
Chronic Antibody-Mediated Rejection and Plasma Cell ER Stress: Opportunities and Challenges with Calcineurin Inhibitors


Abstract
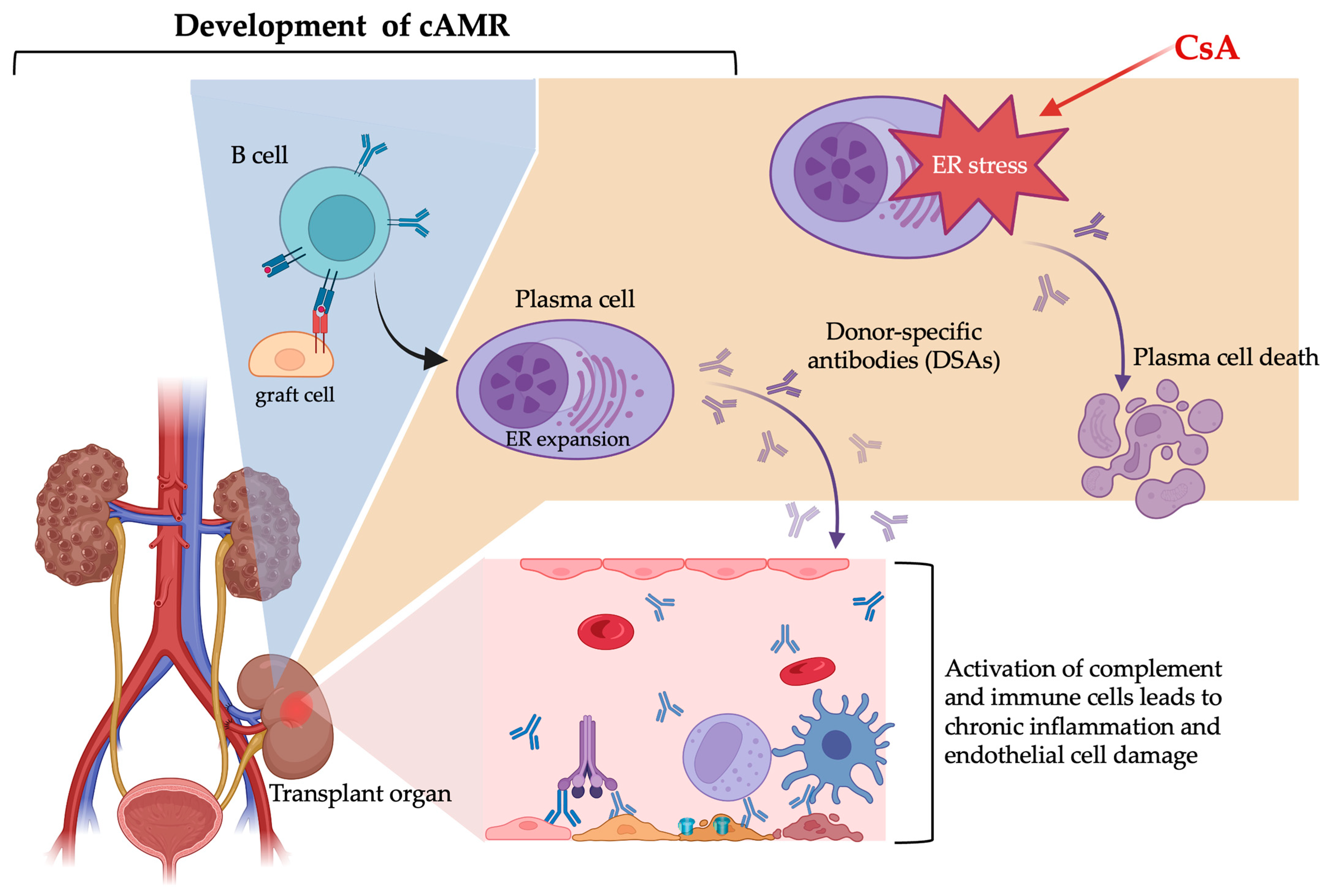
1. Introduction
2. Damage of Donor-Specific Antibodies in Organ Transplantation
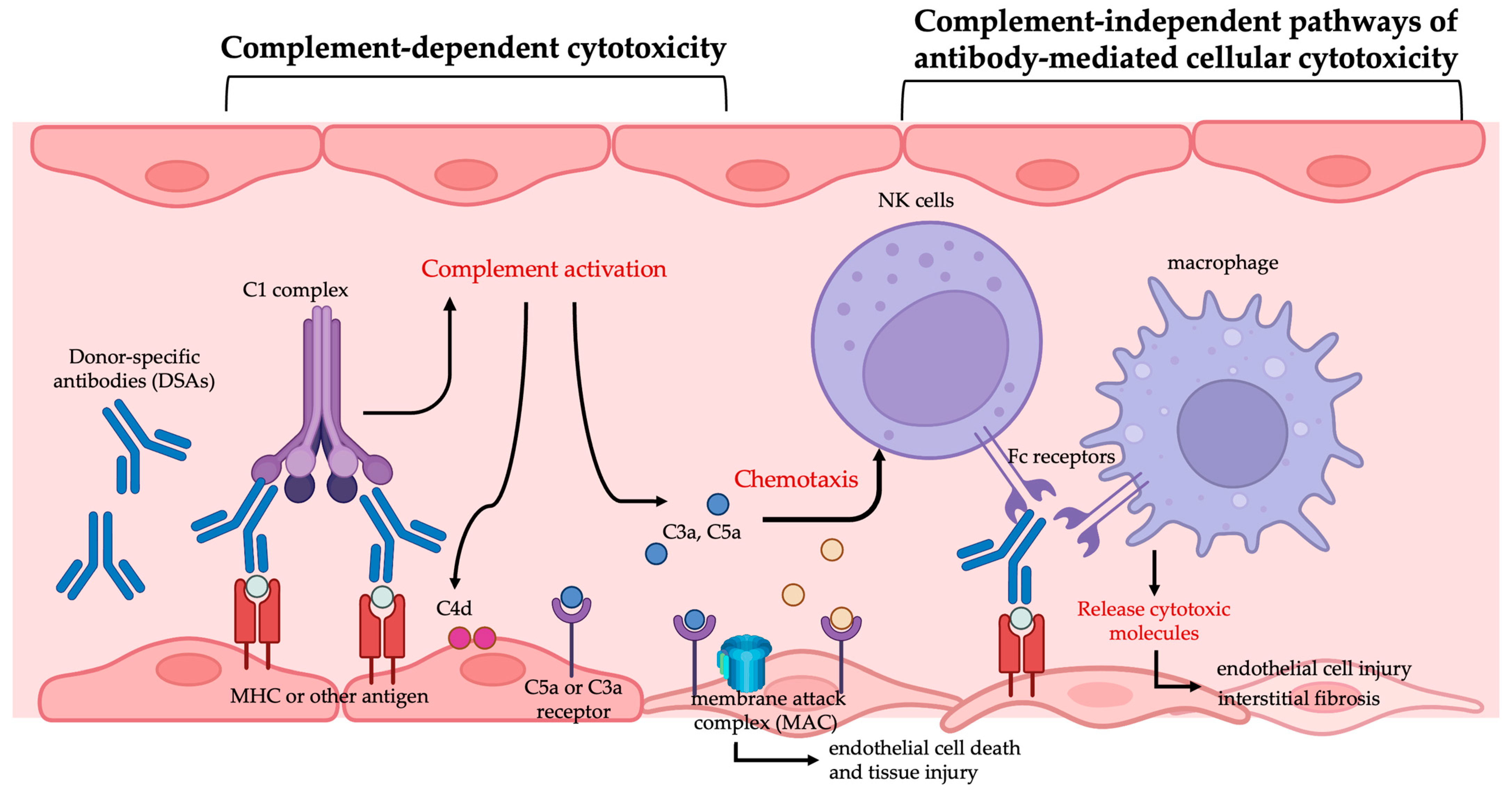
2.1. Antibody-Mediated Rejection
2.2. Donor-Specific Antibodies and AMR
2.3. Diagnosis of AMR
2.4. Treatment Strategies for cAMR
2.4.1. Plasmapheresis
2.4.2. Intravenous Immunoglobulin (IVIG)
2.4.3. Rituximab
2.4.4. Proteasome Inhibitors
2.4.5. Tocilizumab
2.4.6. Eculizumab
3. Targeting Plasma Cells with Conventional Immunosuppressive Drugs: A Strategy to Mitigate Antibody-Mediated Rejection
3.1. The Function of the Endoplasmic Reticulum in Cells
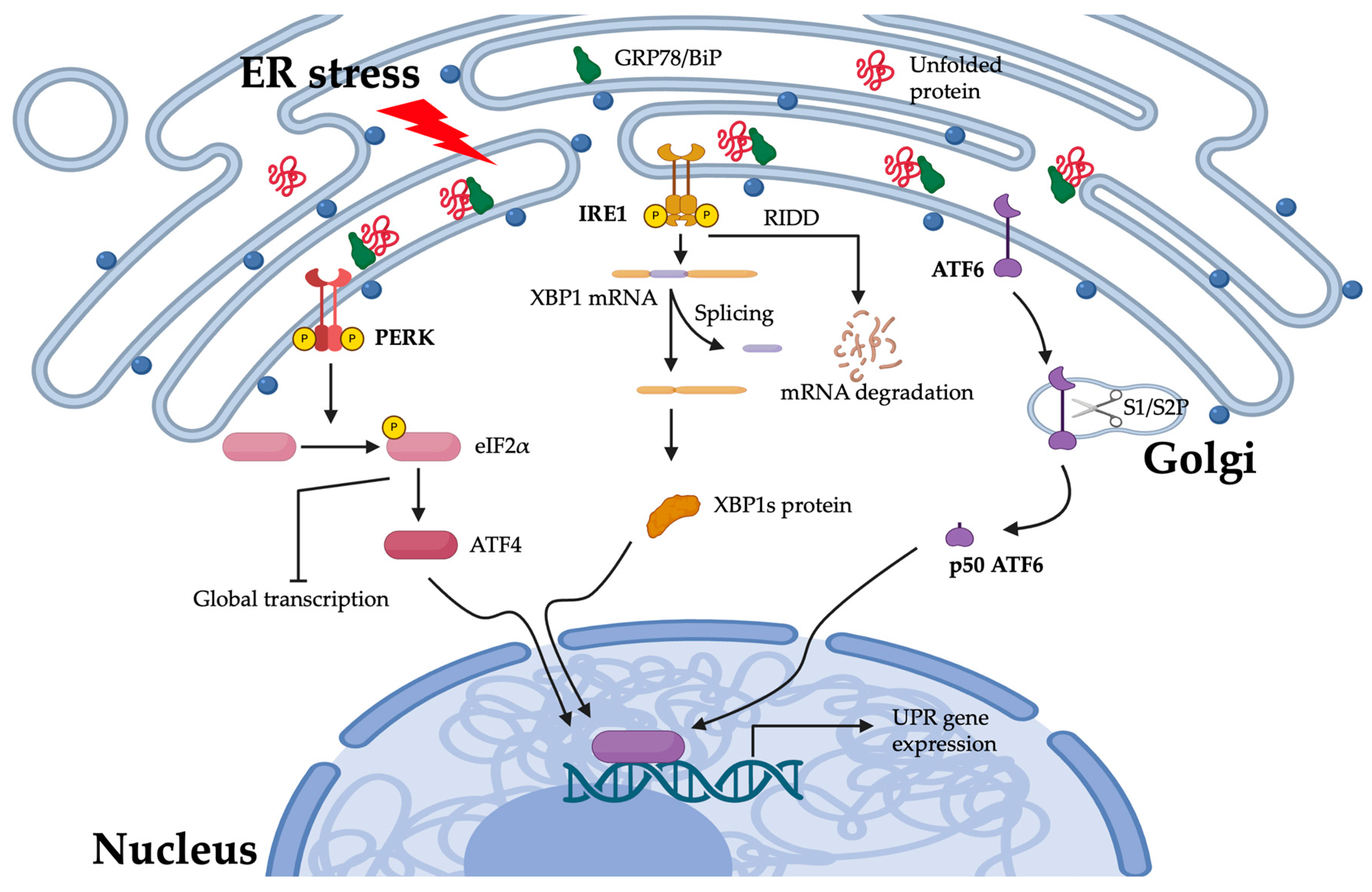
3.1.1. Response to Stress in the Endoplasmic Reticulum
3.1.2. The PERK Pathway
3.1.3. The IRE1 Pathway
3.1.4. The ATF6 Pathway
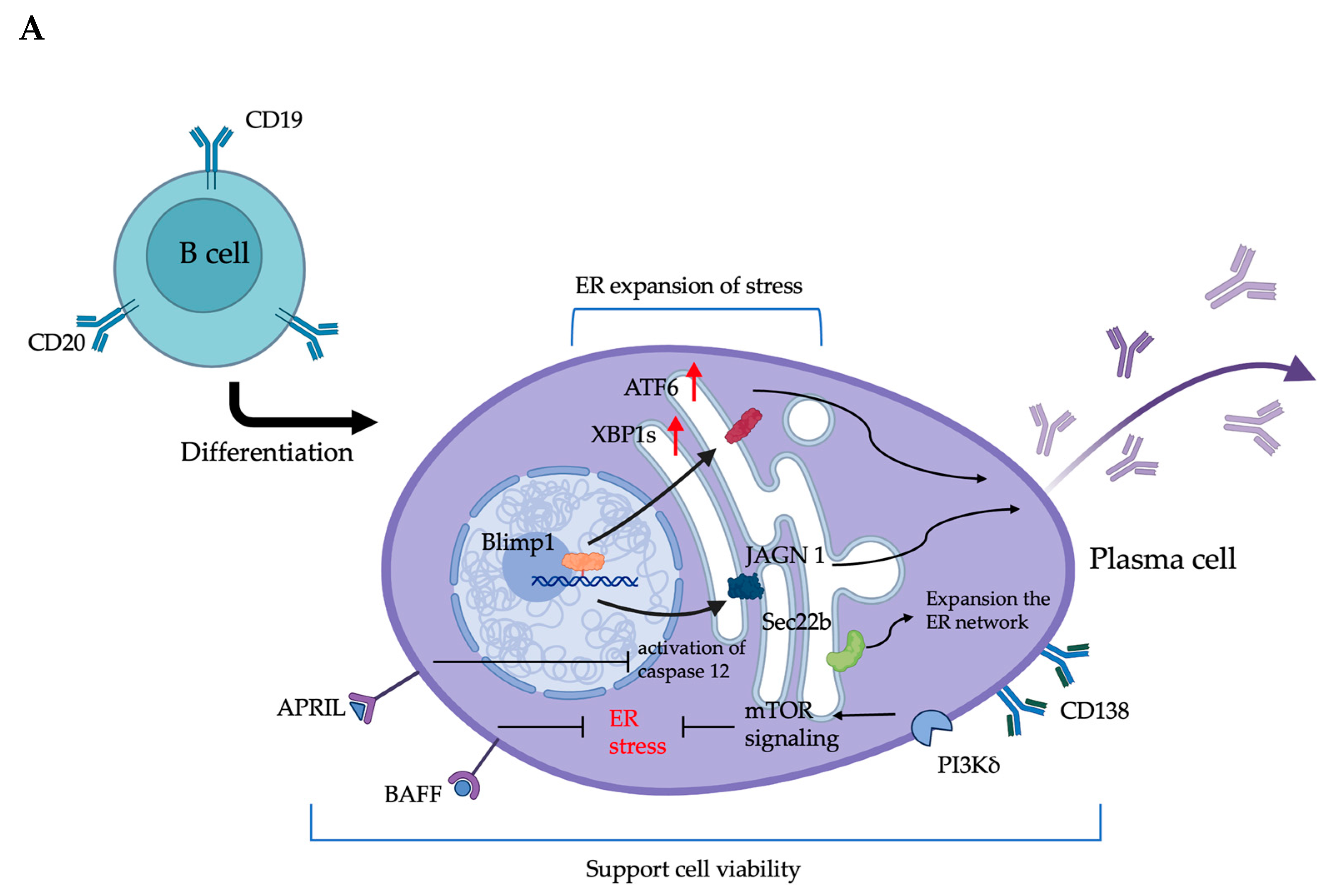
3.2. The ER in Plasma Cells Is Closely Related to Antibody Production and Survival
3.2.1. ER-Associated Molecular Regulation and Its Impact on the Antibody Production of Plasma Cells
3.2.2. Plasma Cell Survival and ER Stress: Molecule Mechanism and Regulation
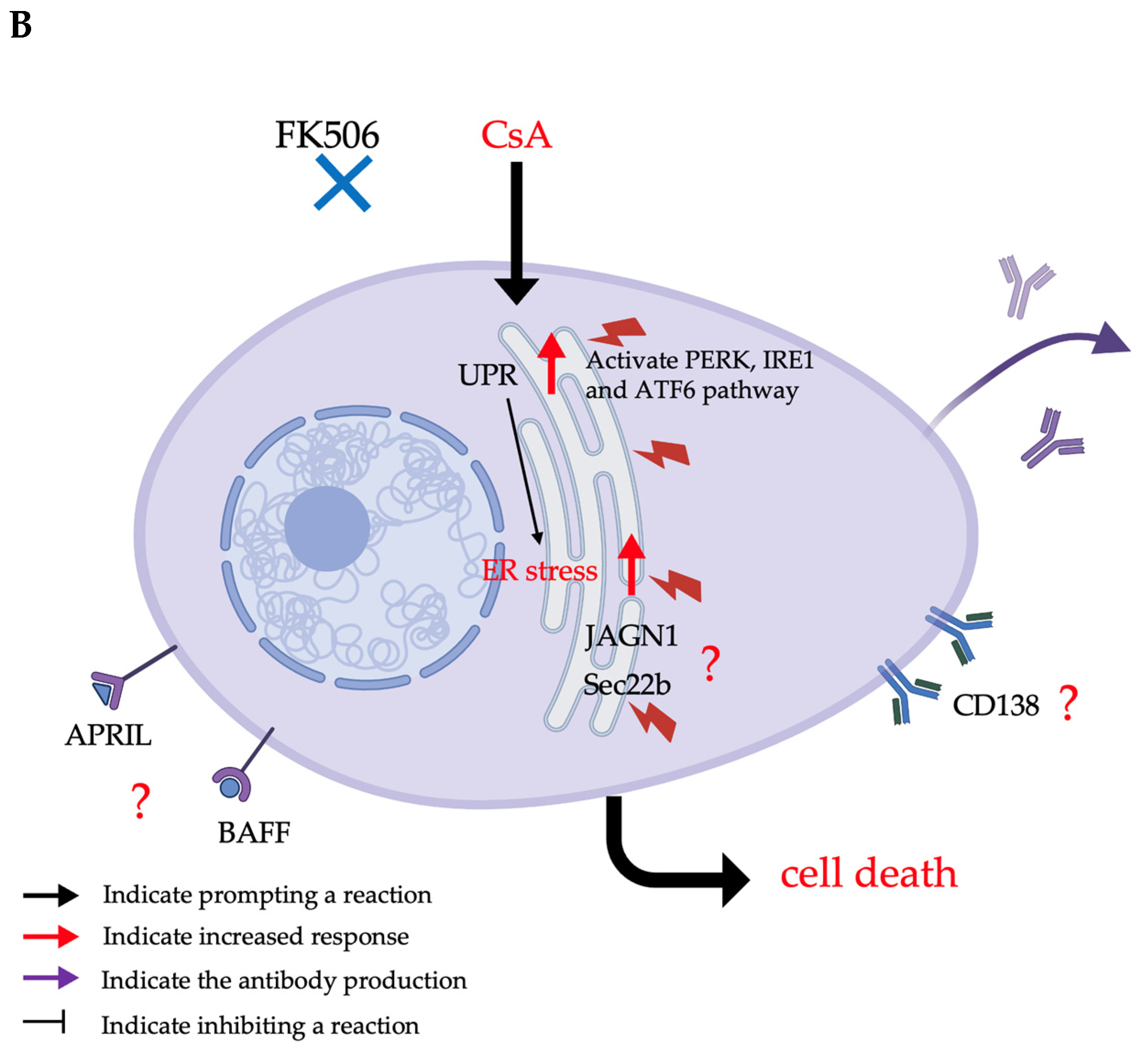
3.3. The Effects of Immunosuppressive Drugs, Particularly Calcineurin Inhibitors, on ER Stress
3.3.1. The Mechanism of Calcineurin Inhibitors
3.3.2. Side Effects of Calcineurin Inhibitors
3.3.3. The Potential cAMR Treatment of CsA Through the ER Stress Effect on Plasma Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| ESKD | Kidney disease or end-stage kidney disease |
| SRTR | Scientific Registry of Transplant Recipients |
| AMR | Antibody-mediated rejection |
| cAMR | Chronic antibody-mediated rejection |
| FDA | U.S. Food and Drug Administration |
| HLA | Human leukocyte antigen A |
| DSAs | Donor-specific antibodies |
| Mac | Membrane attack complex |
| Adcc | Antibody-dependent cellular cytotoxicity |
| Nk | Natural killer |
| IVIG | Intravenous immunoglobulin |
| IL-6 | Interleukin-6 |
| RER | Rough endoplasmic reticulum |
| SER | Smooth endoplasmic reticulum |
| UPR | Unfolded protein response |
| PERK | Protein kinase RNA-like ER kinase |
| IRE1 | Inositol-requiring enzyme 1 |
| CHOP | C/EBP homologous protein |
| Noxa | Phorbol-12-myristate-13-acetate-induced protein 1; PMAIP1 |
| ERAD | ER-associated degradation |
| RIDD | Regulated IRE1-dependent decay |
| BiP/GRP78 | Binding immunoglobulin protein or 78 kDa glucose-regulated protein |
| XBP1 | X-box-binding protein 1 |
| XBP1s | XBP1 splicing protein |
| ATF6 | Activating transcription factor 6 |
| Blimp-1 | B-lymphocyte-induced maturation protein 1 |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex |
| PI3Kδ | Phosphatidylinositol 3-kinase delta isoform |
| Sec22b | SEC22 homolog B recombinant protein |
| APRIL | Proliferation-inducing ligand |
| NF-κB | Nuclear factor kappa B |
| BAFF | B-cell activating factor belonging to the TNF family |
| BCMA | B-cell maturation antigen |
| TACI | Transmembrane activator and CAML interactor |
| CNI | Calcineurin inhibitors |
| CsA | Cyclosporine A |
| NFAT | Nuclear factor of activated T cells |
| TNF-α | Tumor necrosis factor-alpha |
| CYP3A | Cytochrome P450, family 3, subfamily A |
| RAAS | Renin–angiotensin–aldosterone system |
| EPC | Endothelial phenotypic changes |
| GCN2 | General control nonderepressible 2 |
References
- Poggio, E.D.; Augustine, J.J.; Arrigain, S.; Brennan, D.C.; Schold, J.D. Long-term kidney transplant graft survival-Making progress when most needed. Am. J. Transplant. 2021, 21, 2824–2832. [Google Scholar] [CrossRef]
- Berger, M.; Baliker, M.; Van Gelder, T.; Bohmig, G.A.; Mannon, R.B.; Kumar, D.; Chadban, S.; Nickerson, P.; Lee, L.A.; Djamali, A. Chronic Active Antibody-mediated Rejection: Opportunity to Determine the Role of Interleukin-6 Blockade. Transplantation 2024, 108, 1109–1114. [Google Scholar] [CrossRef]
- Schinstock, C.A.; Mannon, R.B.; Budde, K.; Chong, A.S.; Haas, M.; Knechtle, S.; Lefaucheur, C.; Montgomery, R.A.; Nickerson, P.; Tullius, S.G.; et al. Recommended Treatment for Antibody-mediated Rejection After Kidney Transplantation: The 2019 Expert Consensus From the Transplantion Society Working Group. Transplantation 2020, 104, 911–922. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Ricci, D.; Gidalevitz, T.; Argon, Y. The special unfolded protein response in plasma cells. Immunol. Rev. 2021, 303, 35–51. [Google Scholar] [CrossRef]
- Woodle, E.S.; Tremblay, S.; Rossi, A.; Rojas, C.C.; Alloway, R.; Roskin, K.; Allman, D.; Hildeman, D. Plasma cell targeting to prevent antibody-mediated rejection. Am. J. Transplant. 2020, 20, 33–41. [Google Scholar] [CrossRef]
- Blume, O.R.; Yost, S.E.; Kaplan, B. Antibody-mediated rejection: Pathogenesis, prevention, treatment, and outcomes. J. Transplant. 2012, 2012, 201754. [Google Scholar] [CrossRef]
- Halloran, P.F.; Wadgymar, A.; Ritchie, S.; Falk, J.; Solez, K.; Srinivasa, N.S. The significance of the anti-class I antibody response. II. Clinical and pathologic features of renal transplants with anti-class I-like antibody. Transplantation 1990, 49, 85–91. [Google Scholar] [CrossRef]
- Worthington, J.E.; Martin, S.; Al-Husseini, D.M.; Dyer, P.A.; Johnson, R.W. Posttransplantation production of donor HLA-specific antibodies as a predictor of renal transplant outcome. Transplantation 2003, 75, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, P.I.; Ozawa, M.; Castro, R. Four-year Follow-up of a Prospective Trial of HLA and MICA Antibodies on Kidney Graft Survival. Am. J. Transplant. 2007, 7, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Tanabe, T.; Tsuji, T.; Hotta, K. Mechanism and treatment for chronic antibody-mediated rejection in kidney transplant recipients. Int. J. Urol. 2023, 30, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Colvin, R.B. Antibody-mediated renal allograft rejection: Diagnosis and pathogenesis. J. Am. Soc. Nephrol. 2007, 18, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Loupy, A.; Lefaucheur, C. Antibody-Mediated Rejection of Solid-Organ Allografts. N. Engl. J. Med. 2018, 379, 1150–1160. [Google Scholar] [CrossRef]
- Chong, A.S. Mechanisms of organ transplant injury mediated by B cells and antibodies: Implications for antibody-mediated rejection. Am. J. Transplant. 2020, 20 (Suppl. S4), 23–32. [Google Scholar] [CrossRef] [PubMed]
- Solez, K.; Axelsen, R.A.; Benediktsson, H.; Burdick, J.F.; Cohen, A.H.; Colvin, R.B.; Croker, B.P.; Droz, D.; Dunnill, M.S.; Halloran, P.F.; et al. International standardization of criteria for the histologic diagnosis of renal allograft rejection: The Banff working classification of kidney transplant pathology. Kidney Int. 1993, 44, 411–422. [Google Scholar] [CrossRef]
- Racusen, L.C.; Solez, K.; Colvin, R.B.; Bonsib, S.M.; Castro, M.C.; Cavallo, T.; Croker, B.P.; Demetris, A.J.; Drachenberg, C.B.; Fogo, A.B.; et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999, 55, 713–723. [Google Scholar] [CrossRef]
- Solez, K.; Colvin, R.B.; Racusen, L.C.; Sis, B.; Halloran, P.F.; Birk, P.E.; Campbell, P.M.; Cascalho, M.; Collins, A.B.; Demetris, A.J.; et al. Banff ’05 Meeting Report: Differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’). Am. J. Transplant. 2007, 7, 518–526. [Google Scholar] [CrossRef]
- Jeong, H.J. Diagnosis of renal transplant rejection: Banff classification and beyond. Kidney Res. Clin. Pract. 2020, 39, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Loupy, A.; Haas, M.; Roufosse, C.; Naesens, M.; Adam, B.; Afrouzian, M.; Akalin, E.; Alachkar, N.; Bagnasco, S.; Becker, J.U.; et al. The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am. J. Transpl. 2020, 20, 2318–2331. [Google Scholar] [CrossRef]
- BANFF Foundation for Allograft Pathology Banff Classification for Renal Allograft Pathology. 2022. Available online: https://banfffoundation.org/central-repository-for-banff-classification-resources-3/ (accessed on 9 January 2025).
- Tambur, A.R.; Bestard, O.; Campbell, P.; Chong, A.S.; Barrio, M.C.; Ford, M.L.; Gebel, H.M.; Heidt, S.; Hickey, M.; Jackson, A.; et al. Sensitization in transplantation: Assessment of Risk 2022 Working Group Meeting Report. Am. J. Transpl. 2023, 23, 133–149. [Google Scholar] [CrossRef]
- Naesens, M.; Roufosse, C.; Haas, M.; Lefaucheur, C.; Mannon, R.B.; Adam, B.A.; Aubert, O.; Böhmig, G.A.; Callemeyn, J.; Clahsen-van Groningen, M.; et al. The Banff 2022 Kidney Meeting Report: Reappraisal of microvascular inflammation and the role of biopsy-based transcript diagnostics. Am. J. Transplant. 2024, 24, 338–349. [Google Scholar] [CrossRef]
- Tambur, A.R.; Campbell, P.; Chong, A.S.; Feng, S.; Ford, M.L.; Gebel, H.; Gill, R.G.; Kelsoe, G.; Kosmoliaptsis, V.; Mannon, R.B.; et al. Sensitization in transplantation: Assessment of risk (STAR) 2019 Working Group Meeting Report. Am. J. Transplant. 2020, 20, 2652–2668. [Google Scholar] [CrossRef]
- Cornell, L.D. Histopathologic Features of Antibody Mediated Rejection: The Banff Classification and Beyond. Front. Immunol. 2021, 12, 718122. [Google Scholar] [CrossRef]
- Loupy, A.; Mengel, M.; Haas, M. Thirty years of the International Banff Classification for Allograft Pathology: The past, present, and future of kidney transplant diagnostics. Kidney Int. 2022, 101, 678–691. [Google Scholar] [CrossRef]
- Vrielink, H.; le Poole, K. Plasmapheresis in ABO incompatible kidney transplant. Transfus. Apher. Sci. 2023, 62, 103673. [Google Scholar] [CrossRef]
- Tyden, G.; Kumlien, G.; Efvergren, M. Present techniques for antibody removal. Transplantation 2007, 84 (Suppl. S12), S27–S29. [Google Scholar] [CrossRef]
- Clark, W.F.; Huang, S.S.; Walsh, M.W.; Farah, M.; Hildebrand, A.M.; Sontrop, J.M. Plasmapheresis for the treatment of kidney diseases. Kidney Int. 2016, 90, 974–984. [Google Scholar] [CrossRef]
- Chaigne, B.; Mouthon, L. Mechanisms of action of intravenous immunoglobulin. Transfus. Apher. Sci. 2017, 56, 45–49. [Google Scholar] [CrossRef]
- Seite, J.F.; Goutsmedt, C.; Youinou, P.; Pers, J.O.; Hillion, S. Intravenous immunoglobulin induces a functional silencing program similar to anergy in human B cells. J. Allergy Clin. Immunol. 2014, 133, 181–188.e9. [Google Scholar] [CrossRef]
- Hou, Y.B.; Chang, S.; Chen, S.; Zhang, W.J. Intravenous immunoglobulin in kidney transplantation: Mechanisms of action, clinical applications, adverse effects, and hyperimmune globulin. Clin. Immunol. 2023, 256, 109782. [Google Scholar] [CrossRef]
- Moreso, F.; Crespo, M.; Ruiz, J.C.; Torres, A.; Gutierrez-Dalmau, A.; Osuna, A.; Perello, M.; Pascual, J.; Torres, I.B.; Redondo-Pachon, D.; et al. Treatment of chronic antibody mediated rejection with intravenous immunoglobulins and rituximab: A multicenter, prospective, randomized, double-blind clinical trial. Am. J. Transplant. 2018, 18, 927–935. [Google Scholar] [CrossRef]
- Mella, A.; Gallo, E.; Messina, M.; Caorsi, C.; Amoroso, A.; Gontero, P.; Verri, A.; Maletta, F.; Barreca, A.; Fop, F.; et al. Treatment with plasmapheresis, immunoglobulins and rituximab for chronic-active antibody-mediated rejection in kidney transplantation: Clinical, immunological and pathological results. World J. Transplant. 2018, 8, 178–187. [Google Scholar] [CrossRef]
- Macklin, P.S.; Morris, P.J.; Knight, S.R. A systematic review of the use of rituximab for the treatment of antibody-mediated renal transplant rejection. Transplant. Rev. 2017, 31, 87–95. [Google Scholar] [CrossRef]
- Vincenti, F.; Rostaing, L.; Grinyo, J.; Rice, K.; Steinberg, S.; Gaite, L.; Moal, M.C.; Mondragon-Ramirez, G.A.; Kothari, J.; Polinsky, M.S.; et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. N. Engl. J. Med. 2016, 374, 333–343. [Google Scholar] [CrossRef]
- Woodle, E.S.; Tremblay, S.; Brailey, P.; Girnita, A.; Alloway, R.R.; Aronow, B.; Dasgupta, N.; Ebstein, F.; Kloetzel, P.M.; Lee, M.J.; et al. Proteasomal adaptations underlying carfilzomib-resistance in human bone marrow plasma cells. Am. J. Transplant. 2020, 20, 399–410. [Google Scholar] [CrossRef]
- Eskandary, F.; Regele, H.; Baumann, L.; Bond, G.; Kozakowski, N.; Wahrmann, M.; Hidalgo, L.G.; Haslacher, H.; Kaltenecker, C.C.; Aretin, M.B.; et al. A Randomized Trial of Bortezomib in Late Antibody-Mediated Kidney Transplant Rejection. J. Am. Soc. Nephrol. 2018, 29, 591–605. [Google Scholar] [CrossRef]
- Ensor, C.R.; Yousem, S.A.; Marrari, M.; Morrell, M.R.; Mangiola, M.; Pilewski, J.M.; D’Cunha, J.; Wisniewski, S.R.; Venkataramanan, R.; Zeevi, A.; et al. Proteasome Inhibitor Carfilzomib-Based Therapy for Antibody-Mediated Rejection of the Pulmonary Allograft: Use and Short-Term Findings. Am. J. Transplant. 2017, 17, 1380–1388. [Google Scholar] [CrossRef]
- Shin, B.-H.; Everly, M.J.; Zhang, H.; Choi, J.; Vo, A.; Zhang, X.; Huang, E.; Jordan, S.C.; Toyoda, M. Impact of Tocilizumab (Anti–IL-6R) Treatment on Immunoglobulins and Anti-HLA Antibodies in Kidney Transplant Patients With Chronic Antibody-mediated Rejection. Transplantation 2020, 104, 856–863. [Google Scholar] [CrossRef]
- Stegall, M.D.; Diwan, T.; Raghavaiah, S.; Cornell, L.D.; Burns, J.; Dean, P.G.; Cosio, F.G.; Gandhi, M.J.; Kremers, W.; Gloor, J.M. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am. J. Transplant. 2011, 11, 2405–2413. [Google Scholar] [CrossRef]
- Vonbrunn, E.; Buttner-Herold, M.; Amann, K.; Daniel, C. Complement Inhibition in Kidney Transplantation: Where Are We Now? BioDrugs 2023, 37, 5–19. [Google Scholar] [CrossRef]
- Loupy, A.; Hill, G.S.; Jordan, S.C. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat. Rev. Nephrol. 2012, 8, 348–357. [Google Scholar] [CrossRef]
- Clatworthy, M.R. Targeting B cells and antibody in transplantation. Am. J. Transplant. 2011, 11, 1359–1367. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2015, 73, 79–94. [Google Scholar] [CrossRef]
- Tu, M.K.; Levin, J.B.; Hamilton, A.M.; Borodinsky, L.N. Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 2016, 59, 91–97. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47. [Google Scholar] [CrossRef]
- Rana, S.V.S. Endoplasmic Reticulum Stress Induced by Toxic Elements-a Review of Recent Developments. Biol. Trace Elem. Res. 2020, 196, 10–19. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Ingold, K.; Zumsteg, A.; Tardivel, A.; Huard, B.; Steiner, Q.G.; Cachero, T.G.; Qiang, F.; Gorelik, L.; Kalled, S.L.; Acha-Orbea, H.; et al. Identification of proteoglycans as the APRIL-specific binding partners. J. Exp. Med. 2005, 201, 1375–1383. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpedek-Kaminska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Lee, A.-H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Tellier, J.; Shi, W.; Minnich, M.; Liao, Y.; Crawford, S.; Smyth, G.K.; Kallies, A.; Busslinger, M.; Nutt, S.L. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol. 2016, 17, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Hagelkruys, A.; Wirnsberger, G.; Stadlmann, J.; Wohner, M.; Horrer, M.; Vilagos, B.; Jonsson, G.; Kogler, M.; Tortola, L.; Novatchkova, M.; et al. A crucial role for Jagunal homolog 1 in humoral immunity and antibody glycosylation in mice and humans. J. Exp. Med. 2021, 218, e20200559. [Google Scholar] [CrossRef]
- Cornelis, R.; Hahne, S.; Taddeo, A.; Petkau, G.; Malko, D.; Durek, P.; Thiem, M.; Heiberger, L.; Peter, L.; Mohr, E.; et al. Stromal Cell-Contact Dependent PI3K and APRIL Induced NF-kappaB Signaling Prevent Mitochondrial- and ER Stress Induced Death of Memory Plasma Cells. Cell Rep. 2020, 32, 107982. [Google Scholar] [CrossRef]
- Dong, X.; Qin, J.; Ma, J.; Zeng, Q.; Zhang, H.; Zhang, R.; Liu, C.; Xu, C.; Zhang, S.; Huang, S.; et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca(2+)-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell Signal 2019, 53, 68–79. [Google Scholar] [CrossRef]
- Cornelis, R.; Chang, H.D.; Radbruch, A. Keeping up with the stress of antibody production: BAFF and APRIL maintain memory plasma cells. Curr. Opin. Immunol. 2021, 71, 97–102. [Google Scholar] [CrossRef]
- McCarron, M.J.; Park, P.W.; Fooksman, D.R. CD138 mediates selection of mature plasma cells by regulating their survival. Blood 2017, 129, 2749–2759. [Google Scholar] [CrossRef]
- Bonaud, A.; Gargowitsch, L.; Gilbert, S.M.; Rajan, E.; Canales-Herrerias, P.; Stockholm, D.; Rahman, N.F.; Collins, M.O.; Taskiran, H.; Hill, D.L.; et al. Sec22b is a critical and nonredundant regulator of plasma cell maintenance. Proc. Natl. Acad. Sci. USA 2023, 120, e2213056120. [Google Scholar] [CrossRef]
- Goldfinger, M.; Shmuel, M.; Benhamron, S.; Tirosh, B. Protein synthesis in plasma cells is regulated by crosstalk between endoplasmic reticulum stress and mTOR signaling. Eur. J. Immunol. 2011, 41, 491–502. [Google Scholar] [CrossRef]
- Al Qureshah, F.; Sagadiev, S.; Thouvenel, C.D.; Liu, S.; Hua, Z.; Hou, B.; Acharya, M.; James, R.G.; Rawlings, D.J. Activated PI3Kdelta signals compromise plasma cell survival via limiting autophagy and increasing ER stress. J. Exp. Med. 2021, 218, e20211035. [Google Scholar] [CrossRef]
- Borel, J.F.; Feurer CGubler, H.U.; Stähelin, H. Biological effects of cyclosporin A: A new antilymphocytic agent. Agents Actions 1976, 43, 179–186. [Google Scholar] [CrossRef]
- Krönke, M.; Leonard, W.J.; Depper, J.M.; Arya, S.K.; Wong-Staal, F.; Gallo, R.C.; Waldmann, T.A.; Greene, W.C. Cyclosporin A inhibits T-cell growth factor gene expression at the level of mRNA transcription. Proc. Natl. Acad. Sci. USA 1984, 81, 5214–5218. [Google Scholar] [CrossRef] [PubMed]
- Trenn, G.; Taffs, R.; Hohman, R.; Kincaid, R.; Shevach, E.M.; Sitkovsky, M. Biochemical characterization of the inhibitory effect of CsA on cytolytic T lymphocyte effector functions. J. Immunol. 1989, 142, 3796–3802. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Immunosuppressive drugs- the first 50 years and a glance forward. Immunopharmacology 2000, 47, 63–83. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Moriguchi, T.; Koyasu, S.; Nishida, E. T lymphocyte activation signals for interleukin-2 production involve activation of MKK6-p38 and MKK7-SAPK/JNK signaling pathways sensitive to cyclosporin A. J. Biol. Chem. 1998, 273, 12378–12382. [Google Scholar] [CrossRef]
- Kino, T.H.H.; Miyata, S.; Inamura, N.; Nishiyama, M.; Yajima, T.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; Ochiai, T. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J. Antibiot. 1987, 40, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Tocci, M.J.; Matkovich, D.A.; Collier, K.A.; Kwok, P.; Dumont, F.; Lin, S.; Degudicibus, S.; Siekierka, J.J.; Chin, J.; Hutchinson, N.I. The immunosuppressant FK506 selectively inhibits expression of early T cell activation genes. J. Immunol. 1989, 143, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, B.; Barndt, R.J.; Chen, H.; Xin, H.; Yan, G.; Chen, L.; Cheng, D.; Heitman, J.; Zhuang, Y.; et al. FKBP12 is the only FK506 binding protein mediating T-cell inhibition by the immunosuppressant FK506. Transplantation 2002, 73, 1835–1838. [Google Scholar] [CrossRef]
- Szumilas, K.; Wilk, A.; Wisniewski, P.; Gimpel, A.; Dziedziejko, V.; Kipp, M.; Pawlik, A. Current Status Regarding Immunosuppressive Treatment in Patients after Renal Transplantation. Int. J. Mol. Sci. 2023, 24, 10301. [Google Scholar] [CrossRef]
- Karolin, A.; Genitsch, V.; Sidler, D. Calcineurin Inhibitor Toxicity in Solid Organ Transplantation. Pharmacology 2021, 106, 347–355. [Google Scholar] [CrossRef]
- Ekberg, H.; Tedesco-Silva, H.; Demirbas, A.; Vítko, Š.; Nashan, B.; Gürkan, A.; Margreiter, R.; Hugo, C.; Grinyó, J.M.; Frei, U.; et al. Reduced Exposure to Calcineurin Inhibitors in Renal Transplantation. New Engl. J. Med. 2007, 357, 2562–2575. [Google Scholar] [CrossRef]
- Bentata, Y. Tacrolimus: 20 years of use in adult kidney transplantation. What we should know about its nephrotoxicity. Artif. Organs 2020, 44, 140–152. [Google Scholar] [CrossRef]
- Zhang, W.; Egashira, N.; Masuda, S. Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities. Int. J. Mol. Sci. 2019, 20, 3210. [Google Scholar] [CrossRef]
- Wagle Shukla, A.; Lunny, C.; Mahboob, O.; Khalid, U.; Joyce, M.; Jha, N.; Nagaraja, N.; Shukla, A.M. Tremor Induced by Cyclosporine, Tacrolimus, Sirolimus, or Everolimus: A Review of the Literature. Drugs R D 2023, 23, 301–329. [Google Scholar] [CrossRef]
- King, C.P.; Cossart, A.R.; Isbel, N.M.; Campbell, S.B.; Staatz, C.E. The association between tacrolimus exposure and tremor, headache and insomnia in adult kidney transplant recipients: A systematic review. Transplant. Rev. 2024, 38, 100815. [Google Scholar] [CrossRef]
- Opalka, B.; Zolnierczuk, M.; Grabowska, M. Immunosuppressive Agents-Effects on the Cardiovascular System and Selected Metabolic Aspects: A Review. J. Clin. Med. 2023, 12, 6935. [Google Scholar] [CrossRef] [PubMed]
- Elezaby, A.; Dexheimer, R.; Sallam, K. Cardiovascular effects of immunosuppression agents. Front. Cardiovasc. Med. 2022, 9, 981838. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Myoung, H.; Kim, S.M. Review of two immunosuppressants: Tacrolimus and cyclosporine. J. Korean Assoc. Oral. Maxillofac. Surg. 2023, 49, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Hiramatsu, N.; Hayakawa, K.; Kasai, A.; Okamura, M.; Huang, T.; Yao, J.; Takeda, M.; Araki, I.; Sawada, N.; et al. Suppression of NF-kappaB by cyclosporin a and tacrolimus (FK506) via induction of the C/EBP family: Implication for unfolded protein response. J. Immunol. 2009, 182, 7201–7211. [Google Scholar] [CrossRef]
- Bouvier, N.; Flinois, J.P.; Gilleron, J.; Sauvage, F.L.; Legendre, C.; Beaune, P.; Thervet, E.; Anglicheau, D.; Pallet, N. Cyclosporine triggers endoplasmic reticulum stress in endothelial cells: A role for endothelial phenotypic changes and death. Am. J. Physiol. Ren. Physiol. 2009, 296, F160–F169. [Google Scholar] [CrossRef]
- Fedele, A.O.; Carraro, V.; Xie, J.; Averous, J.; Proud, C.G. Cyclosporin A but not FK506 activates the integrated stress response in human cells. J. Biol. Chem. 2020, 295, 15134–15143. [Google Scholar] [CrossRef]
- Yilmaz, D.E.; Kirschner, K.; Demirci, H.; Himmerkus, N.; Bachmann, S.; Mutig, K. Immunosuppressive calcineurin inhibitor cyclosporine A induces proapoptotic endoplasmic reticulum stress in renal tubular cells. J. Biol. Chem. 2022, 298, 101589. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Banff Lesion Score | Pathological Indicator | Scoring Criteria |
|---|---|---|
| i | Interstitial Inflammation | i0—No inflammation or < 10% of unscarred cortical parenchyma. i1—Inflammation in 10-25% of unscarred cortical parenchyma. i2—Inflammation in 26-50% of unscarred cortical parenchyma. i3—Inflammation in more than 50% of unscarred cortical parenchyma. |
| t | Tubulitis | t0—No tubulitis. t1—1~4 mononuclear cells per tubular cross-section or per 10 tubular epithelial cells. t2—5~10 mononuclear cells per tubular cross-section or per 10 tubular epithelial cells. t3—>10 mononuclear cells per tubular cross-section or per 10 tubular epithelial cells. |
| v | Intimal Arteritis | v0—No arteritis. v1—Mild to moderate intimal arteritis in at least 1 arterial cross-section. v2—Severe intimal arteritis with at least 25% luminal area lost in at least 1 arterial cross-section. v3—Transmural arteritis and/or arterial fibrinoid change and medial smooth muscle necrosis with lymphocytic infiltrate in vessel. |
| g | Glomerulitis | g0—No glomerulitis. g1—Glomerulitis in <25% of glomeruli. g2—Segmental or global glomerulitis in about 25–75% of glomeruli. g3—Glomerulitis (mostly global) in >75% of glomeruli. |
| ptc | Peritubular Capillaritis | ptc0—Maximum number of leukocytes < 3. ptc1—At least 1 leukocyte cell in ≥10% of cortical PTCs with 3–4 leukocytes in the most severely involved PTC. ptc2—At least 1 leukocyte in ≥10% of cortical PTCs with 5–10 leukocytes in the most severely involved PTC. ptc3—At least 1 leukocyte in ≥10% of cortical PTCs with >10 leukocytes in the most severely involved PTC. |
| C4d | C4d Deposition | C4d0—No staining of PTC and medullary vasa recta (0%). C4d1—Minimal C4d staining (>0 but <10% of PTC and medullary vasa recta). C4d2—Focal C4d staining (10–50% of PTC and medullary vasa recta). C4d3—Diffuse C4d staining (>50% of PTC and medullary vasa recta). |
| ci | Interstitial Fibrosis | ci0—Interstitial fibrosis in up to 5% of cortical area. ci1—Interstitial fibrosis in 6 to 25% of cortical area (mild interstitial fibrosis). ci2—Interstitial fibrosis in 26 to 50% of cortical area (moderate interstitial fibrosis). ci3—Interstitial fibrosis in >50% of cortical area (severe interstitial fibrosis). |
| ct | Tubular Atrophy | ct0—No tubular atrophy (defined as tubules with a thickened basement membrane or a reduction of greater than 50% in tubular diameter). ct1—Tubular atrophy (see ct0) involving up to 25% of the area of cortical tubules. ct2—Tubular atrophy (see ct0) involving 26 to 50% of the area of cortical tubules. ct3—Tubular atrophy (see ct0) involving >50% of the area of cortical tubules. |
| cv | Vascular Fibrous Intimal Thickening | cv0—No chronic vascular changes. cv1—Vascular narrowing of up to 25% luminal area by fibrointimal thickening. cv2—Vascular narrowing of 26 to 50% luminal area by fibrointimal thickening. cv3—Vascular narrowing of more than 50% luminal area by fibrointimal thickening. |
| cg | Glomerular Basement Membrane Double Contours | cg0—No double contours of glomerular basement membrane (GBM). cg1a—No double contours by light microscopy (LM), but present in ≥3 capillaries by transmission (EM) with endothelial swelling or subendothelial widening. cg1b—Double contours in 1~25% of capillary loops in the most affected glomerulus by LM. cg2—Double contours in 26–50% of capillary loops in the most affected glomerulus. cg3—Double contours in > 50% of capillary loops in the most affected glomerulus. |
| mm | Mesangial Matrix Expansion | mm0—Mild increase in mesangial matrix. mm1—Moderate increase in up to 25% of non-sclerotic glomeruli. mm2—Moderate increase in 26–50% of non-sclerotic glomeruli. mm3—Moderate increase in >50% of non-sclerotic glomeruli. |
| ah | Arteriolar Hyalinosis | ah0—No hyaline thickening. ah1—Mild to moderate thickening in ≥1 arteriole. ah2—Moderate to severe thickening in >1 arteriole. ah3—Severe thickening in many arterioles. |
| aah | Hyaline Arteriolar Thickening | aah0—No typical lesions of calcineurin inhibitor-related arteriolopathy. aah1—Hyaline deposits in 1 arteriole, no circumferential involvement. aah2—Hyaline deposits in >1 arteriole, no circumferential involvement. aah3—Circumferential hyaline deposits in arterioles, regardless of number. |
| ti | Total Inflammation | ti0—No or minimal inflammation (<10%). ti1—10–25% of cortex inflamed. ti2—26~50% of cortex inflamed. ti3—>50% of cortex inflamed due to interstitial inflammation and tubulitis |
| i-IFTA | Inflammation in Areas of Interstitial Fibrosis and Tubular Atrophy | i-IFTA0—No inflammation or <10% of cortex with fibrosis and atrophy. i-IFTA1—Inflammation in 10~25% of cortex with fibrosis and atrophy. i-IFTA2—Inflammation in 26~50% of cortex with fibrosis and atrophy. i-IFTA3—Inflammation in >50% of cortex with fibrosis and atrophy. |
| t-IFTA | Tubulitis in Areas of Interstitial Fibrosis | t-IFTA0—No mononuclear cells or single focus of tubulitis. t-IFTA1—2+ foci with 1~4 mononuclear cells/tubule in the most affected focus. t-IFTA2—2+ foci with 5~10 mononuclear cells/tubule in the most affected focus. t-IFTA3—2+ foci with >10 mononuclear cells/tubule in the most affected focus. |
| pvi | Polyomavirus Load | pvi0—No positive nuclei in any tubules/ducts. pvi1—≤1% of all tubules/ducts. pvi2—>1% to ≤10% of all tubules/ducts. pvi3—>10% of all tubules/ducts. |
| Treatment Strategy | Representative Drug/Method | Purpose | Effectiveness | Side Effects |
|---|---|---|---|---|
| Antibody Removal Therapy | Plasma exchange | Remove circulating antibodies, reduce antibody levels in the short term | Effective in the short term | Temporary effect, requires repeated procedures |
| IVIG | Neutralize antibodies, inhibit complement response | |||
| B-cell Inhibitors | Rituximab (anti-CD20 antibody) | Reduce B cells, lower antibody production | Effective for some patients | Increased risk of infection |
| Emerging Therapies | Bortezomib, carfilzomib | Suppress plasma cells, reduce antibody production | Effectively reduces inflammation and tissue damage | Increased infection risk, peripheral neuropathy, gastrointestinal discomfort |
| Tocilizumab (anti-IL-6) | Inhibit IL-6 activation, reduce inflammatory response | Increased infection risk | ||
| Complement Inhibitor | Eculizumab | Inhibit complement activation, reduce immune response | Effective for reducing antibody-mediated rejection and atypical hemolytic uremic syndrome | Increased risk of infections |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-Y.; Lee, C.-Y.; Chen, J.-H.; Chiang, C.-K. Chronic Antibody-Mediated Rejection and Plasma Cell ER Stress: Opportunities and Challenges with Calcineurin Inhibitors. Int. J. Mol. Sci. 2025, 26, 2711. https://doi.org/10.3390/ijms26062711
Tsai C-Y, Lee C-Y, Chen J-H, Chiang C-K. Chronic Antibody-Mediated Rejection and Plasma Cell ER Stress: Opportunities and Challenges with Calcineurin Inhibitors. International Journal of Molecular Sciences. 2025; 26(6):2711. https://doi.org/10.3390/ijms26062711
Chicago/Turabian StyleTsai, Ching-Yi, Chih-Yuan Lee, Jia-Huang Chen, and Chih-Kang Chiang. 2025. "Chronic Antibody-Mediated Rejection and Plasma Cell ER Stress: Opportunities and Challenges with Calcineurin Inhibitors" International Journal of Molecular Sciences 26, no. 6: 2711. https://doi.org/10.3390/ijms26062711
APA StyleTsai, C.-Y., Lee, C.-Y., Chen, J.-H., & Chiang, C.-K. (2025). Chronic Antibody-Mediated Rejection and Plasma Cell ER Stress: Opportunities and Challenges with Calcineurin Inhibitors. International Journal of Molecular Sciences, 26(6), 2711. https://doi.org/10.3390/ijms26062711