dependent regulation of insulin signaling and action in human skeletal muscle cells. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E173–E183. [Google Scholar] [CrossRef]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef]
- Santoro, A.; McGraw, T.E.; Kahn, B.B. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab. 2021, 33, 748–757. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koh, H.E.; van Vliet, S.; Pietka, T.A.; Meyer, G.A.; Razani, B.; Laforest, R.; Gropler, R.J.; Mittendorfer, B. Subcutaneous Adipose Tissue Metabolic Function and Insulin Sensitivity in People With Obesity. Diabetes 2021, 70, 2225–2236. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, S.; Tan, J.; Zhang, H.; Hou, D.X.; He, J. Tissue-specific mechanisms of fat metabolism that focus on insulin actions. J. Adv. Res. 2023, 53, 187–198. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wasserman, D.H. Insulin, Muscle Glucose Uptake, and Hexokinase: Revisiting the Road Not Taken. Physiology 2022, 37, 115–127. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tan, S.X.; Ng, Y.; Burchfield, J.G.; Ramm, G.; Lambright, D.G.; Stöckli, J.; James, D.E. The Rab GTPase-activating protein TBC1D4/AS160 contains an atypical phosphotyrosine-binding domain that interacts with plasma membrane phospholipids to facilitate GLUT4 trafficking in adipocytes. Mol. Cell. Biol. 2012, 32, 4946–4959. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takenaka, N.; Araki, N.; Satoh, T. Involvement of the protein kinase Akt2 in insulin-stimulated Rac1 activation leading to glucose uptake in mouse skeletal muscle. PLoS ONE 2019, 14, e0212219. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Katz, A. The role of glycogen phosphorylase in glycogen biogenesis in skeletal muscle after exercise. Sports Med. Health Sci. 2022, 5, 29–33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agius, L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol. Aspects Med. 2015, 46, 34–45. [Google Scholar] [CrossRef]
- Uehara, K.; Santoleri, D.; Whitlock, A.E.G.; Titchenell, P.M. Insulin Regulation of Hepatic Lipid Homeostasis. Compr. Physiol. 2023, 13, 4785–4809. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jung, I.; Koo, D.J.; Lee, W.Y. Insulin Resistance, Non-Alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Clinical and Experimental Perspective. Diabetes Metab. J. 2024, 48, 327–339. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mann, V.; Sundaresan, A.; Shishodia, S. Overnutrition and Lipotoxicity: Impaired Efferocytosis and Chronic Inflammation as Precursors to Multifaceted Disease Pathogenesis. Biology 2024, 13, 241. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cheng, X.; Jiang, S.; Pan, B.; Xie, W.; Meng, J. Ectopic and visceral fat deposition in aging, obesity, and idiopathic pulmonary fibrosis: An interconnected role. Lipids Health Dis. 2023, 22, 201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Snel, M.; Jonker, J.T.; Schoones, J.; Lamb, H.; de Roos, A.; Pijl, H.; Smit, J.W.; Meinders, A.E.; Jazet, I.M. Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Int. J. Endocrinol. 2012, 2012, 983814. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jayasinghe, S.U.; Tankeu, A.T.; Amati, F. Reassessing the Role of Diacylglycerols in Insulin Resistance. Trends Endocrinol. Metab. 2019, 30, 618–635. [Google Scholar] [CrossRef]
- Xiao, K.; Liu, C.; Tu, Z.; Xu, Q.; Chen, S.; Zhang, Y.; Wang, X.; Zhang, J.; Hu, C.A.; Liu, Y. Activation of the NF-κB and MAPK Signaling Pathways Contributes to the Inflammatory Responses, but Not Cell Injury, in IPEC-1 Cells Challenged with Hydrogen Peroxide. Oxidative Med. Cell. Longev. 2020, 2020, 5803639. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Scarano, F.; Gliozzi, M.; Zito, M.C.; Guarnieri, L.; Carresi, C.; Macrì, R.; Nucera, S.; Scicchitano, M.; Bosco, F.; Ruga, S.; et al. Potential of Nutraceutical Supplementation in the Modulation of White and Brown Fat Tissues in Obesity-Associated Disorders: Role of Inflammatory Signalling. Int. J. Mol. Sci. 2021, 22, 3351. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, Z.; Li, G.; Tong, T.; Chen, J. Micheliolide suppresses LPS-induced neuroinflammatory responses. PLoS ONE 2017, 12, e0186592. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yi, Y.S. Caspase-11 non-canonical inflammasome: A critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunol. 2017, 152, 207–217. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin.Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- Kojta, I.; Chacińska, M.; Błachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adams, J.M., 2nd; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant, humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef]
- Kasumov, T.; Solomon, T.P.; Hwang, C.; Huang, H.; Haus, J.M.; Zhang, R.; Kirwan, J.P. Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity 2015, 23, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Razak Hady, H.; Błachnio-Zabielska, A.U.; Szczerbiński, Ł.; Zabielski, P.; Imierska, M.; Dadan, J.; Krętowski, A.J. Ceramide Content in Liver Increases Along with Insulin Resistance in Obese Patients. J. Clin. Med. 2019, 8, 2197. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism Reduced Hepatic, Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef]
- Hage Hassan, R.; Pacheco de Sousa, A.C.; Mahfouz, R.; Hainault, I.; Blachnio-Zabielska, A.; Bourron, O.; Koskas, F.; Górski, J.; Ferré, P.; Foufelle, F.; et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells: Implication Of The Double-Stranded Rna-Activated Protein Kinase. J. Biol. Chem. 2016, 291, 3019–3029. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke et, a.l. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef]
- Turpin-Nolan, S.M.; Hammerschmidt, P.; Chen, W.; Jais, A.; Timper, K.; Awazawa, M.; Brodesser, S. Brüning JCCerS1-Derived C18:0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin resistance. Cell Rep. 2019, 26, 1–10.e7. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A.V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef]
- He, W.; Wang, H.; Yang, G.; Zhu, L.; Liu, X. The Role of Chemokines in Obesity and Exercise-Induced Weight Loss. Biomolecules 2024, 14, 1121. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.P.; Gupta, S.; Sarangi, P.P. Monocytes and macrophages: Origin, homing, differentiation, and functionality during inflammation. Heliyon 2024, 10, e29686. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, W.; Qi, Y.; Yi, H.; Mao, C.; Meng, Q.; Wang, H.; Zheng, C. The Roles of Adipose Tissue Macrophages in Human Disease. Front. Immunol. 2022, 13, 908749. [Google Scholar] [CrossRef]
- Li, X.; Ren, Y.; Chang, K.; Wu, W.; Griffiths, H.R.; Lu, S.; Gao, D. Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Front. Immunol. 2023, 14, 1153915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Woo, J.R.; Bae, S.H.; Wales, T.E.; Engen, J.R.; Lee, J.; Jang, H.; Park, S. The serine phosphorylations in the IRS-1 PIR domain abrogate IRS-1 and IR interaction. Proc. Natl. Acad. Sci. USA 2024, 121, e2401716121. [Google Scholar] [CrossRef] [PubMed]
- Sarvas, J.L.; Khaper, N.; Lees, S.J. The IL-6 Paradox: Context Dependent Interplay of SOCS3 and AMPK. J. Diabetes Metab. 2013, (Suppl. S13). [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cipriano, A.; Viviano, M.; Feoli, A.; Milite, C.; Sarno, G.; Castellano, S.; Sbardella, G. NADPH Oxidases: From Molecular Mechanisms to Current Inhibitors. J. Med. Chem. 2023, 66, 11632–11655. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Vallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; McAllister, M.J.; Slusher, A.L.; Webb, H.E.; Mock, J.T.; Acevedo, E.O. Obesity-Related Oxidative Stress: The Impact of Physical Activity and Diet Manipulation. Sports Med. Open 2015, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Cota-Magaña, A.I.; Vazquez-Moreno, M.; Rocha-Aguado, A.; Ángeles-Mejía, S.; Valladares-Salgado, A.; Díaz-Flores, M.; López-Díazguerrero, N.E.; Cruz, M. Obesity Is Associated with Oxidative Stress Markers and Antioxidant Enzyme Activity in Mexican Children. Antioxidants 2024, 13, 457. [Google Scholar] [CrossRef]
- Li, N.; Li, B.; Brun, T.; Deffert-Delbouille, C.; Mahiout, Z.; Daali, Y.; Ma, X.J.; Krause, K.H.; Maechler, P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012, 61, 2842–2850. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef]
- Yu, T.; Wang, L.; Zhang, L.; Deuster, P.A. Mitochondrial Fission as a Therapeutic Target for Metabolic Diseases: Insights into Antioxidant Strategies. Antioxidants 2023, 12, 1163. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xia, W.; Veeragandham, P.; Cao, Y.; Xu, Y.; Rhyne, T.E.; Qian, J.; Hung, C.W.; Zhao, P.; Jones, Y.; Gao, H.; et al. Obesity causes mitochondrial fragmentation and dysfunction in white adipocytes due to RalA activation. Nat. Metab. 2024, 6, 273–289. [Google Scholar] [CrossRef]
- Mann, J.P.; Duan, X.; Patel, S.; Tábara, L.C.; Scurria, F.; Alvarez-Guaita, A.; Haider, A.; Luijten, I.; Page, M.; Protasoni, M.; et al. A mouse model of human mitofusin-2-related lipodystrophy exhibits adipose-specific mitochondrial stress and reduced leptin secretion. eLife 2023, 12, e82283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, P.; Ma, W.; Gu, Y.; Wu, H.; Bian, Z.; Liu, N.; Yang, D.; Chen, X. High-fat diet causes mitochondrial damage and downregulation of mitofusin-2 and optic atrophy-1 in multiple organs. J. Clin. Biochem. Nutr. 2023, 73, 61–76. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial oxidative function in NAFLD: Friend or foe? Mol. Metab. 2021, 50, 101134. [Google Scholar] [CrossRef]
- Li, F.; Guan, Z.; Gao, Y.; Bai, Y.; Zhan, X.; Ji, X.; Xu, J.; Zhou, H.; Rao, Z. ER stress promotes mitochondrial calcium overload and activates the ROS/NLRP3 axis to mediate fatty liver ischemic injury. Hepatol. Commun. 2024, 8, e0399. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Virgilio, L.; Silva-Lucero, M.D.; Flores-Morelos, D.S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.M.; Zacapala-Gómez, A.E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Zhou, X.; Li, D.L.; Ye, J.M. Role of the mTOR-autophagy-ER stress pathway in high fructose-induced metabolic-associated fatty liver disease. Acta Pharmacol. Sin. 2022, 43, 10–14. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef]
- Sinha, R.A. Autophagy: A Cellular Guardian against Hepatic Lipotoxicity. Genes 2023, 14, 553. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013, 15, 647–658, Erratum in Nat. Cell Biol. 2013, 15, 1016. [Google Scholar] [CrossRef]
- Menikdiwela, K.R.; Ramalingam, L.; Rasha, F.; Wang, S.; Dufour, J.M.; Kalupahana, N.S.; Sunahara, K.K.S.; Martins, J.O.; Moustaid-Moussa, N. Autophagy in metabolic syndrome: Breaking the wheel by targeting the renin-angiotensin system. Cell Death Dis. 2020, 11, 87. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef]
- Diane, A.; Allouch, A.; Mu-U.-Min, R.B.A.; Al-Siddiqi, H.H. Endoplasmic reticulum stress in pancreatic β-cell dysfunctionality and diabetes mellitus: A promising target for generation of functional hPSC-derived β-cells in vitro. Front. Endocrinol. 2024, 15, 1386471. [Google Scholar] [CrossRef]
- Biwer, L.A.; Isakson, B.E. Endoplasmic reticulum-mediated signalling in cellular microdomains. Acta Physiol. 2017, 219, 162–175. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Liu, S.; Klionsky, D.J.; Lip, G.Y.H.; Tuomilehto, J.; Kavalakatt, S.; Pereira, D.M.; Samali, A.; Ren, J.E.R. stress in obesity pathogenesis and management. Trends Pharmacol. Sci. 2022, 43, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, Y.B.; Pandey, V. Obesity and endoplasmic reticulum (ER) stresses. Front. Immunol. 2012, 3, 240. [Google Scholar] [CrossRef]
- Li, H.; Min, Q.; Ouyang, C.; Lee, J.; He, C.; Zou, M.H.; Xie, Z. AMPK activation prevents excess nutrient-induced hepatic lipid accumulation by inhibiting mTORC1 signaling and endoplasmic reticulum stress response. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef]
- Mubarak, S.A.; Otaibi, A.A.; Qarni, A.A.; Lee jhBakillah, A.; Iqbal, J. Reduction in Insulin Mediated ERK Phosphorylation by Palmitate in Liver Cells Is Independent of Fatty Acid Induced ER Stress. Nutrients 2022, 14, 3641. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Hainault, I.; Vilquin, J.T.; Samama, C.; Lasnier, F.; Ferré, P.; Foufelle, F.; Hajduch, E. Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells. Diabetologia 2012, 55, 204–214. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef] [PubMed]
- Caputo, T.; Gilardi, F.; Desvergne, B. From chronic overnutrition to metaflammation and insulin resistance: Adipose tissue and liver contributions. FEBS Lett. 2017, 591, 3061–3088. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Li, W.; Jin, K.; Luo, J.; Xu, W.; Wu, Y.; Zhou, J.; Wang, Y.; Xu, R.; Jiao, L.; Wang, T.; et al. NF-κB and its crosstalk with endoplasmic reticulum stress in atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 988266. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Brown, A.E.; Walker, M. Genetics of Insulin Resistance and the Metabolic Syndrome. Curr. Cardiol. Rep. 2016, 18, 75. [Google Scholar] [CrossRef]
- Oliveri, A.; Rebernick, R.J.; Kuppa, A.; Pant, A.; Chen, Y.; Du, X.; Cushing, K.C.; Bell, H.N.; Raut, C.; Prabhu, P. Comprehensive genetic study of the insulin resistance marker TG:HDL-C in the UK Biobank. Nat. Genet. 2024, 56, 212–221. [Google Scholar] [CrossRef]
- Islam, M.A.; Bhayye, S.; Adeniyi, A.A.; Soliman, M.E.; Pillay, T.S. Diabetes mellitus caused by mutations in human insulin: Analysis of impaired receptor binding of insulins Wakayama, Los Angeles and Chicago using pharmacoinformatics. J. Biomol. Struct. Dyn. 2017, 35, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction-Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. [Google Scholar] [CrossRef] [PubMed]
- Elangeeb, M.E.; Elfaki, I.; Elkhalifa, M.A.; Adam, K.M.; Alameen, A.O.; Elfadl, A.K.; Albalawi, I.A.; Almasoudi, K.S.; Almotairi, R.; Alsaedi, B.S.O.; et al. In Silico Investigation of AKT2 Gene and Protein Abnormalities Reveals Potential Association with Insulin Resistance and Type 2 Diabetes. Curr. Issues Mol. Biol. 2023, 45, 7449–7475. [Google Scholar] [CrossRef]
- Velayutham, K.; Ramanathan, B.; Murugan, J.; Murugan, A.; Thavamani, V.; Gomathinayagam, R. Carriers of the TCF7L2 rs7903146, rs12255372 Risk Alleles in the South Tamil Nadu T2DM Patients Present with Early Incidence and Insulin Dependence. Indian J. Endocrinol. Metab. 2019, 23, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Arrona-Cardoza, P.; Labonté, K.; Cisneros-Franco, J.M.; Nielsen, D.E. The Effects of Food Advertisements on Food Intake and Neural Activity: A Systematic Review and Meta-Analysis of Recent Experimental Studies. Adv. Nutr. 2023, 14, 339–351. [Google Scholar] [CrossRef]
- Hanifah, L.; Nasrulloh, N.; Sufyan, D.L. Sedentary Behavior and Lack of Physical Activity among Children in Indonesia. Children 2023, 10, 1283. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Habibi, P.; Razmjouei, J.; Moradi, A.; Mahdavi, F.; Fallah-Aliabadi, S.; Heydari, A. Climate change and heat stress resilient outdoor workers: Findings from systematic literature review. BMC Public Health 2024, 24, 1711. [Google Scholar] [CrossRef]
- Popkin, B.M.; Ng, S.W. The nutrition transition to a stage of high obesity and noncommunicable disease prevalence dominated by ultra-processed foods is not inevitable. Obes. Rev. 2022, 23, e13366. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Hao, T.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Changes in diet and lifestyle and long-term weight gain in women and men. N. Engl. J. Med. 2011, 364, 2392–2404. [Google Scholar] [CrossRef] [PubMed]
- Sanyaolu, A.; Okorie, C.; Qi, X.; Locke, J.; Rehman, S. Childhood and Adolescent Obesity in the United States: A Public Health Concern. Glob. Pediatr. Health 2019, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Liu, C.H.; Chen, F.Y.; Kuo, C.H.; Pitrone, P.; Liu, J.S. Aging Affects Insulin Resistance, Insulin Secretion, and Glucose Effectiveness in Subjects with Normal Blood Glucose and Body Weight. Diagnostics 2023, 13, 2158. [Google Scholar] [CrossRef] [PubMed]
- Leon, A.S. Attenuation of Adverse Effects of Aging on Skeletal Muscle by Regular Exercise and Nutritional Support. Am. J. Lifestyle Med. 2016, 11, 4–16. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef]
- Long, Y.C.; Cheng, Z.; Copps, K.D.; White, M.F. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol Cell Biol. 2011, 31, 430–441, Erratum in Mol. Cell. Biol. 2017, 37, e00232-17. [Google Scholar] [CrossRef]
- Sasako, T.; Umehara, T.; Soeda, K.; Kaneko, K.; Suzuki, M.; Kobayashi, N.; Okazaki, Y.; Tamura-Nakano, M.; Chiba, T.; Accili, D.; et al. Deletion of skeletal muscle Akt1/2 causes osteosarcopenia and reduces lifespan in mice. Nat. Commun. 2022, 13, 5655. [Google Scholar] [CrossRef]
- Okuma, H.; Tsuchiya, K. Tissue-specific activation of insulin signaling as a potential target for obesity-related metabolic disorders. Pharmacol. Ther. 2024, 262, 108699. [Google Scholar] [CrossRef]
- Lai, K.M.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell. Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Bo, T.; Gao, L.; Yao, Z.; Shao, S.; Wang, X.; Proud, C.G.; Zhao, J. Hepatic selective insulin resistance at the intersection of insulin signaling and metabolic dysfunction-associated steatotic liver disease. Cell Metab. 2024, 36, 947–968. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Jiang, Q.; Wang, Q. Adipose tissue macrophages in remote modulation of hepatic glucose production. Front. Immunol. 2022, 13, 998947. [Google Scholar] [CrossRef] [PubMed]
- Torikai, H.; Chen, M.H.; Jin, L.; He, J.; Angle, J.F.; Shi, W. Atherogenesis in Apoe−/− and Ldlr−/− Mice with a Genetically Resistant Background. Cells 2023, 12, 1255. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nakamura, K.; Taguchi, Y.; Tokita, R.; Takeuchi, S.; Osawa, H.; Teramoto, N.; Sugihara, H.; Yoshizawa, F.; Yamanouchi, K.; et al. Deletion of IRS-1 leads to growth failure and insulin resistance with downregulation of liver and muscle insulin signaling in rats. Sci. Rep. 2025, 15, 649. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Martín-Rodríguez, A.; Martínez-Guardado, I.; Navarro-Jiménez, E.; Laborde-Cárdenas, C.C.; Tornero-Aguilera, J.F. The Role of Adipokines in Health and Disease. Biomedicines 2023, 11, 1290. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Hocking, S.; Samocha-Bonet, D.; Milner, K.L.; Greenfield, J.R.; Chisholm, D.J. Adiposity and insulin resistance in humans: The role of the different tissue and cellular lipid depots. Endocr Rev. 2013, 34, 463–500. [Google Scholar] [CrossRef]
- Caturano, A.; Galiero, R.; Vetrano, E.; Sardu, C.; Rinaldi, L.; Russo, V.; Monda, M.; Marfella, R.; Sasso, F.C. Insulin-Heart Axis: Bridging Physiology to Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 8369. [Google Scholar] [CrossRef]
- Ono, H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1317. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Thomas, P.; Pemberton, S.; Babin, A.; Noonan, C.; Weaver, R.; Banks, W.A.; Rhea, E.M. Central nervous system insulin signaling can influence the rate of insulin influx into brain. Fluids Barriers CNS 2023, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 2017, 66, 1560–1571. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Tobe, K.; Yano, W.; Suzuki, R.; Ueki, K.; Takamoto, I.; Satoh, H.; Maki, T.; Kubota, T. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J. Clin. Investig. 2004, 114, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Skovsø, S.; Panzhinskiy, E.; Kolic, J.; Cen, H.H.; Dionne, D.A.; Dai, X.Q.; Sharma, R.B.; Elghazi, L.; Ellis, C.E.; Faulkner, K.; et al. Beta-cell specific Insr deletion promotes insulin hypersecretion and improves glucose tolerance prior to global insulin resistance. Nat. Commun. 2022, 13, 735. [Google Scholar] [CrossRef]
- Park, K.; Mima, A.; Li, Q.; Rask-Madsen, C.; He, P.; Mizutani, K.; Katagiri, S.; Maeda, Y.; Wu, I.H.; Khamaisi, M.; et al. Insulin decreases atherosclerosis by inducing endothelin receptor B expression. JCI Insight 2016, 1, e86574. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Tanaka, J.; Shuiqing, Y.; Welch, C.L.; DePinho, R.A.; Tabas, I.; Tall, A.R.; Goldberg, I.J.; Accili, D. FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. 2012, 15, 372–381. [Google Scholar] [CrossRef]
- Luo, S.; Li, L.; Chen, H.; Wei, J.; Yang, D. Glucose Metabolism Reprogramming of Vascular Endothelial Cells and Its Implication in Development of Atherosclerosis. Rev. Cardiovasc. Med. 2024, 25, 423. [Google Scholar] [CrossRef]
- Blagov, A.V.; Markin, A.M.; Bogatyreva, A.I.; Tolstik, T.V.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Macrophages in Pathogenesis of Atherosclerosis. Cells 2023, 12, 522. [Google Scholar] [CrossRef]
- Li, H.; Meng, Y.; He, S.; Tan, X.; Zhang, Y.; Zhang, X.; Wang, L.; Zheng, W. Macrophages, Chronic Inflammation, and Insulin Resistance. Cells 2022, 11, 3001. [Google Scholar] [CrossRef]
- Mir, M.M.; Alfaifi, J.; Sohail, S.K.; Rizvi, S.F.; Akhtar, M.T.; Alghamdi, M.A.A.; Mir, R.; Wani, J.I.; Sabah, Z.U.; Alhumaydhi, F.A.; et al. The Role of Pro-Inflammatory Chemokines CCL-1, 2, 4, and 5 in the Etiopathogenesis of Type 2 Diabetes Mellitus in Subjects from the Asir Region of Saudi Arabia: Correlation with Different Degrees of Obesity. J. Pers. Med. 2024, 14, 743. [Google Scholar] [CrossRef]
- Pop, A.; Clenciu, D.; Anghel, M.; Radu, S.; Socea, B.; Mota, E.; Mota, M.; Panduru, N.M. Insulin resistance is associated with all chronic complications in type 1 diabetes. J. Diabetes. 2016, 8, 220–228. [Google Scholar] [CrossRef]
- Bjornstad, P.; Eckel, R.H. Pathogenesis of Lipid Disorders in Insulin Resistance: A Brief Review. Curr. Diabetes Rep. 2018, 18, 127. [Google Scholar] [CrossRef]
- Kerr, A.G.; Andersson, D.P.; Dahlman, I.; Rydén, M.; Arner, P. Adipose Insulin Resistance Associates With Dyslipidemia Independent of Liver Resistance and Involves Early Hormone Signaling. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Eddy, D.; Schlessinger, L.; Kahn, R.; Peskin, B.; Schiebinger, R. Relationship of insulin resistance and related metabolic variables to coronary artery disease: A mathematical analysis. Diabetes Care 2009, 32, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Agbaje, A.O.; Zachariah, J.P.; Bamsa, O.; Odili, A.N.; Tuomainen, T.P. Cumulative insulin resistance and hyperglycemia with arterial stiffness and carotid IMT progression in 1,779 adolescents: A 9-yr longitudinal cohort study. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E268–E278. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.; Redfors, B.; Maehara, A.; McAndrew, T.; Ben-Yehuda, O.; De Bruyne, B.; Mehran, R.; Vogel, B.; Giustino, G.; Serruys, P.W.; et al. Relationship between insulin resistance, coronary plaque, and clinical outcomes in patients with acute coronary syndromes: An analysis from the PROSPECT study. Cardiovasc. Diabetol. 2021, 20, 10. [Google Scholar] [CrossRef]
- Zhou, X.; Kang, C.; Hu, Y.; Wang, X. Study on insulin resistance and ischemic cerebrovascular disease: A bibliometric analysis via CiteSpace. Front. Public Health 2023, 11, 1021378. [Google Scholar] [CrossRef]
- Lee, J.E.; Shin, D.W.; Yun, J.M.; Kim, S.H.; Nam, Y.S.; Cho, B.; Lim, J.S.; Jeong, H.Y.; Kwon, H.M.; Park, J.H. Insulin Resistance Is a Risk Factor for Silent Lacunar Infarction. Stroke 2016, 47, 2938–2944. [Google Scholar] [CrossRef]
- Someya, Y.; Tamura, Y.; Kaga, H.; Sugimoto, D.; Kadowaki, S.; Suzuki, R.; Aoki, S.; Hattori, N.; Motoi, Y.; Shimada, K.; et al. Insulin resistance and muscle weakness are synergistic risk factors for silent lacunar infarcts: The Bunkyo Health Study. Sci. Rep. 2021, 11, 21093. [Google Scholar] [CrossRef]
- Feng, X.; Yao, Y.; Wu, L.; Cheng, C.; Tang, Q.; Xu, S. Triglyceride-Glucose Index and the Risk of Stroke: A Systematic Review and Dose-Response Meta-Analysis. Horm. Metab. Res. 2022, 54, 175–186. [Google Scholar] [CrossRef]
- Nogueira, J.P.; Cusi, K. Role of Insulin Resistance in the Development of Nonalcoholic Fatty Liver Disease in People With Type 2 Diabetes: From Bench to Patient Care. Diabetes Spectr. 2024, 37, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Begum, G.S.; Almashaikhi, N.A.T.; Albalushi, M.Y.; Alsalehi, H.M.; Alazawi, R.S.; Goud, B.K.M.; Dube, R. Prevalence of Polycystic Ovary Syndrome (PCOS) and Its Associated Risk Factors among Medical Students in Two Countries. Int. J. Environ. Res. Public Health 2024, 21, 1165. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Chakole, S. Prevalence of Polycystic Ovarian Syndrome and Its Link to Obesity in Adolescent Girls. Cureus 2023, 15, e45405. [Google Scholar] [CrossRef]
- Olatunde, A.; Nigam, M.; Singh, R.K.; Panwar, A.S.; Lasisi, A.; Alhumaydhi, F.A.; Jyoti Kumar, V.; Mishra, A.P.; Sharifi-Rad, J. Cancer and diabetes: The interlinking metabolic pathways and repurposing actions of antidiabetic drugs. Cancer Cell Int. 2021, 21, 499. [Google Scholar] [CrossRef]
- Zhang, A.M.Y.; Wellberg, E.A.; Kopp, J.L.; Johnson, J.D. Hyperinsulinemia in Obesity, Inflammation, and Cancer. Diabetes Metab J. 2021, 45, 285–311, Erratum in Diabetes Metab. J. 2021, 45, 622. [Google Scholar] [CrossRef]
- Buono, G.; Crispo, A.; Giuliano, M.; De Angelis, C.; Schettini, F.; Forestieri, V.; Lauria, R.; De Laurentiis, M.; De Placido, P.; Rea, C.G.; et al. Metabolic syndrome and early-stage breast cancer outcome: Results from a prospective observational study. Breast Cancer Res. Treat. 2020, 182, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, E.; Mirabelli, M.; La Vignera, S.; Tanyolaç, S.; Foti, D.P.; Aversa, A.; Brunetti, A. Insulin Resistance and Cancer: In Search for a Causal Link. Int. J. Mol. Sci. 2021, 22, 11137. [Google Scholar] [CrossRef]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-Activated Protein Kinase (MAPK) and Obesity-Related Cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef]
- Nallamothu, P.; Nimmanapalli, H.D.; Sachan, A.; Srinivasa Rao, P.V.; Vishnubotla, S. Insulin resistance in nondiabetic chronic kidney disease patients. Saudi J. Kidney Dis. Transpl. 2021, 32, 1300–1309. [Google Scholar] [CrossRef]
- Clarembeau, F.; Bale, G.; Lanthier, N. Cirrhosis and insulin resistance: Current knowledge, pathophysiological mechanisms, complications and potential treatments. Clin. Sci. 2020, 134, 2117–2135. [Google Scholar] [CrossRef]
- Fu, Y.H.; Liu, W.J.; Lee, C.L.; Wang, J.S. Associations of insulin resistance and insulin secretion with bone mineral density and osteoporosis in a general population. Front. Endocrinol. 2022, 13, 971960. [Google Scholar] [CrossRef]
- Balta, I.; Ekiz, O.; Ozuguz, P.; Ustun, I.; Karaca, S.; Dogruk Kacar, S.; Eksioglu, M. Insulin resistance in patients with post-adolescent acne. Int. J. Dermatol. 2015, 54, 662–666. [Google Scholar] [CrossRef]
- Wang, T.; Laher, I.; Li, S. Exercise snacks and physical fitness in sedentary populations. Sports Med. Health Sci. 2024, 7, 1–7. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Barrea, L.; Caprio, M.; Ceriani, F.; Chavez, A.O.; El Ghoch, M.; Frias-Toral, E.; Mehta, R.J.; Mendez, V.; Paschou, S.A.; et al. Nutritional guidelines for the management of insulin resistance. Crit. Rev. Food Sci. Nutr. 2022, 62, 6947–6960. [Google Scholar] [CrossRef]
- Weinberg Sibony, R.; Segev, O.; Dor, S.; Raz, I. Drug Therapies for Diabetes. Int. J. Mol. Sci. 2023, 24, 17147. [Google Scholar] [CrossRef]
- Elian, V.; Popovici, V.; Karampelas, O.; Pircalabioru, G.G.; Radulian, G.; Musat, M. Risks and Benefits of SGLT-2 Inhibitors for Type 1 Diabetes Patients Using Automated Insulin Delivery Systems-A Literature Review. Int. J. Mol. Sci. 2024, 25, 1972. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 234. [Google Scholar] [CrossRef]
- Yin, R.; Xu, Y.; Wang, X.; Yang, L.; Zhao, D. Role of Dipeptidyl Peptidase 4 Inhibitors in Antidiabetic Treatment. Molecules 2022, 27, 3055. [Google Scholar] [CrossRef]
- Consoli, A.; Czupryniak, L.; Duarte, R.; Jermendy, G.; Kautzky-Willer, A.; Mathieu, C.; Melo, M.; Mosenzon, O.; Nobels, F.; Papanas, N.; et al. Positioning sulphonylureas in a modern treatment algorithm for patients with type 2 diabetes: Expert opinion from a European consensus panel. Diabetes Obes. Metab. 2020, 22, 1705–1713. [Google Scholar] [CrossRef]
- Holst, J.J. The incretin system in healthy humans: The role of GIP and GLP-1. Metabolism 2019, 96, 46–55. [Google Scholar] [CrossRef]
- Rangraze, I.; Patoulias, D.; Karakasis, P.; El-Tanani, M.; Rizzo, M. Tirzepatide, a novel, dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 receptor agonist for the ongoing diabesity epidemic: The dawn of a new era? Expert. Rev. Clin. Pharmacol. 2024, 17, 853–856. [Google Scholar] [CrossRef]
- Varney, M.J.; Benovic, J.L. The Role of G Protein-Coupled Receptors and Receptor Kinases in Pancreatic β-Cell Function and Diabetes. Pharmacol. Rev. 2024, 76, 267–299. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.W.; Wang, H.; Kim, R.H.; Park, H.K.; Lee, H.; Kang, E.S. DA-1241, a Novel GPR119 Agonist, Improves Hyperglycaemia by Inhibiting Hepatic Gluconeogenesis and Enhancing Insulin Secretion in Diabetic Mice. Diabetes Metab. J. 2022, 46, 337–348. [Google Scholar] [CrossRef]
- Parnova, R.G. GPR40/FFA1 Free Fatty Acid Receptors and Their Functional Role. Neurosci. Behav. Physiol. 2021, 51, 256–264. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Q.; Ma, Y.; Lin, L.; Liu, W.; Ding, A.; Wang, C.; Zhou, S.; Cai, J.; Tang, H. Recent Developments in Drug Design of Oral Synthetic Free Fatty Acid Receptor 1 Agonists. Drug Des. Dev. Ther. 2024, 18, 5961–5983. [Google Scholar] [CrossRef]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Z. Effects of Melatonin Supplementation on Insulin Levels and Insulin Resistance: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Horm. Metab. Res. 2021, 53, 616–624. [Google Scholar] [CrossRef]
- Dhankhar, S.; Chauhan, S.; Mehta, D.K.; Nitika Saini, K.; Saini, M.; Das, R.; Gupta, S.; Gautam, V. Novel targets for potential therapeutic use in Diabetes mellitus. Diabetol. Metab. Syndr. 2023, 15, 17. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. 11β-hydroxysteroid dehydrogenases: A growing multi-tasking family. Mol. Cell Endocrinol. 2021, 526, 111210. [Google Scholar] [CrossRef]
- Mir, M.M.; Mir, R.; Alghamdi, M.A.A.; Wani, J.I.; Sabah, Z.U.; Jeelani, M.; Marakala, V.; Sohail, S.K.; O’haj, M.; Alharthi, M.H.; et al. Differential Association of Selected Adipocytokines, Adiponectin, Leptin, Resistin, Visfatin and Chemerin, with the Pathogenesis and Progression of Type 2 Diabetes Mellitus (T2DM) in the Asir Region of Saudi Arabia: A Case Control Study. J. Pers. Med. 2022, 12, 735. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liang, Y.; Ma, Y.; Wu, J.; Luo, H.; Yi, B. The Variation and Correlation of Serum Adiponectin, Nesfatin-1, IL-6, and TNF-α Levels in Prediabetes. Front. Endocrinol. 2022, 13, 774272. [Google Scholar] [CrossRef] [PubMed]
- Chekol Abebe, E.; Tilahun Muche, Z.; Behaile T/Mariam, A.; Mengie Ayele, T.; Mekonnen Agidew, M.; Teshome Azezew, M.; Abebe Zewde, E.; Asmamaw Dejenie, T.; Asmamaw Mengstie, M. The structure, biosynthesis, and biological roles of fetuin-A: A review. Front. Cell Dev. Biol. 2022, 10, 945287. [Google Scholar] [CrossRef]
- Hsu, M.C.; Chen, C.H.; Wang, M.C.; Chen, W.H.; Hu, P.A.; Guo, B.C.; Chang, R.W.; Wang, C.H.; Lee, T.S. Apigenin targets fetuin-A to ameliorate obesity-induced insulin resistance. Int. J. Biol. Sci. 2024, 20, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- Semerena, E.; Nencioni, A.; Masternak, K. Extracellular nicotinamide phosphoribosyltransferase: Role in disease pathophysiology and as a biomarker. Front. Immunol. 2023, 14, 1268756. [Google Scholar] [CrossRef]
- Dong, W.S.; Hu, C.; Hu, M.; Gao, Y.P.; Hu, Y.X.; Li, K.; Ye, Y.J.; Zhang, X. Metrnl: A promising biomarker and therapeutic target for cardiovascular and metabolic diseases. Cell Commun. Signal. 2024, 22, 389. [Google Scholar] [CrossRef]
- Miao, Z.W.; Hu, W.J.; Li, Z.Y.; Miao, C.Y. Involvement of the secreted protein Metrnl in human diseases. Acta Pharmacol. Sin. 2020, 41, 1525–1530. [Google Scholar] [CrossRef]
- Huang, K.T.; Lin, C.C.; Tsai, M.C.; Chen, K.D.; Chiu, K.W. Pigment epithelium-derived factor in lipid metabolic disorders. Biomed. J. 2018, 41, 102–108. [Google Scholar] [CrossRef]
- Carnagarin, R.; Dharmarajan, A.M.; Dass, C.R. PEDF-induced alteration of metabolism leading to insulin resistance. Mol. Cell. Endocrinol. 2015, 401, 98–104. [Google Scholar] [CrossRef]
- Dimova, R.; Tankova, T. The role of vaspin in the development of metabolic and glucose tolerance disorders and atherosclerosis. Biomed. Res. Int. 2015, 2015, 823481. [Google Scholar] [CrossRef]
- Pilarski, Ł.; Pelczyńska, M.; Koperska, A.; Seraszek-Jaros, A.; Szulińska, M.; Bogdański, P. Association of Serum Vaspin Concentration with Metabolic Disorders in Obese Individuals. Biomolecules 2023, 13, 508. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef]
- Tryggestad, J.B.; Wang, J.J.; Zhang, S.X.; Thompson, D.M.; Short, K.R. Elevated plasma pigment epithelium-derived factor in children with type 2 diabetes mellitus is attributable to obesity. Pediatr. Diabetes 2015, 16, 600–605. [Google Scholar] [CrossRef]
- Barton, M.; Prossnitz, E.R. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol. Metab. 2015, 26, 185–192. [Google Scholar] [CrossRef]
- Khartabil, N.; Avoundjian, A. Gene Therapy and Diabetes: A Narrative Review of Recent Advances and the Role of Multidisciplinary Healthcare Teams. Genes 2025, 16, 107. [Google Scholar] [CrossRef]
- Rohban, R.; Martins, C.P.; Esni, F. Advanced therapy to cure diabetes: Mission impossible is now possible? Front. Cell Dev. Biol. 2024, 12, 1484859. [Google Scholar] [CrossRef] [PubMed]
- Callejas, D.; Mann, C.J.; Ayuso, E.; Lage, R.; Grifoll, I.; Roca, C.; Andaluz, A.; Ruiz-de Gopegui, R.; Montané, J.; Muñoz, S.; et al. Treatment of diabetes and long-term survival after insulin and glucokinase gene therapy. Diabetes 2013, 62, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Ropper, A.H.; Gorson, K.C.; Gooch, C.L.; Weinberg, D.H.; Pieczek, A.; Ware, J.H.; Kershen, J.; Rogers, A.; Simovic, D.; Schratzberger, P.; et al. Vascular endothelial growth factor gene transfer for diabetic polyneuropathy: A randomized, double-blinded trial. Ann. Neurol. 2009, 65, 386–393. [Google Scholar] [CrossRef]
- Kupczyńska, D.; Lubieniecki, P.; Antkiewicz, M.; Barć, J.; Frączkowska-Sioma, K.; Dawiskiba, T.; Dorobisz, T.; Małodobra-Mazur, M.; Baczyńska, D.; Pańczak, K.; et al. Complementary Gene Therapy after Revascularization with the Saphenous Vein in Diabetic Foot Syndrome. Genes 2023, 14, 1968. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Wan, X.X.; Kambey, P.A.; Luo, Y.; Hu, X.M.; Liu, Y.F.; Shan, J.Q.; Chen, Y.W.; Xiong, K. Therapeutic role of growth factors in treating diabetic wound. World J. Diabetes 2023, 14, 364–395. [Google Scholar] [CrossRef]
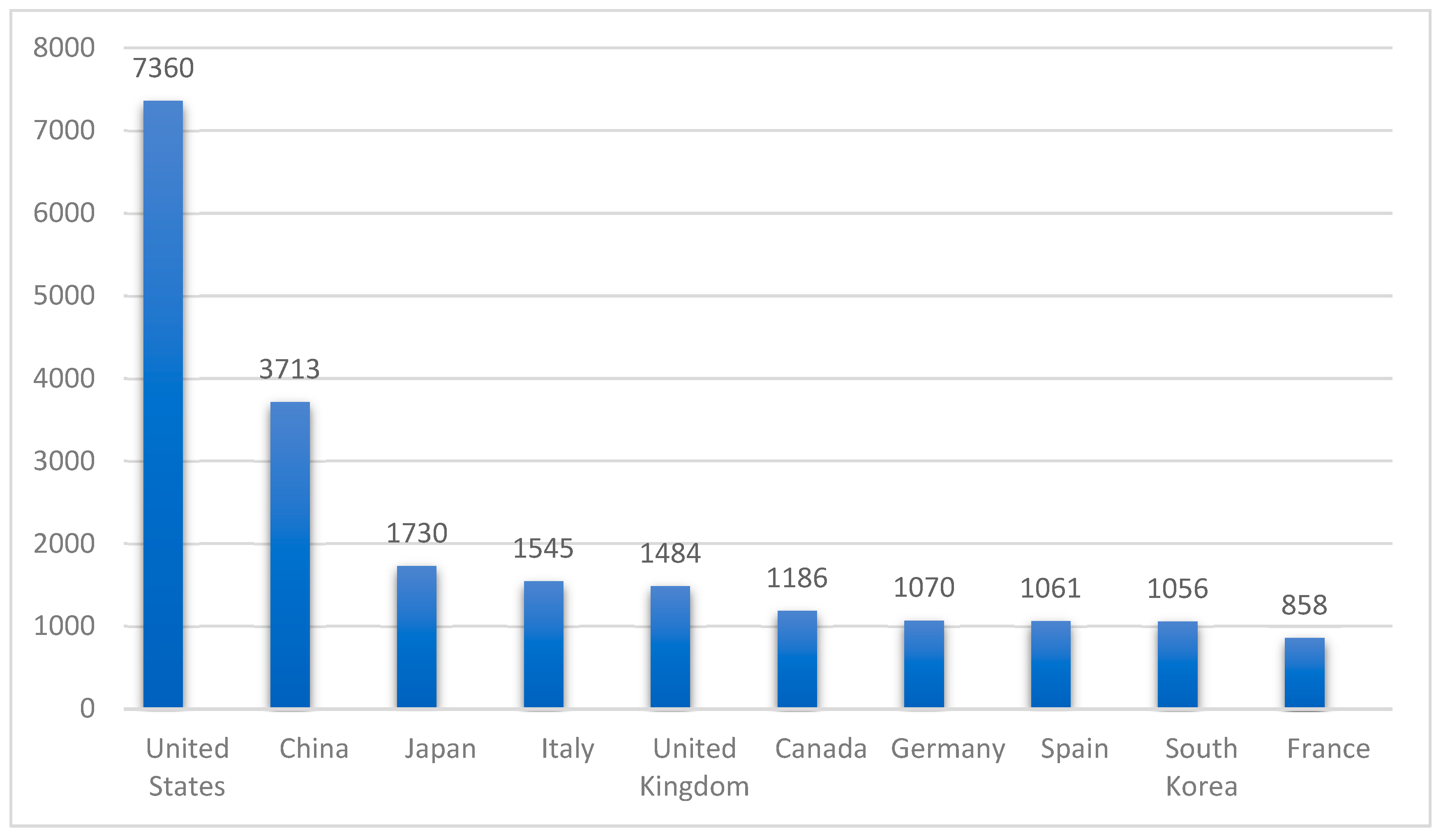
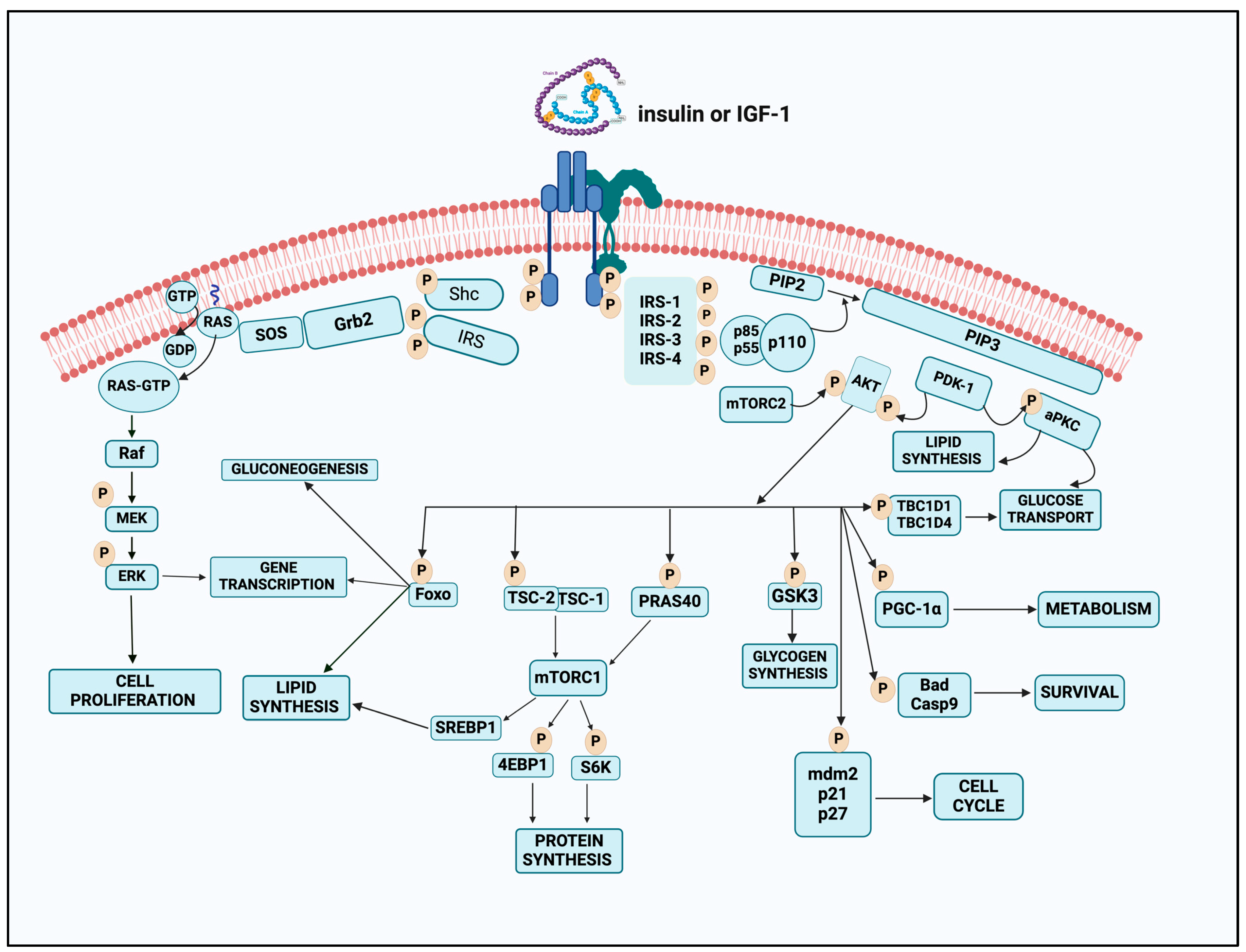
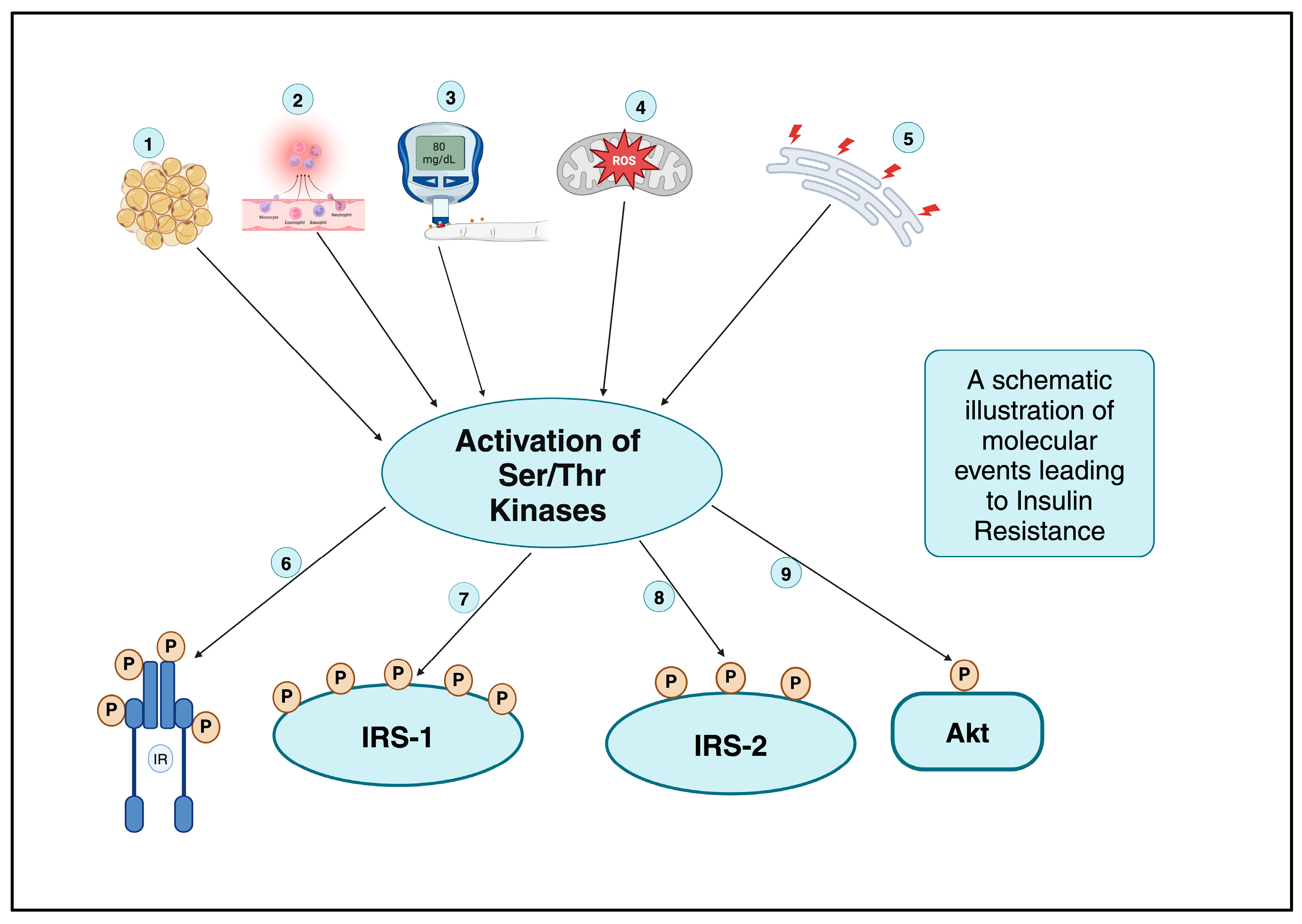
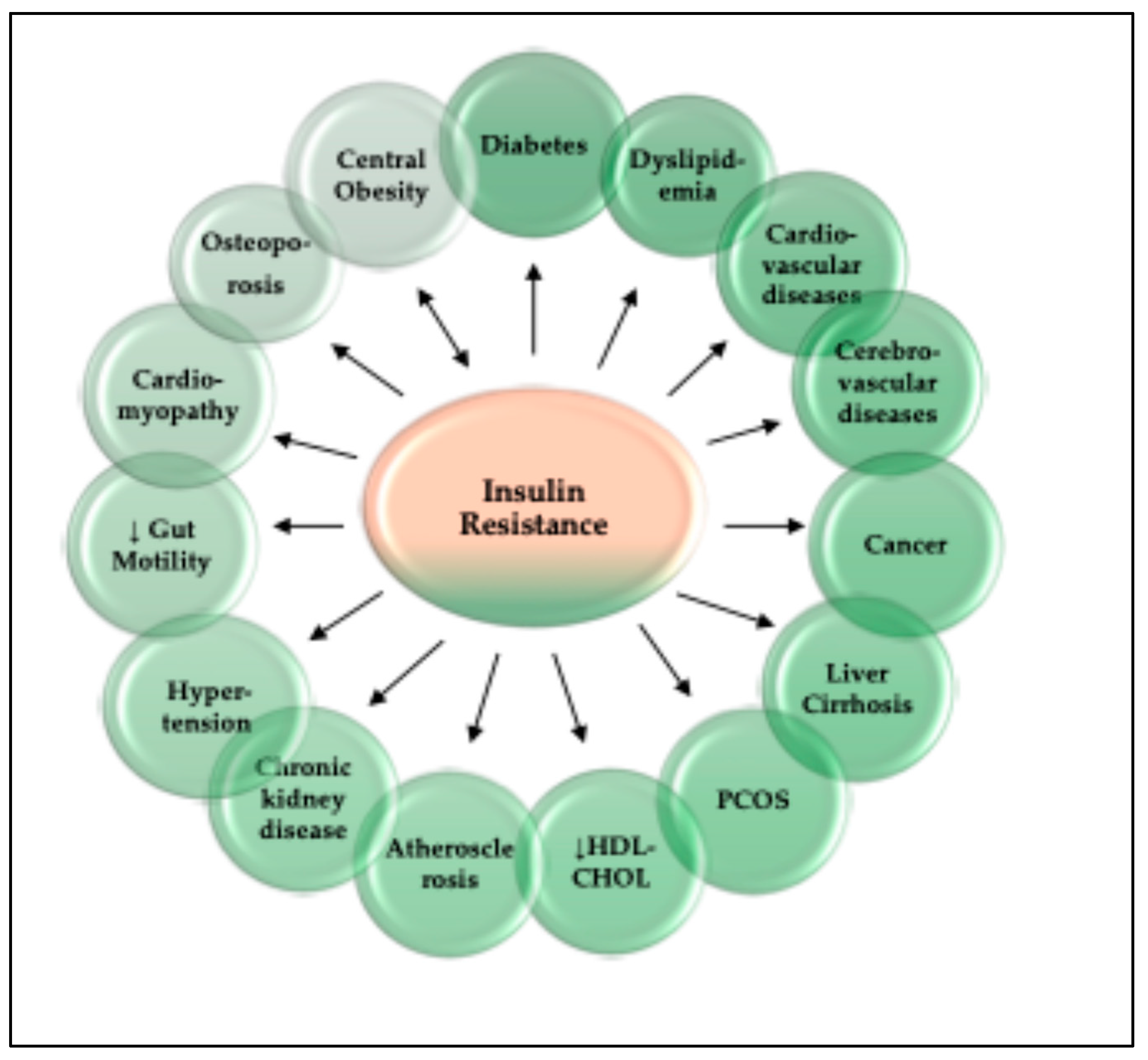

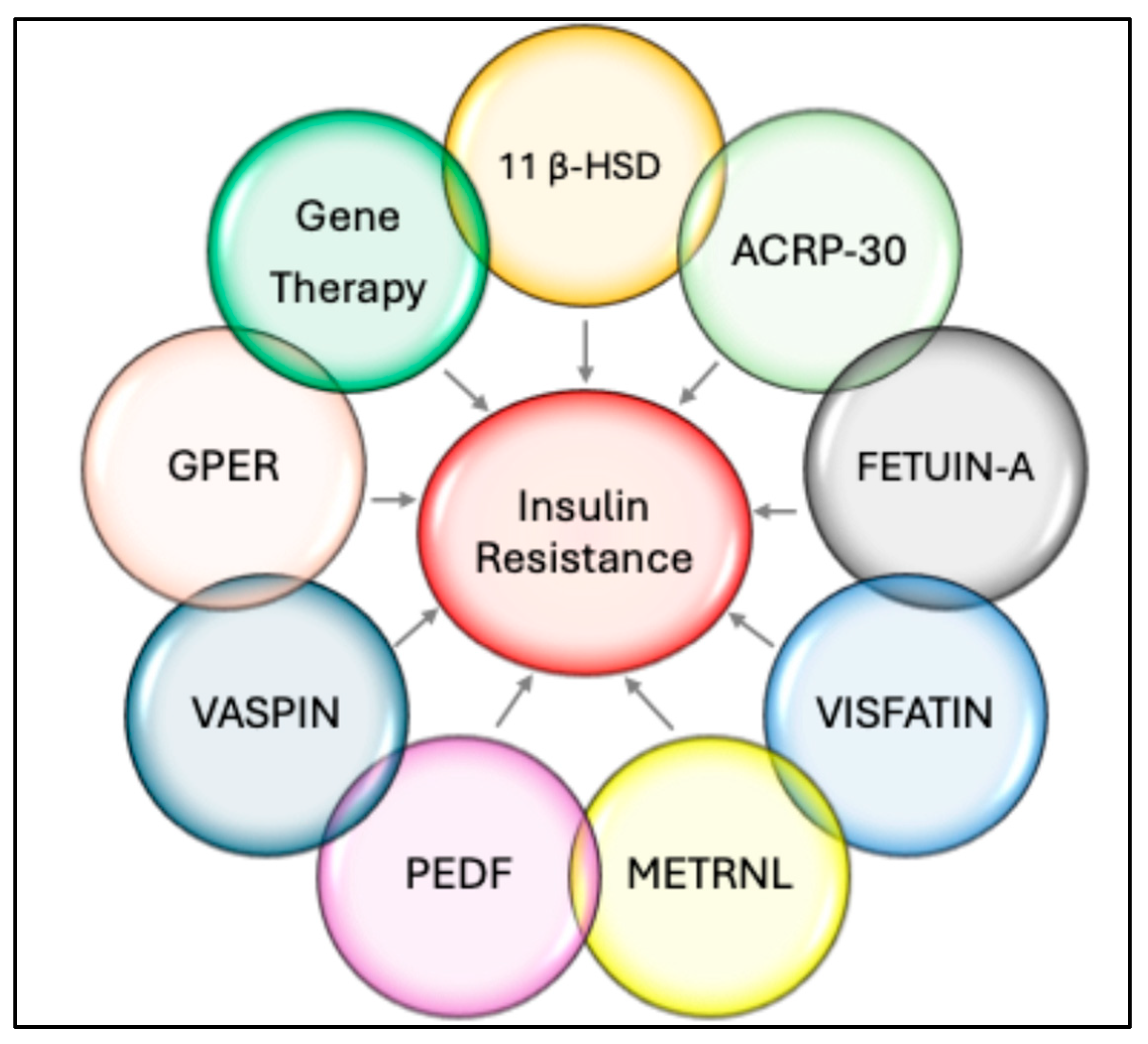

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role | Drug Class | Examples | Mechanism | Citation |
|---|---|---|---|---|
| Decrease Hepatic glucose production | Biguanides | Metformin | The precise mechanism of metformin is still elusive and is thought to reduce HGP, a process that is facilitated by the stimulation of mitochondrial activity or the suppression of glucagon signaling via AMPK activation and increased expression of the GLUT4 glucose transporter. | [268] |
| Increase insulin sensitivity | Thiazolidinediones | Rosiglitazone Pioglitazone | Thiazolidinediones function through their interaction with the PPAR-γ to enhance the sensitivity of adipose muscle and liver to insulin. | [268] |
| Inhibit renal glucose reabsorption | Sodium-Glucose Cotransporter Inhibitors (SGLT-2i) | Empagliflozin Dapagliflozin | SGLT-2is facilitate insulin-independent glucose reduction by inhibiting glucose reabsorption in the proximal renal tubules, thereby decreasing blood glucose levels. Additionally, These medications are linked to reliable and well documented weight loss and decreases in blood pressure | [268,269] |
| Increase insulin sensitivity | Glucagon-like Peptide-1 Receptor Agonists (GLP 1 RA) | Semaglutide Dulaglutide Liraglutide Exenatide | GLP 1RAs increase insulin sensitivity in peripheral tissues and also have notable anti-inflammatory and anti-obesity effects, protective benefits for lung health, and favorable impact on gut microbiome composition. However, GLP-1RAs are linked to prevalent gastrointestinal adverse effects, impacting over one-third of patients and other complications. | [268,270] |
| Increase insulin secretion | Dipeptidyl Peptidase-4 Inhibitors (DPP-4i) | Vildagliptin Alogliptin Linagliptin Gemigliptin Teneligliptin Trelagliptin Saxagliptin | DPP-4is inhibit incretin degradation and facilitates postprandial insulin secretion. Their advantages include the reduction in HbA1c levels, renal microalbuminuria, and inflammation. | [268,271] |
| Increase insulin secretion | Sulfonylureas | Glimepiride Gliclazide | Sulfonylureas reduce blood glucose levels by enhancing insulin secretion from beta cells through the inhibition of KATP channels. They also inhibit gluconeogenesis and lipid breakdown into fatty acids. They also promote insulin sensitivity. | [268,272] |
| Role | Drug Target | Examples | Mechanism | Citation |
|---|---|---|---|---|
| Increase insulin secretion | Glucose-dependent insulinotropic polypeptide (GIP) | Tirzepatide | GIP is present in β-cells, adipose tissue, and the brain and increases intracellular cAMP by binding to its receptor. High cAMP levels activate PKA, and exchange protein-activated cAMP2. Depolarizing voltage-gated calcium channels raises intracellular Ca2+ and promotes insulin release from β-cells. Recently, Tirzepatide, a novel dual GIP/GLP 1 receptor agonist, not only achieved significantly improved glycemic control but also allowed the majority of participants to attain a mean weight reduction exceeding 10% from baseline, which is a notable outcome in the realm of current pharmacotherapy. Its safety profile is being investigated, and it offers a lot of promise as of now. | [273,274] |
| ↑ Insulin release ↓ HGP | G-Protein coupled receptor (GPCR 119) |
GSK1292263, MBX-2982 DS-8500a APD668 BMS-903452 | GPR119, a Class-I G protein coupled receptor, is found in muscles, liver, and pancreatic β-cells. Similar to incretin hormones, GPR119 activation may enhance insulin synthesis and secretion when agonists bind to its binding site. GPR119 enhances glucose homeostasis via direct β-cell insulin release and indirect GLP-1 and GIP release in enteroendocrine cells. More than 40 GPR 119 agonists have been reported to show promising effects on glucoses homeostasis by depressing HGP and increasing insulin synthesis in both humans and/or animal models. The efficacy and the safety profile of these agonists is under continuous scrutiny. | [275,276] |
| ↑Incretin hormone Release ↑ Insulin release | Free-fatty acid receptor-1 agonists | TAK-875 TSL1806 | G-protein-coupled receptor-40 (FFA1) is a Class-A receptor and is expressed in the mammalian pancreas, gut, taste buds, and CNS. FFA1 affects blood glucose levels by increasing incretin hormones and promoting insulin release from pancreatic β-cells. Synthetic GPR40/FFA1 receptor agonists, such as TAK-875 and TSL1806, have been tried in the last many years, but their side effects, including hepatotoxicity, are a matter of concern, which is being investigated. | [277,278] |
| Target Fatty acid oxidation | PPAR full agonists | Chiglitazar Sodium | Chiglitazar Sodium is a peroxisome proliferator-activated receptor (PPAR) full agonist simultaneously activates three subtypes of PPAR receptors (α, γ, and δ). It can induce the expression of downstream target genes related to insulin sensitivity, fatty acid oxidation, energy conversion and lipid transport, and inhibit the phosphorylation of PPARγ receptors associated with insulin resistance. | [279] |
| ↑ Insulin release | Melatonin (neuroendocrine hormone) | Melatonin | Melatonin modulates glucose levels via its melatonin receptors MT1 and MT2 in diverse cells. Melatonin supplementation has been reported to ameliorate hyperinsulinemia, insulin resistance, and insulin sensitivity by many investigators and there is enough evidence to use it as an adjuvant therapy. | [280] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mir, M.M.; Jeelani, M.; Alharthi, M.H.; Rizvi, S.F.; Sohail, S.K.; Wani, J.I.; Sabah, Z.U.; BinAfif, W.F.; Nandi, P.; Alshahrani, A.M.; et al. Unraveling the Mystery of Insulin Resistance: From Principle Mechanistic Insights and Consequences to Therapeutic Interventions. Int. J. Mol. Sci. 2025, 26, 2770. https://doi.org/10.3390/ijms26062770
Mir MM, Jeelani M, Alharthi MH, Rizvi SF, Sohail SK, Wani JI, Sabah ZU, BinAfif WF, Nandi P, Alshahrani AM, et al. Unraveling the Mystery of Insulin Resistance: From Principle Mechanistic Insights and Consequences to Therapeutic Interventions. International Journal of Molecular Sciences. 2025; 26(6):2770. https://doi.org/10.3390/ijms26062770
Chicago/Turabian StyleMir, Mohammad Muzaffar, Mohammed Jeelani, Muffarah Hamid Alharthi, Syeda Fatima Rizvi, Shahzada Khalid Sohail, Javed Iqbal Wani, Zia Ul Sabah, Waad Fuad BinAfif, Partha Nandi, Abdullah M. Alshahrani, and et al. 2025. "Unraveling the Mystery of Insulin Resistance: From Principle Mechanistic Insights and Consequences to Therapeutic Interventions" International Journal of Molecular Sciences 26, no. 6: 2770. https://doi.org/10.3390/ijms26062770
APA StyleMir, M. M., Jeelani, M., Alharthi, M. H., Rizvi, S. F., Sohail, S. K., Wani, J. I., Sabah, Z. U., BinAfif, W. F., Nandi, P., Alshahrani, A. M., Alfaifi, J., Jehangir, A., & Mir, R. (2025). Unraveling the Mystery of Insulin Resistance: From Principle Mechanistic Insights and Consequences to Therapeutic Interventions. International Journal of Molecular Sciences, 26(6), 2770. https://doi.org/10.3390/ijms26062770