
Identification of Elite Alleles and Candidate Genes for the Cotton Boll Opening Rate via a Genome-Wide Association Study

 ,
,
Abstract
1. Introduction
2. Results
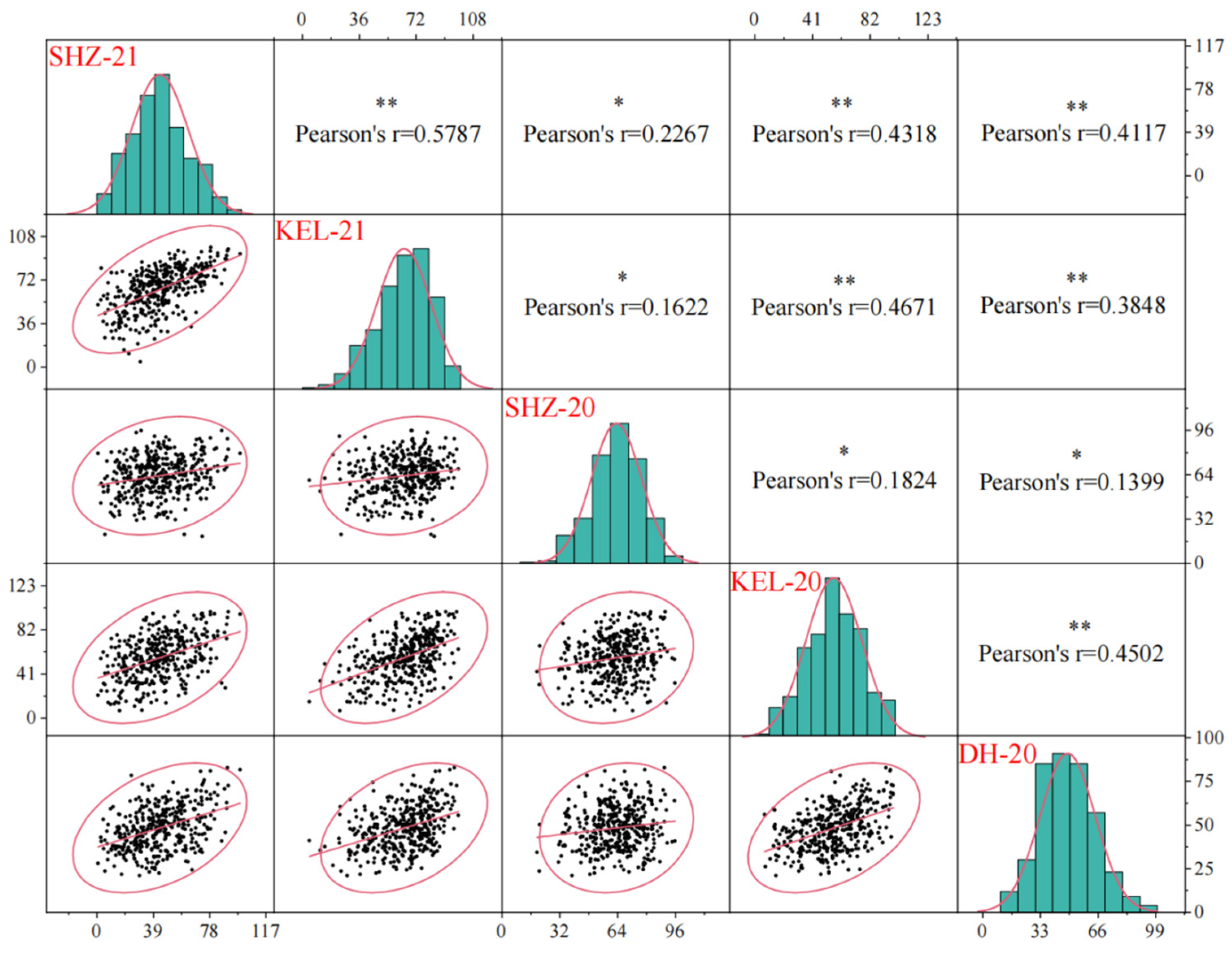
2.1. BOR Varies Widely in the Natural Population
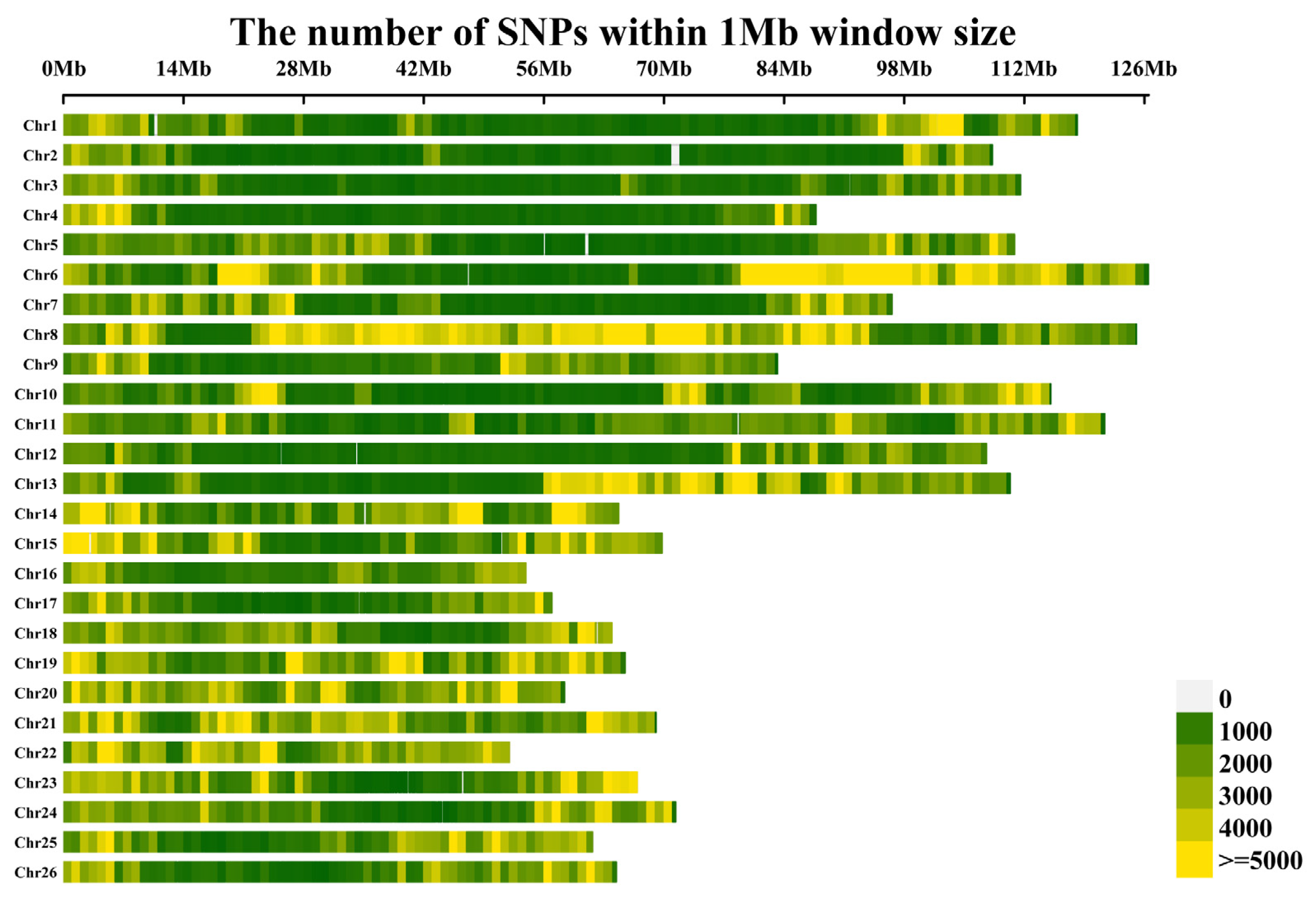
2.2. SNP Genotyping
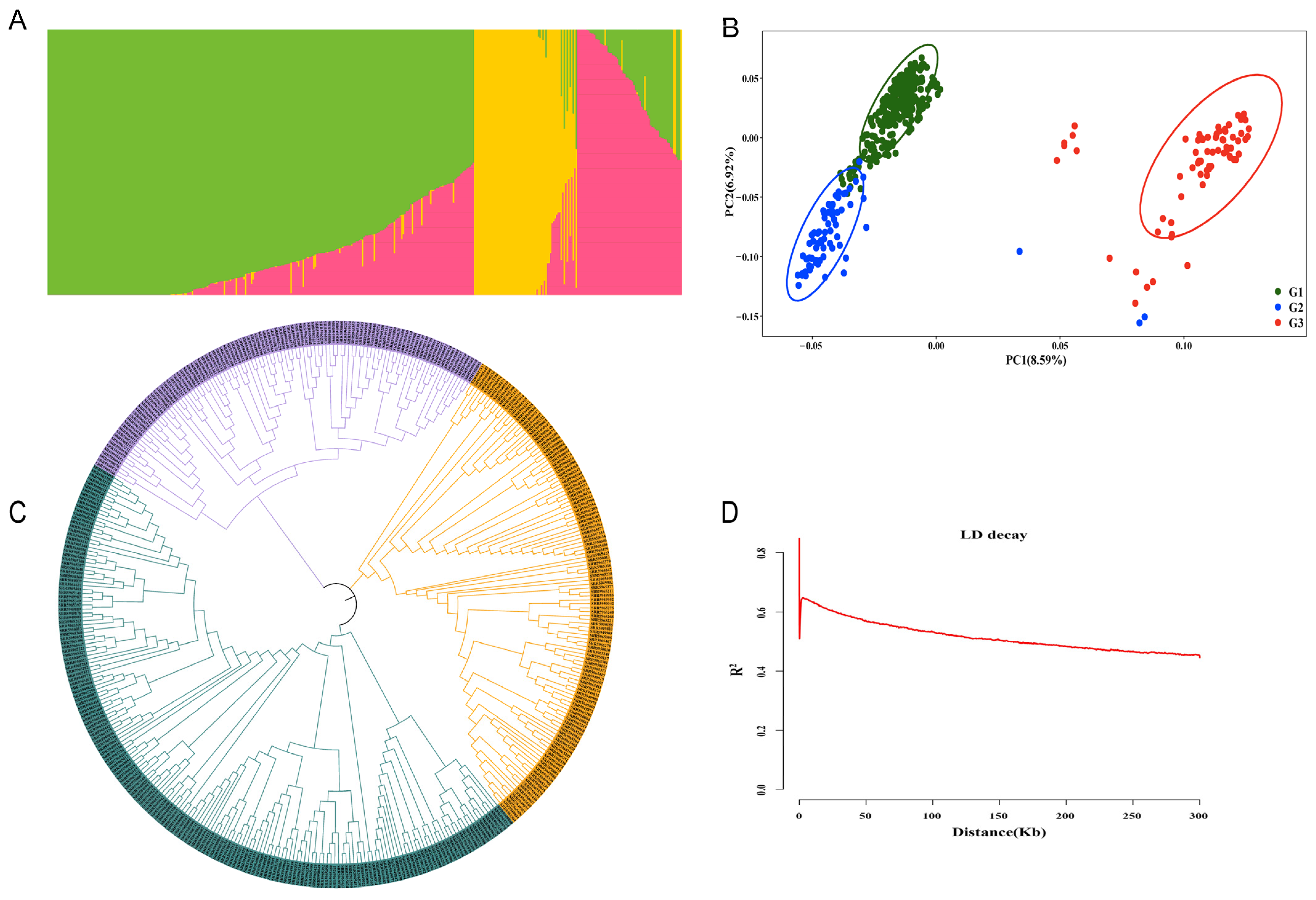
2.3. Population Structure and LD Analysis
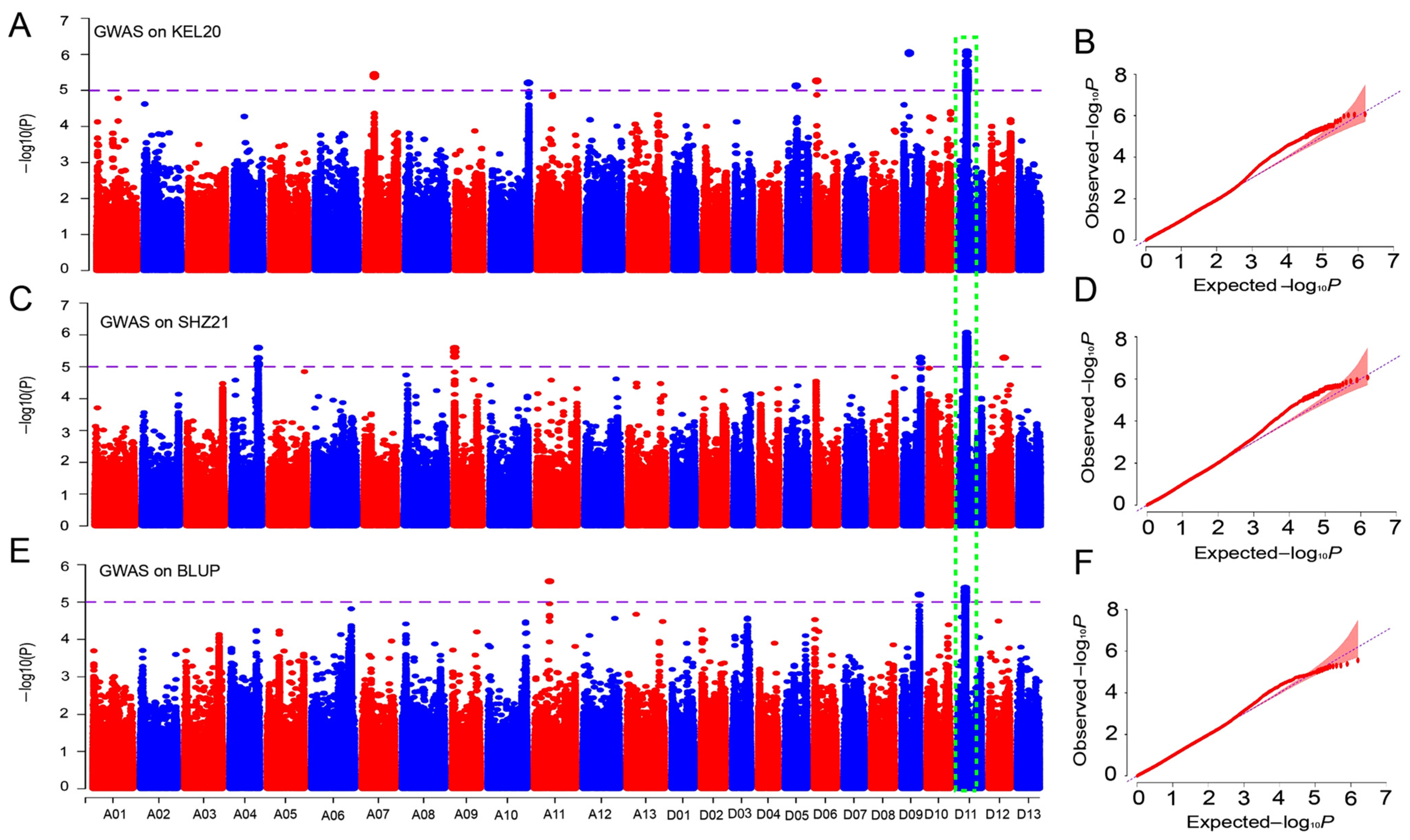
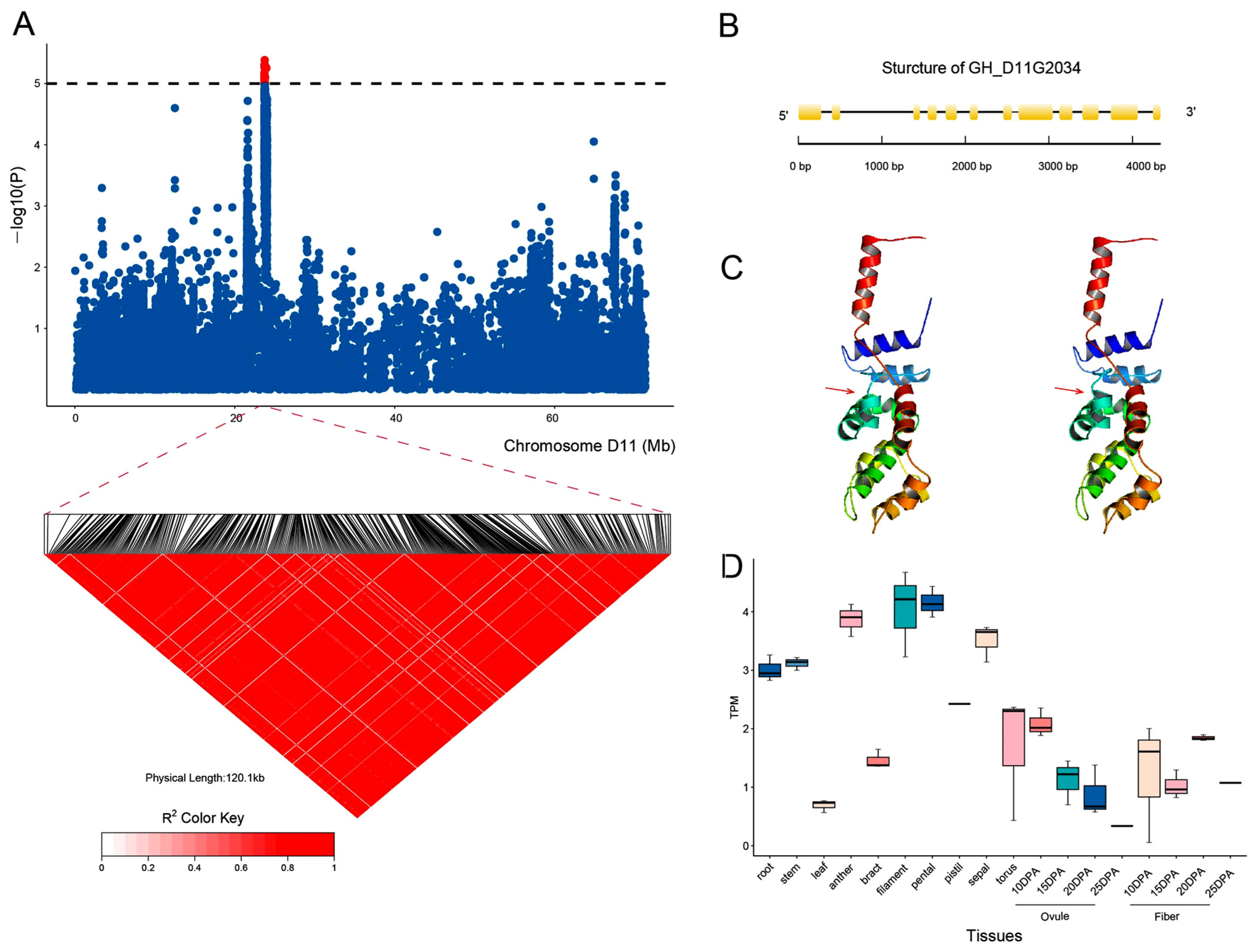
2.4. GWAS for the BOR Trait in Upland Cotton
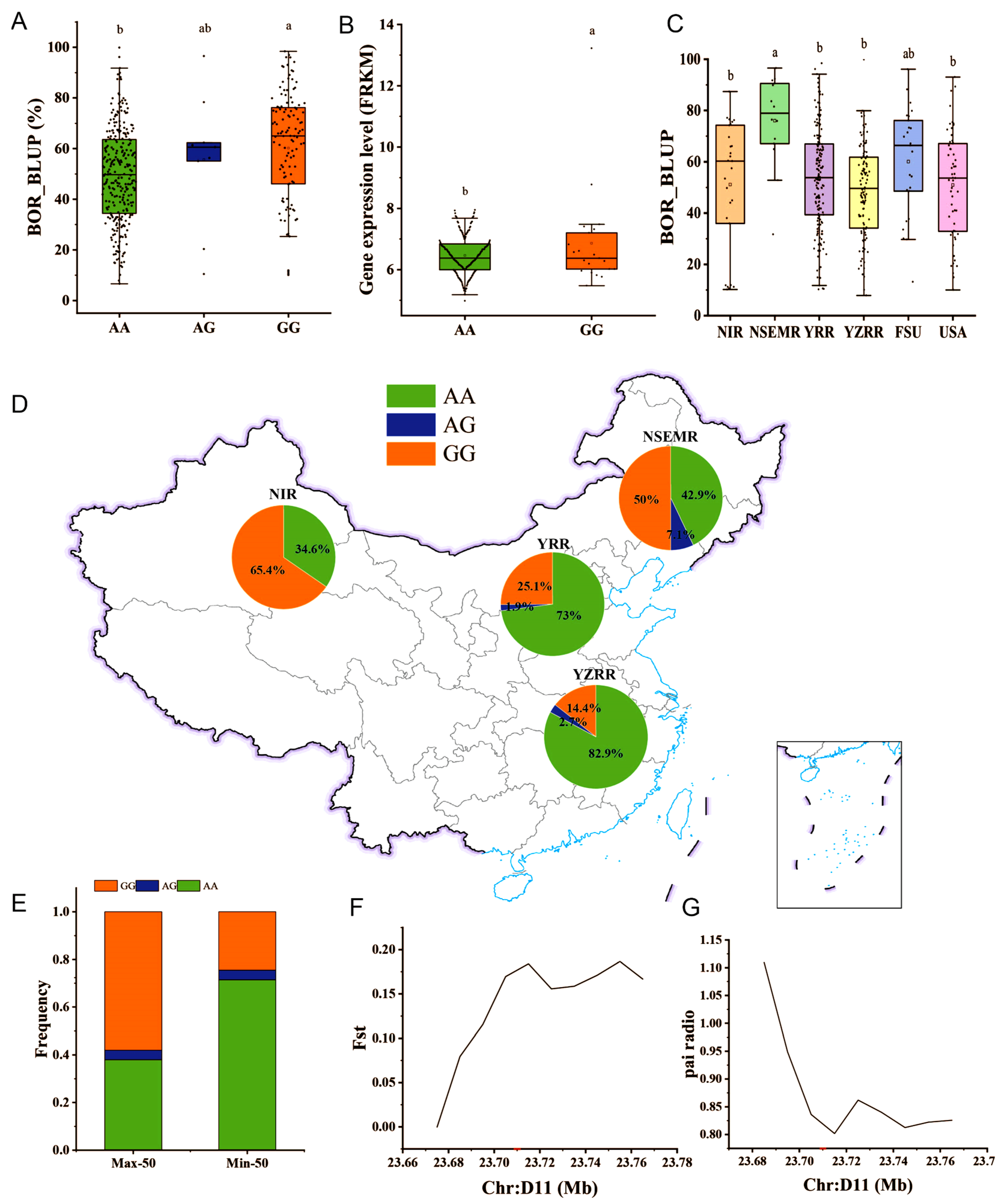
2.5. Identification of Candidate Genes for the BOR Trait
2.6. GH_D11G2034 May Undergo Artificial Selection in Upland Cotton
3. Discussion
4. Materials and Methods
4.1. Experimental Materials
4.2. Field Experimental Design
4.3. Statistical Analysis of Phenotypic Data
4.4. SNP Identification
4.5. Population Structure and LD Analyses
4.6. GWAS for BOR in Upland Cotton
4.7. Identification of Elite Alleles and Candidate Genes for the BOR Trait
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mounica, D.; Swaroopa, V.J. Innovative frontline demonstrations in tribal areas to enhance Bt cotton yield and income through integrated pest management. Int. J. Farm Sci. 2016, 6, 177–182. [Google Scholar]
- Song, X.-L.; Zhang, T.-Z. Identification of quantitative trait loci controlling seed physical and nutrient traits in cotton. Cotton Sci. Res. 2008, 20, 32. [Google Scholar] [CrossRef]
- Wang, C.-X.; Yuan, W.-M.; Liu, J.-J.; Xie, X.-Y.; Ma, Q.; Ju, J.-S.; Chen, D.; Wang, N.; Feng, K.-Y.; Su, J.-J. Comprehensive evaluation and breeding evolution of early maturing upland cotton varieties in the Northwest Inland of China. Sci. Agric. Sin. 2023, 56, 1–16. [Google Scholar]
- Lu, X.-R.; Jia, X.-Y.; Niu, J.-H. The present situation and prospects of cotton industry development in China. Sci. Agric. Sin. 2018, 51, 26–36. [Google Scholar]
- Yu, S.-X. Breeding direction in Xinjiang cotton area at the beginning of 21st century. China Cotton 1999, 26, 2–3. [Google Scholar]
- Dong, C.-G.; Wang, J.; Zhou, X.-F.; Ma, X.-M.; Li, S.-X.; Wang, X.-W.; Xiao, G.-S.; Li, B.-C. Inheritance of earliness traits in Xinjiang early-maturity upland cotton (G. hirsutum L.). Acta Agric. Boreali-Occident. Sin. 2014, 23, 96–101. [Google Scholar]
- Xie, X.-Y.; Wang, K.-H.; Qin, X.-X.; Wang, C.-X.; Shi, C.-H.; Ning, X.-Z.; Yang, Y.-L.; Qin, J.-H.; Li, C.-Z.; Ma, Q.; et al. Restricted two-stage multi-locus genome-wide association analysis and candidate gene prediction of boll opening rate in upland cotton. Sci. Agric. Sin. 2022, 55, 248–264. [Google Scholar]
- Feng, L.; Dai, J.-L.; Tian, L.-W.; Zhang, H.-J.; Li, W.-J.; Dong, H.-Z. Review of the technology for high-yielding and efficient cotton cultivation in the northwest inland cotton-growing region of China. Field Crops Res. 2017, 208, 18–26. [Google Scholar] [CrossRef]
- Gao, L.-L. Study of Defoliants Spraying Time on Cotton Physiological Mechanism; Xinjiang Agricultural University: Ürümqi, China, 2016. [Google Scholar]
- Wang, X.-R.; Zhang, H.-H.; Hu, L.-T.; Pang, N.-C.; Dong, Q.; Gui, H.-P.; Song, M.-Z.; Zhang, X.-L. Screening for suitable cotton harvest aids in Xinjiang. China Cotton 2018, 45, 8–14. [Google Scholar]
- Ma, Q.; Li, J.-L.; Ning, X.-Z.; Liu, P.; Deng, F.-J.; Lin, H. Analysis on the effect of chemical defoliation and ripening of Xinluzao 60 under two kinds of mechanical cotton-picking planting models. J. Chin. Agric. Mech. 2020, 41, 139–144. [Google Scholar]
- Li, L.-B.; Zhang, C.; Huang, J.-Q.; Liu, Q.-B.; Wei, H.-L.; Wang, H.-T.; Liu, G.-Y.; Gu, L.-J.; Yu, S.-X. Genomic analyses reveal the genetic basis of early maturity and identification of loci and candidate genes in upland cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2021, 19, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Lin, H.; Su, J.-J.; Li, J.-L.; Ning, X.-Z.; Xu, S.-Z.; Liu, W.-H.; Zhao, F.-X. Flower-boll two-stage accurate identification method for cotton variety boll opening concentration degree. China Patent 113940248B, 13 May 2022. [Google Scholar]
- Risch, N.; Merikangas, K. The future of genetic studies of complex human diseases: An epidemiologic perspective. Epidemiology 1998, 9, 350–354. [Google Scholar]
- Larsson, S.J.; Lipka, A.E.; Buckler, E.S. Lessons from dwarf 8 on the strengths and weaknesses of structured association mapping. PLoS Genet. 2013, 9, e1003246. [Google Scholar] [CrossRef]
- Sun, Z.-W.; Wang, X.-F.; Liu, Z.-W.; Gu, Q.-S.; Zhang, Y.; Li, Z.-K.; Yang, J.; Wu, J.-H.; Wu, L.-Q.; Zhang, G.-Y.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Atwell, S.; Huang, Y.-S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.-T.; et al. Genome-wide association study of 107 phenotypes in a conlinon set of Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Singh, S.; Sehgal, D.; Kumar, S.; Arif, M.A.R.; Vikram, P.; Sansaloni, C.P.; Fuentes-Dávila, G.; Ortiz, C. GWAS revealed a novel resistance locus on chromosome 4D for the quarantine disease Karnal bunt in diverse wheat pre-breeding germplasm. Sci. Rep. 2020, 10, 5999. [Google Scholar] [CrossRef]
- Li, Q.; Lu, X.-L.; Wang, C.-J.; Shen, L.; Dai, L.-P.; He, J.-L.; Yang, L.; Li, P.-Y.; Hong, Y.; Zhang, Q. Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen-deficiency tolerance in rice. Crop J. 2021, 10, 942–951. [Google Scholar] [CrossRef]
- Yang, Y.-X.; Zhang, C.; Zhu, D.; He, H.-Y.; Wei, Z.-R.; Yuan, Q.-L.; Li, X.-X.; Gao, X.; Zhang, B.; Gao, H.-S.; et al. Identifying candidate genes and patterns of heat-stress response in rice using a genome-wide association study and transcriptome analyses. Crop J. 2022, 10, 1633–1643. [Google Scholar] [CrossRef]
- He, K.-H.; Xu, S.-T.; Zhang, X.-H.; Li, Y.-A.; Chang, L.-G.; Wang, Y.-H.; Shi, Y.-Q.; Cui, T.-T.; Dong, Y.; Lan, T.-R.; et al. Mining of candidate genes for nitrogen use efficiency in maize based on genome-wide association study. Mol. Breed. 2020, 40, 83. [Google Scholar] [CrossRef]
- Ruanjaichon, V.; Khammona, K.; Thunnom, B.; Suriharn, K.; Kerdsri, C.; Aesomnuk, W.; Yongsuwan, A.; Chaomueang, N.; Thammapichai, P.; Arikit, S.; et al. Identification of gene associated with sweetness in corn (Zea mays L.) by genome-wide association study (GWAS) and development of a functional SNP marker for predicting sweet corn. Plants 2021, 10, 1239. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.-C.; Fan, Y.-H.; Li, Y.-H.; Liu, M.; Sun, W.; Duan, H.-C.; Yu, M.-N.; Chang, W.; Niu, Y.; Li, X.-D.; et al. Petal size in rapeseed: Novel QTL and candidate genes detected through genome-wide association study and transcriptome comparison. J. Exp. Bot. 2021, 72, 3597–3610. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Su, B.; Ibrahim, S.; Kuang, L.-Q.; Tian, Z.; Wang, X.-F.; Wang, H.-Z.; Dun, X.-L. Deciphering the genetic basis of root and biomass traits in rapeseed (Brassica napus L.) through the integration of GWAS and RNA-seq under nitrogen stress. Int. J. Mol. Sci. 2022, 23, 7958. [Google Scholar] [CrossRef]
- Wen, Z.-X.; Tan, R.-J.; Zhang, S.-C.; Collins, P.J.; Yuan, J.-Z.; Du, W.-Y.; Gu, C.-H.; Ou, S.-J.; Song, Q.-J.; CharlesAn, Y.Q.; et al. Integrating GWAS and gene expression data for functional characterization of resistance to white mold in soybean. Plant Biotechnol. J. 2018, 16, 1825–1835. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.-Y.; Tian, R.; Kong, Y.-B.; Chu, J.-H.; Xing, X.-Z.; Du, H.; Jin, H.; Li, X.-H.; Zhang, C.-Y. Genome-wide association and linkage mapping strategies reveal genetic loci and candidate genes of phosphorus utilization in soybean. J. Integr. Agric. 2022, 21, 2521–2537. [Google Scholar] [CrossRef]
- Lee, Y.J.; Yang, B.; Kim, W.; Kim, J.; Kwon, S.J.; Kim, J.H.; Ahn, J.W.; Kim, S.H.; Rha, E.; Ha, B. Genome-wide association study (GWAS) of the agronomic traits and phenolic content in sorghum (Sorghum bicolor L.) genotypes. Agronomy 2023, 13, 1449. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, X.; Wang, X.; Hathorn, A.; Hunt, C.; Cruickshank, A.W.; Oosterom, E.J.V.; Godwin, I.D.; Mace, E.S.; Jordan, D.R. Large-scale GWAS in sorghum reveals common genetic control of grain size among cereals. Plant Biotechnol. J. 2019, 18, 1093–1105. [Google Scholar] [CrossRef]
- Wang, C.-X.; Liu, J.-J.; Xie, X.-Y.; Wang, J.; Ma, Q.; Chen, P.-Y.; Yang, D.-L.; Ma, X.-F.; Hao, F.-S.; Su, J.-J. GhAP1-D3 positively regulates flowering time and early maturity with no yield and fiber quality penalties in upland cotton. J. Integr. Plant Biol. 2022, 65, 985–1002. [Google Scholar] [CrossRef]
- Li, C.-Q.; Wang, Y.-Y.; Ai, N.-J.; Li, Y.; Song, J.-F. A genome-wide association study of early-maturation traits in upland cotton based on the cottonSNP80K array. J. Integr. Plant Biol. 2018, 60, 970–985. [Google Scholar] [CrossRef]
- Ma, Z.-Y.; He, S.-P.; Wang, X.-F.; Sun, J.-L.; Zhang, Y.; Zhang, G.-Y.; Wu, L.-Q.; Li, Z.-K.; Liu, Z.-H.; Sun, G.-F.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Wang, N.; Huang, C.; Wang, M.-J.; Zhang, X.-L.; Lin, Z.-X. Population genomics reveals a fine-scale recombination landscape for genetic improvement of cotton. Plant J. 2019, 99, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Dong, C.-G.; Wang, J.; Wang, Y.-Y.; Li, C.-Q. Genome-wide association study reveals the genetic control underlying node of the first fruiting branch and its height in upland cotton (Gossypium hirsutum L.). Euphytica 2019, 215, 35. [Google Scholar] [CrossRef]
- Cao, C.; Yang, P.; Tang, C.; Liang, F.; Tian, J.; Zhang, Y.; Zhang, W. Morphological characteristic extraction of unopened cotton bolls using image analysis and geometric modeling methods. Comput. Electron. Agric. 2025, 232, 110094. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, Q.; Tian, J.; Zhang, Y.; Jiang, C.; Zhang, W. The high genetic yield of Xinjiang cotton is associated with improvements in boll-leaf system photosynthesis. Field Crops Res. 2023, 304, 109176. [Google Scholar] [CrossRef]
- Shui, G.; Lin, H.; Ma, X.; Zhu, B.; Han, P.; Aini, N.; Guo, C.; Wu, Y.; Pan, Z.; You, C.; et al. Identification of SSR markers linked to the abscission of cotton boll traits and mining germplasm in Cotton. J. Cotton Res. 2024, 7, 20. [Google Scholar] [CrossRef]
- Han, Z.-G.; Chen, H.; Cao, Y.-W.; He, L.; Si, Z.-F.; Hu, Y.; Lin, H.; Ning, X.-Z.; Li, J.-L.; Ma, Q. Genomic insights into genetic improvement of upland cotton in the world’s largest growing region. Ind. Crops Prod. 2022, 183, 114929. [Google Scholar] [CrossRef]
- Tian, X.M.; Li, X.Y.; Lv, X.; Li, B.C.; Chen, G.W. Xinjiang Cotton Theory and Modern Cotton Technology; Science Press: Beijing, China, 2016. [Google Scholar]
- Li, L.-B.; Zhao, S.-Q.; Su, J.-J.; Fan, S.-L.; Pang, C.-Y.; Wei, H.-L.; Wang, H.-T.; Gu, L.-J.; Zhang, C.; Liu, G.-Y. High-density genetic linkage map construction by F2 populations and QTL analysis of early-maturity traits in upland cotton (Gossypium hirsutum L.). PLoS ONE 2017, 12, e0182918. [Google Scholar] [CrossRef]
- Wang, F.-Y.; Han, H.-Y.; Lin, H.; Chen, B.; Kong, X.-H.; Ning, X.-Z.; Wang, X.-W.; Yu, Y.; Liu, J.-D. Effects of planting patterns on yield, quality, and defoliation in machine-harvested cotton. J. Integr. Agric. 2019, 18, 2019–2028. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.-H.; Chen, J.-D.; Liu, B.-L.; Zhang, Z.-Y.; Guan, X.-Y.; Chen, S.-Q.; Zhou, B.-L.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Y.; Chen, P.-Y.; Zhou, J.; Zhang, C.-Y.; Song, Z.-Q.; Huo, X.-H.; Du, Z.-H.; Gong, J.-W.; Zhao, C.J.; et al. Genome-wide association study reveals novel quantitative trait loci and candidate genes of lint percentage in upland cotton based on the CottonSNP80K array. Theor. Appl. Genet. 2022, 135, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-J.; Wang, C.-X.; Yang, D.-L.; Shi, C.-H.; Zhang, A.; Ma, Q.; Liu, J.-J.; Zhang, X.-L.; Huang, L.; Ma, X.-F. Decryption of favourable haplotypes and potential candidate genes for five fibre quality properties using a relatively novel genome-wide association study procedure in upland cotton. Ind. Crops Prod. 2020, 158, 113004. [Google Scholar] [CrossRef]
- Wen, T.-W.; Dai, B.-S.; Wang, T.; Liu, X.-X.; You, C.-Y.; Lin, Z.-X. Genetic variations in plant architecture traits in cotton (Gossypium hirsutum) revealed by a genome-wide association study. Crop J. 2019, 7, 209–216. [Google Scholar] [CrossRef]
- Su, J.-J.; Li, D.-D.; Yuan, W.-M.; Li, Y.; Ju, J.-S.; Wang, N.; Ling, P.-J.; Feng, K.-Y.; Wang, C.-X. Integrating RTM-GWAS and meta-QTL data revealed genomic regions and candidate genes associated with the first fruit branch node and its height in upland cotton. Theor. Appl. Genet. 2024, 137, 207. [Google Scholar] [CrossRef]
- Abdelraheem, A.; Elassbli, H.; Zhu, Y.; Kuraparthy, V.; Zhang, J. A genome-wide association study uncovers consistent quantitative trait loci for resistance to Verticillium wilt and Fusarium wilt race 4 in the US Upland cotton. Theor. Appl. Genet. 2020, 133, 563–577. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Buckler, E.S. TASSEL, software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Li, C.-Q.; Wang, X.-Y.; Dong, N.; Zhao, H.-H.; Xia, Z.; Wang, R.; Converse, R.L.; Wang, Q.-L. QTL analysis for early-maturing traits in cotton using two upland cotton (Gossypium hirsutum L.) crosses. Breed. Sci. 2013, 63, 154–163. [Google Scholar] [CrossRef]
- Jia, X.-Y.; Pang, C.-Y.; Wei, H.-L.; Wang, H.-T.; Ma, Q.-F.; Yang, J.-L.; Cheng, S.-S.; Su, J.-J.; Fan, S.-L.; Song, M.-Z. High-density linkage map construction and QTL analysis for earliness-related traits in Gossypium hirsutum L. BMC Genom. 2016, 17, 909. [Google Scholar] [CrossRef]
- Moose, S.P.; Dudley, J.W.; Rocheford, T.R. Maize selection passes the century mark: A unique resource for 21st century genomics. Trends Plant Sci. 2004, 9, 358–364. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, H.-S.; Fu, Z.-J.; Chen, H.; Lin, Y.-A.; Yan, P.-S.; Li, W.-H.; Xie, H.-L.; Guo, Z.-Y.; Zhang, X.-H. Genetic-based dissection of arsenic accumulation in maize using a genome-wide association analysis method. Plant Biotechnol. J. 2018, 16, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.-D.; Fang, L.; Zhang, Z.-Y.; Ma, W.; Niu, Y.-C.; Ju, L.-Z.; Deng, J.-Q.; Zhao, T.; Lian, J.-M. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows- Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Petr, D.; Adam, A.; Goncalo, A.; Cornelis, A.A.; Eric, B.; Mark, A.D.; Robert, E.H.; Gerton, L.; Gabor, T.M.; Stephen, T.S. The variant call format and VCF tools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zhou, H.; Alexander, D.; Lange, K. A quasi-Newton acceleration for highdimensional optimization algorithms. Stat. Comput. 2011, 21, 261–273. [Google Scholar] [CrossRef]
- Lee, T.H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef]
- Yu, G.-C.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Yang, J.; Lee, H.S.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Chang, C.-C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Sorrce Softw. 2018, 3, 731. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environments a | Max (%) | Min (%) | Mean (%) | SD b | CV c (%) | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|
| SHZ-20 | 95.77 | 20.77 | 63.77 | 13.89 | 21.78 | −0.27 | −0.23 |
| KEL-20 | 99.02 | 7.06 | 57.12 | 20.36 | 35.64 | −0.13 | −0.52 |
| DH-20 | 95.19 | 12.85 | 49.1 | 15.73 | 32.04 | 0.26 | −0.18 |
| SHZ-21 | 98.99 | 0.86 | 44.92 | 20.13 | 44.81 | 0.23 | −0.42 |
| KEL-21 | 98.77 | 12.98 | 62.57 | 17.4 | 27.81 | −0.17 | −0.68 |
| BLUP | 83.57 | 28.44 | 55.48 | 9.64 | 17.38 | 0.2 | −0.1 |
| Chr. | SNP Number | Chr Length (Mb) | SNP Density (Kb) | Chr. | SNP Number | Chr. Length (Mb) | SNP Density (Kb) |
|---|---|---|---|---|---|---|---|
| A01 | 174,575 | 118.15 | 1.48 | D01 | 183,419 | 64.70 | 2.84 |
| A02 | 122,573 | 108.27 | 1.13 | D02 | 179,130 | 69.78 | 2.57 |
| A03 | 245,146 | 111.59 | 2.2 | D03 | 94,075 | 53.9 | 1.75 |
| A04 | 91,660 | 87.7 | 1.05 | D04 | 84,976 | 56.93 | 1.49 |
| A05 | 162,903 | 110.84 | 1.47 | D05 | 141,897 | 63.92 | 2.22 |
| A06 | 351,101 | 126.49 | 2.78 | D06 | 177,358 | 65.46 | 2.71 |
| A07 | 144,608 | 96.6 | 1.5 | D07 | 169,896 | 58.42 | 2.91 |
| A08 | 397,496 | 125.06 | 3.18 | D08 | 183,719 | 69.08 | 2.66 |
| A09 | 140,054 | 83.22 | 1.68 | D09 | 169,952 | 52.00 | 3.27 |
| A10 | 177,383 | 115.1 | 1.54 | D10 | 143,111 | 66.88 | 2.14 |
| A11 | 184,666 | 121.38 | 1.52 | D11 | 130,046 | 71.36 | 1.82 |
| A12 | 119,840 | 107.57 | 1.11 | D12 | 136,328 | 61.69 | 2.21 |
| A13 | 229,546 | 110.37 | 2.08 | D13 | 117,171 | 64.45 | 1.82 |
| A01~A13 | 2,541,551 | D01~D13 | 1,911,078 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Q.; Zhang, X.; Li, J.; Ning, X.; Xu, S.; Liu, P.; Guo, X.; Yuan, W.; Xie, B.; Wang, F.; et al. Identification of Elite Alleles and Candidate Genes for the Cotton Boll Opening Rate via a Genome-Wide Association Study. Int. J. Mol. Sci. 2025, 26, 2697. https://doi.org/10.3390/ijms26062697
Ma Q, Zhang X, Li J, Ning X, Xu S, Liu P, Guo X, Yuan W, Xie B, Wang F, et al. Identification of Elite Alleles and Candidate Genes for the Cotton Boll Opening Rate via a Genome-Wide Association Study. International Journal of Molecular Sciences. 2025; 26(6):2697. https://doi.org/10.3390/ijms26062697
Chicago/Turabian StyleMa, Qi, Xueli Zhang, Jilian Li, Xinzhu Ning, Shouzhen Xu, Ping Liu, Xuefeng Guo, Wenmin Yuan, Bin Xie, Fuxiang Wang, and et al. 2025. "Identification of Elite Alleles and Candidate Genes for the Cotton Boll Opening Rate via a Genome-Wide Association Study" International Journal of Molecular Sciences 26, no. 6: 2697. https://doi.org/10.3390/ijms26062697
APA StyleMa, Q., Zhang, X., Li, J., Ning, X., Xu, S., Liu, P., Guo, X., Yuan, W., Xie, B., Wang, F., Wang, C., Su, J., & Lin, H. (2025). Identification of Elite Alleles and Candidate Genes for the Cotton Boll Opening Rate via a Genome-Wide Association Study. International Journal of Molecular Sciences, 26(6), 2697. https://doi.org/10.3390/ijms26062697