
Exploiting Paradoxical Activation of Oncogenic MAPK Signaling by Targeting Mitochondria to Sensitize NRAS Mutant-Melanoma to Vemurafenib

 , , , , ,
, , , , ,  , and
, and 
Abstract

1. Introduction
2. Results
2.1. Vemurafenib Has Low Cytotoxicity in NRASQ61R-Mutated SK-MEL-147 Melanoma Cells and Conversely Induces Its Proliferation
2.2. Vemurafenib Induces Paradoxical Activation of the MAPK/ERK Pathway in NRASQ61R-Mutated Melanoma Cells
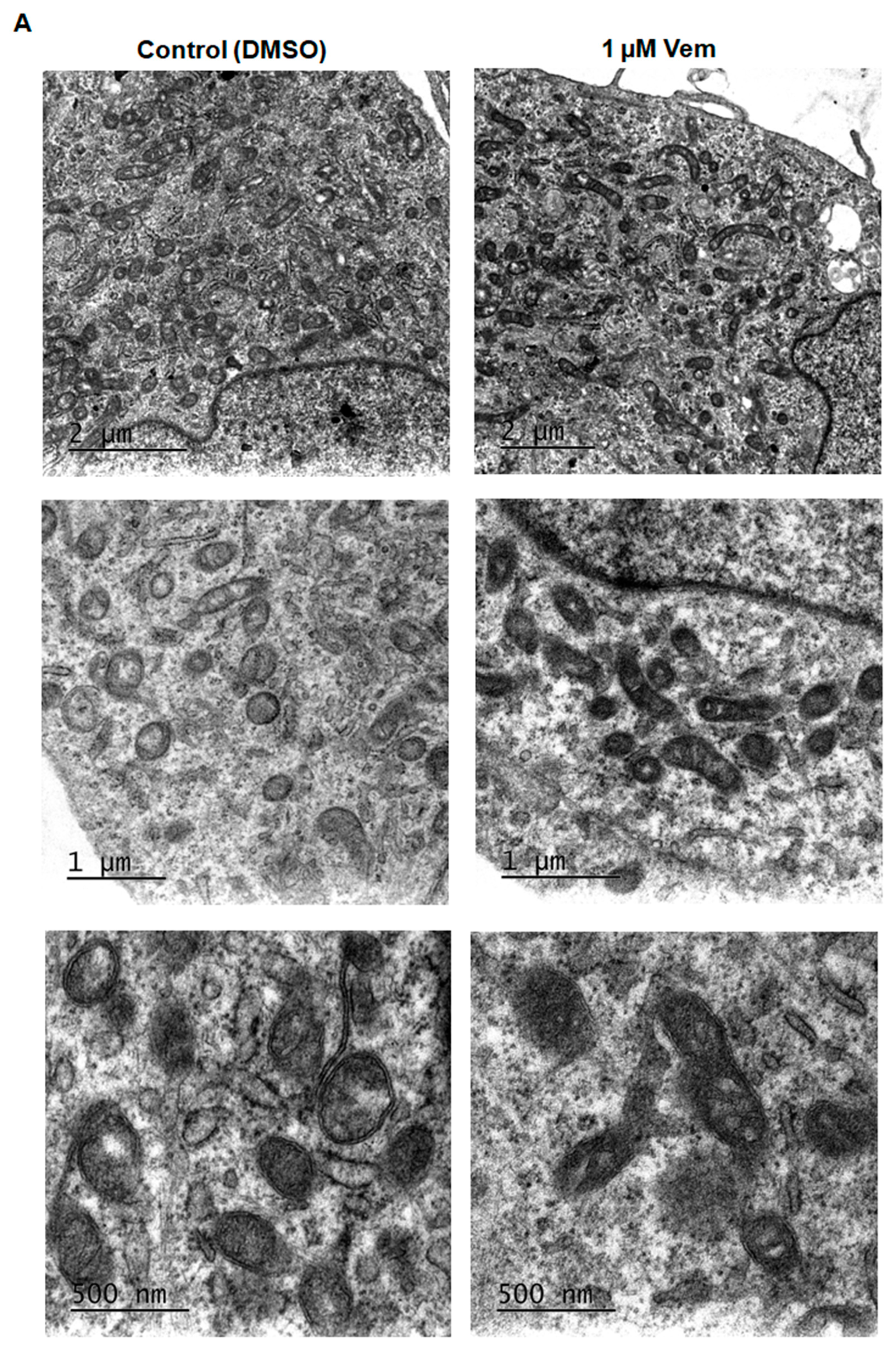
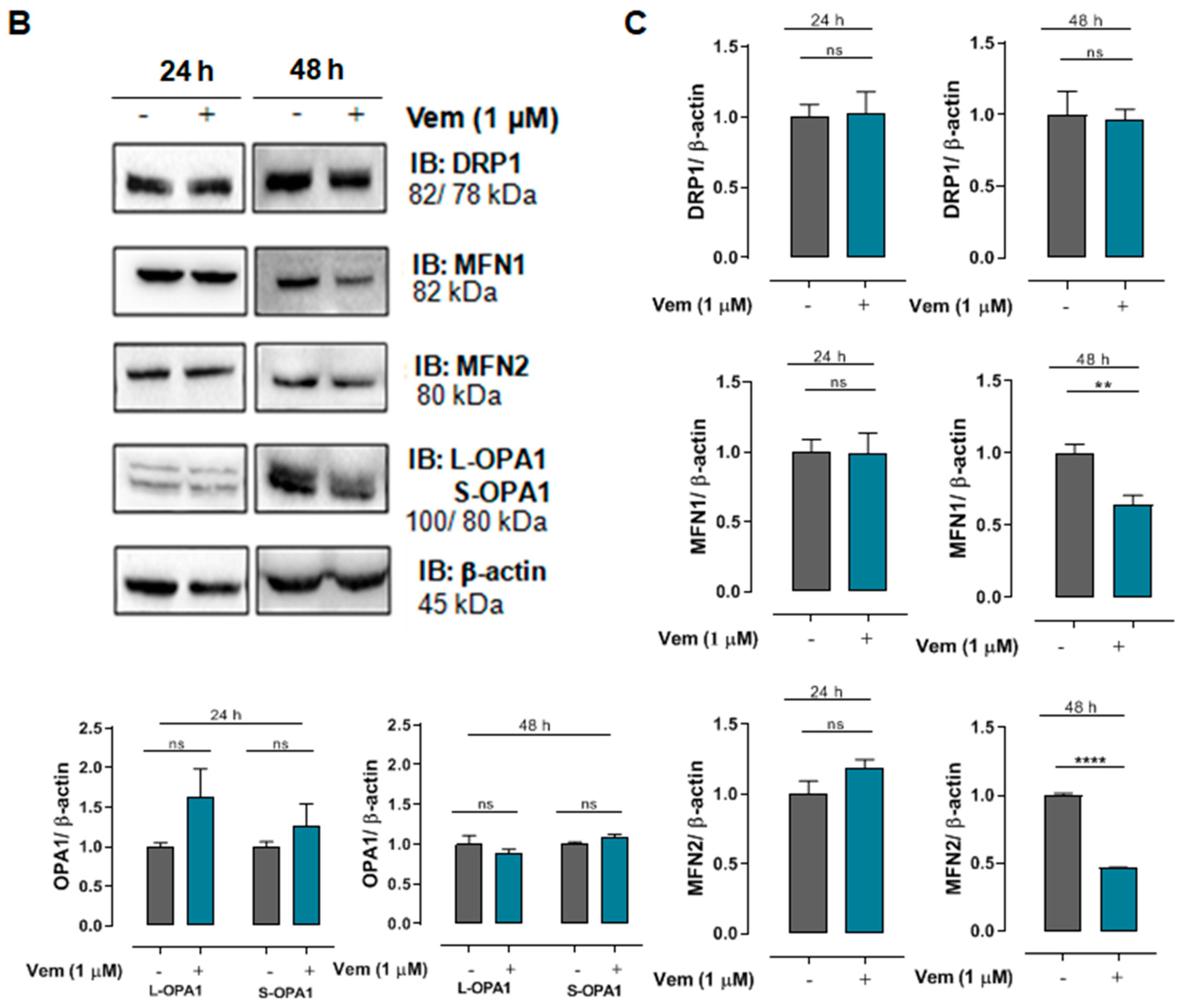
2.3. Vemurafenib Does Not Cause Extensive Mitochondrial Fusion in NRAS-Mutated Melanoma Cells
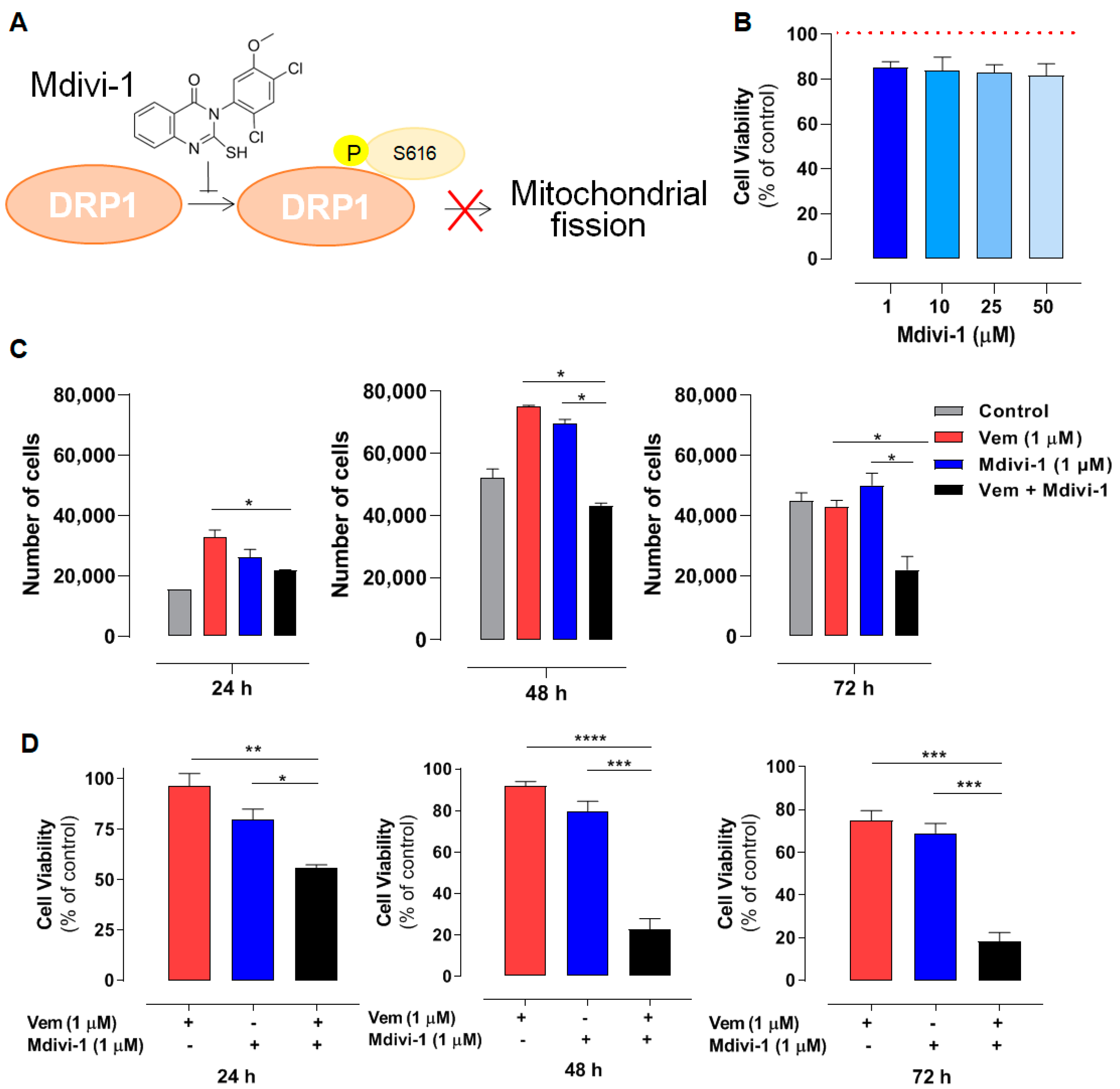
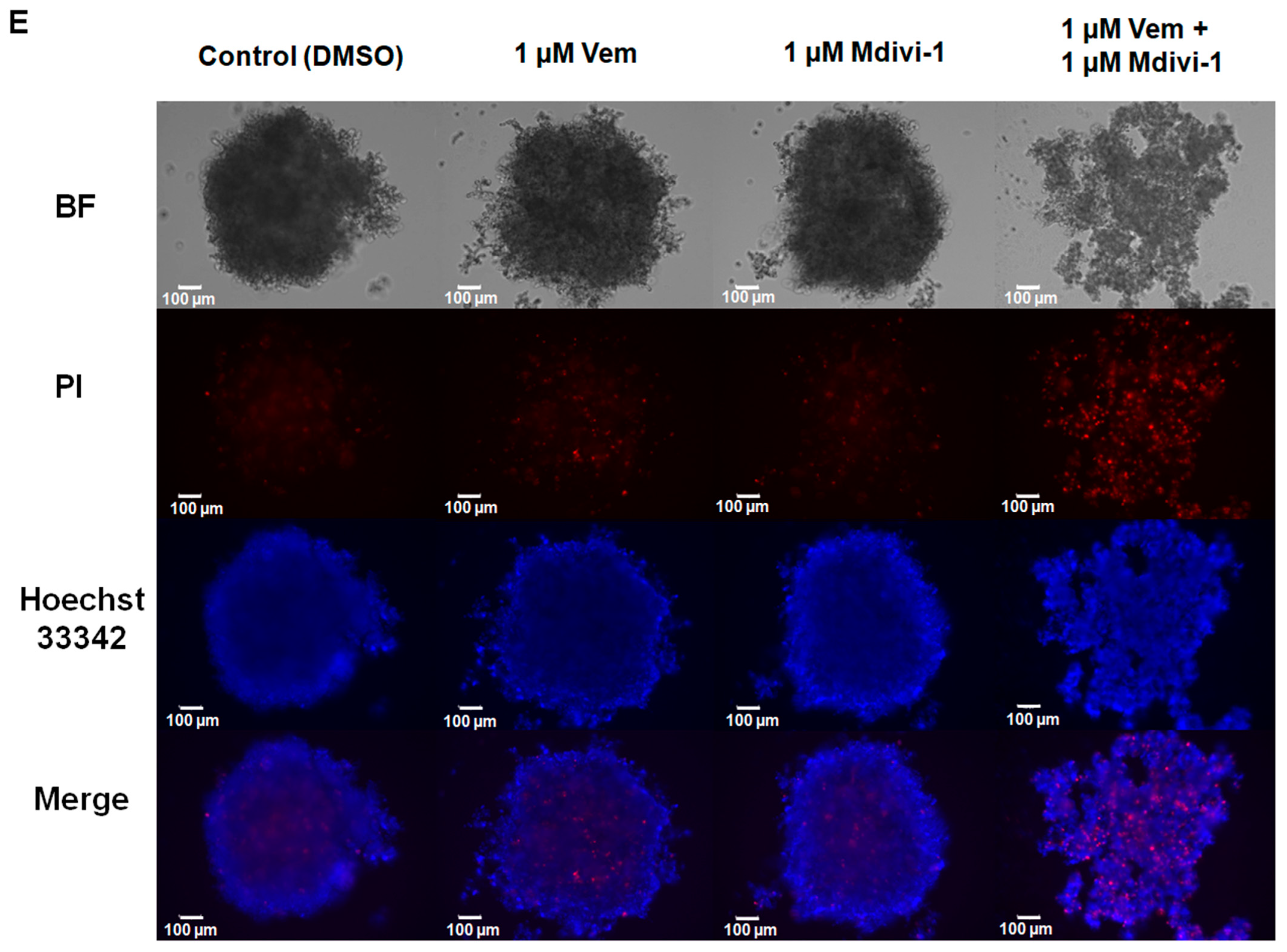
2.4. Combined Treatment with Mdivi-1 Synergistically Sensitizes NRASQ61R Melanoma Cells to Vemurafenib
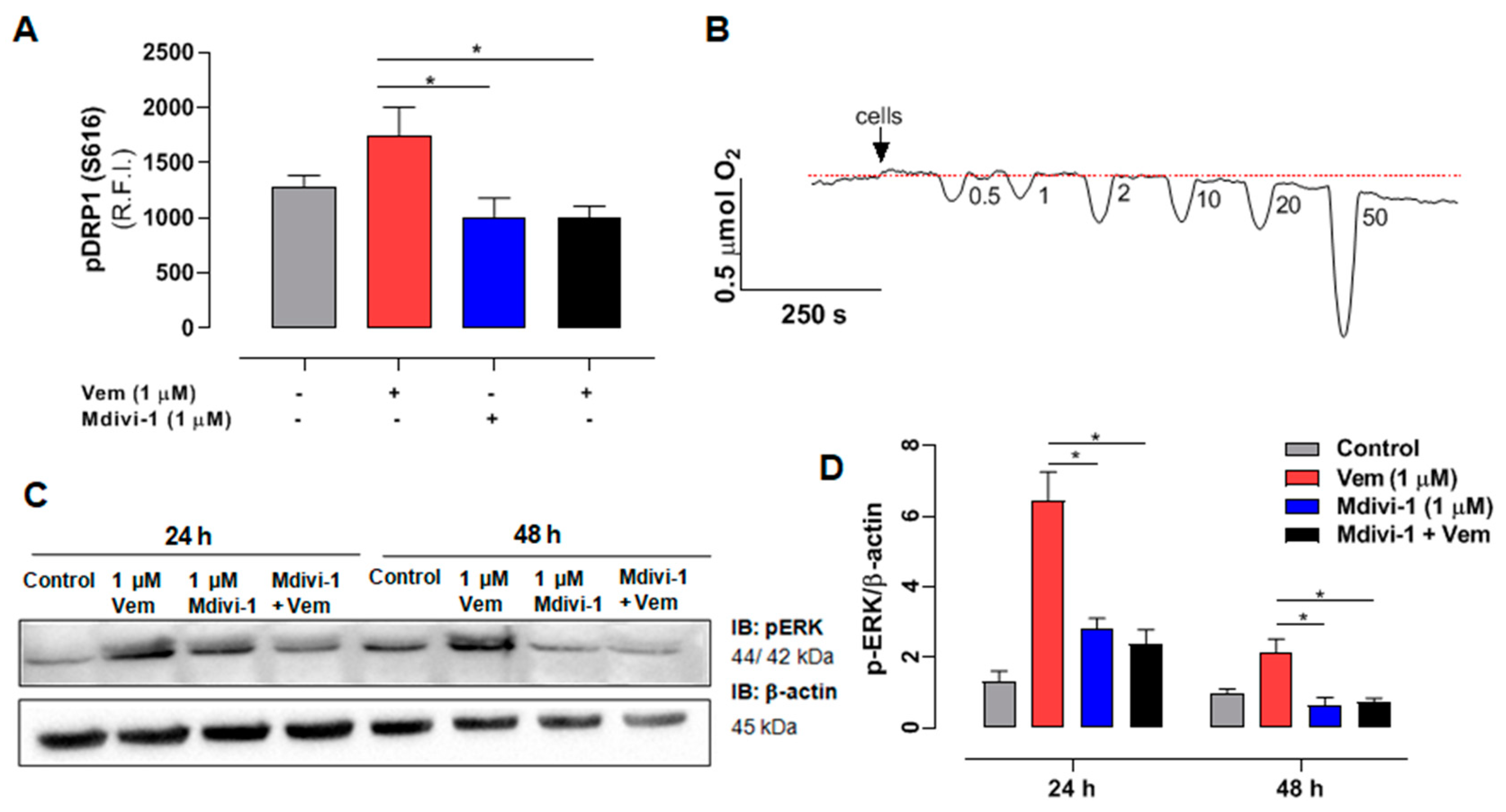
2.5. Mdivi-1 Reverts the Paradoxical Activation of MAPK/ERK Pathway Elicited by Vemurafenib in SK-MEL-147 Cells
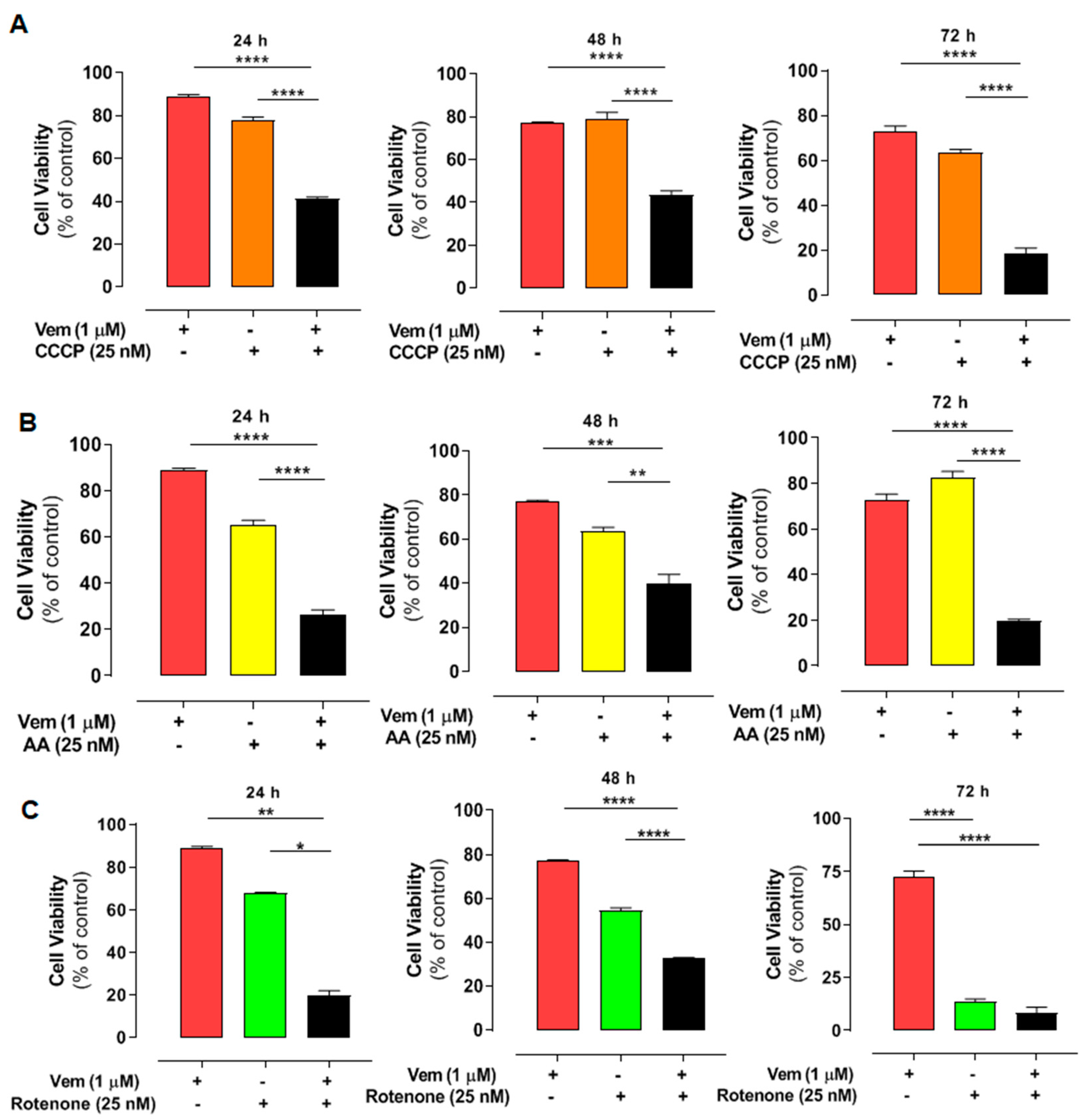
2.6. Interference with Mitochondrial Bioenergetics Also Sensitizes NRASQ61R Melanoma Cells to Vemurafenib
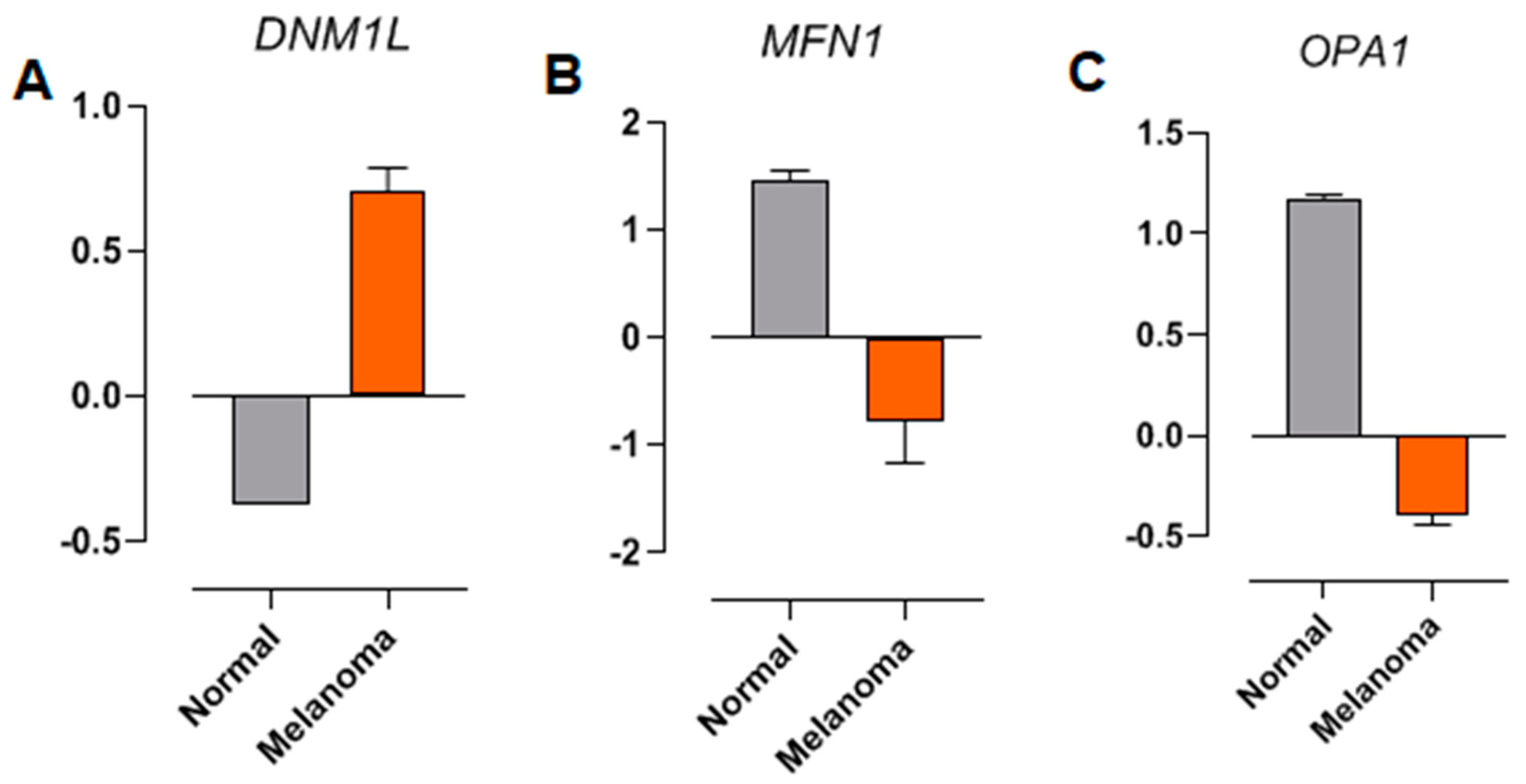
2.7. The Expression of Genes Involved in Mitochondrial Dynamics Is Altered in NRAS-Mutated Melanoma Samples from Patients
3. Discussion
4. Materials and Methods
4.1. Three-Dimensional Cell Culture and Standard Incubation Conditions with Vemurafenib
4.2. Three-DimensionalCell Culture (Spheroids) and Cell Viability Analysis
4.3. Cytotoxicity Assays
4.4. Growth Curves for Cell Proliferation
4.5. Annexin V-FITC/PI Double Staining Assay
4.6. High-Resolution Respirometry
4.7. Western Blotting
4.8. Quantification of pDRP1 ser616
4.9. Mitochondrial Morphology Evaluated by Transmission Electron Microscopy
4.10. Drug Combination
4.11. Bioinformatics
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BRAF | Rapidly Accelerated Fibrosarcoma B-type |
| NRAS | Neuroblastoma RAS Viral Oncogene Homolog |
| MAPK | Mitogen-Activated Protein Kinase |
| ERK | Extracellular Signal-Regulated Kinase |
| MEK | Mitogen-activated Extracellular Signal-regulated Kinase |
| FDA | Food and Drug Administration |
| DRP1 | Dynamin-Related Protein 1 |
| GTP | Guanosine Triphosphate |
| GDP | Guanosine Diphosphate |
| DMSO | Dimethyl Sulfoxide |
| Vem | Vemurafenib |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium Bromide |
| FITC | Fluorescein Isothiocyanate |
| AN | Annexin |
| PI | Propidium Iodide |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| OPA1 | Optic Atrophy 1 |
| Mdivi-1 | Mitochondrial Division Inhibitor 1 |
| CI | Combination Index |
| CCCP | Carbonyl Cyanide 3-chlorophenylhydrazone |
| AA | Antimycin A |
| DE | Differential Expression |
| DNML1 | Dynamin-Like Protein 1 |
| DUSP4 | Dual Specificity Phosphatase 4 |
| DUSP6 | Dual Specificity Phosphatase 6 |
| ROS | Reactive Oxygen Species |
| PRDX2 | Peroxiredoxin 2 |
| OXPHOS | Oxidative Phosphorylation |
| AMPK | AMP-Activated Protein Kinase |
| AICAR | 5-aminoimidazole-4-carboxamide-ribonucleoside |
| DMEM | Dulbecco’s Modified Eagle’s medium |
| PMSF | Phenylmethylsulfonyl Fluoride |
| SDS-PAGE | Sodium Dodecyl Sulfate–polyacrylamide Gel Electrophoresis |
| HRP | Horseradish Peroxidase |
| ANOVA | One-way Analysis of Variance |
References
- Chaiprasongsuk, A.; Panich, U. Role of Phytochemicals in Skin Photoprotection via Regulation of Nrf2. Front. Pharmacol. 2022, 13, 823881. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA. Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Bolick, N.L.; Geller, A.C. Epidemiology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Ossio, R.; Roldán-Marín, R.; Martínez-Said, H.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma: A Global Perspective. Nat. Rev. Cancer 2017, 17, 393–394. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS Mutation Frequencies among Primary Tumors and Metastases in Patients with Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From Mutations to Medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef]
- McArthur, G.A.; Ribas, A. Targeting Oncogenic Drivers and the Immune System in Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 499–506. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas Acquire Resistance to B-RAF(V600E) Inhibition by RTK or N-RAS Upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-Existence of BRAF and NRAS Driver Mutations in the Same Melanoma Cells Results in Heterogeneity of Targeted Therapy Resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Investig. Dermatol. 2021, 141, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choi, J.-W.; Kim, Y.-S. Frequencies of BRAF and NRAS Mutations Are Different in Histological Types and Sites of Origin of Cutaneous Melanoma: A Meta-Analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.-Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.-B. Reactivation of Mitogen-Activated Protein Kinase (MAPK) Pathway by FGF Receptor 3 (FGFR3)/Ras Mediates Resistance to Vemurafenib in Human B-RAF V600E Mutant Melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef]
- Solit, D.B.; Rosen, N. Resistance to BRAF Inhibition in Melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells with Wild-Type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef]
- Holderfield, M.; Merritt, H.; Chan, J.; Wallroth, M.; Tandeske, L.; Zhai, H.; Tellew, J.; Hardy, S.; Hekmat-Nejad, M.; Stuart, D.D.; et al. RAF Inhibitors Activate the MAPK Pathway by Relieving Inhibitory Autophosphorylation. Cancer Cell 2013, 23, 594–602. [Google Scholar] [CrossRef]
- Ferraz, L.S.; Costa, R.T.d.; Costa, C.A.d.; Ribeiro, C.A.J.; Arruda, D.C.; Maria-Engler, S.S.; Rodrigues, T. Targeting Mitochondria in Melanoma: Interplay between MAPK Signaling Pathway and Mitochondrial Dynamics. Biochem. Pharmacol. 2020, 178, 114104. [Google Scholar] [CrossRef]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-Mutant Melanoma: Current Challenges and Future Prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Eskandarpour, M.; Huang, F.; Reeves, K.A.; Clark, E.; Hansson, J. Oncogenic NRAS Has Multiple Effects on the Malignant Phenotype of Human Melanoma Cells Cultured in Vitro. Int. J. Cancer 2009, 124, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.M.; Bastholt, L.; Robert, C.; Sosman, J.; Larkin, J.; Hersey, P.; Middleton, M.; Cantarini, M.; Zazulina, V.; Kemsley, K.; et al. Phase II, Open-Label, Randomized Trial of the MEK1/2 Inhibitor Selumetinib as Monotherapy versus Temozolomide in Patients with Advanced Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 555–567. [Google Scholar] [CrossRef]
- Salzmann, M.; Pawlowski, J.; Loquai, C.; Rafei-Shamsabadi, D.A.; Meiss, F.; Ugurel, S.; Schadendorf, D.; Meier, F.; Enk, A.H.; Hassel, J.C. MEK Inhibitors for Pre-Treated, NRAS-Mutated Metastatic Melanoma: A Multi-Centre, Retrospective Study. Eur. J. Cancer Oxf. Engl. 1990 2022, 166, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef]
- Gopal, Y.N.V.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 Overcomes Resistance to MAPK Pathway Inhibitors Mediated by PGC1α and Oxidative Phosphorylation in Melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef]
- McQuade, J.L.; Vashisht Gopal, Y. Counteracting Oxidative Phosphorylation-Mediated Resistance of Melanomas to MAPK Pathway Inhibition. Mol. Cell. Oncol. 2015, 2, e991610. [Google Scholar] [CrossRef]
- Nikolaou, V.A.; Stratigos, A.J.; Flaherty, K.T.; Tsao, H. Melanoma: New Insights and New Therapies. J. Investig. Dermatol. 2012, 132, 854–863. [Google Scholar] [CrossRef]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF Inhibitor Resistance: A Multicenter Meta-Analysis of the Spectrum and Frequencies, Clinical Behaviour, and Phenotypic Associations of Resistance Mechanisms. Eur. J. Cancer Oxf. Engl. 1990 2015, 51, 2792–2799. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-rot087288. [Google Scholar] [CrossRef]
- Amin, A.D.; Rajan, S.S.; Groysman, M.J.; Pongtornpipat, P.; Schatz, J.H. Oncogene Overdose: Too Much of a Bad Thing for Oncogene-Addicted Cancer Cells. Biomark. Cancer 2015, 7, BIC-S29326. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Prat, N.; Zuberer, H.L.; Mangano, L.; Karimaddini, Z.; Wolf, L.; Tyanova, S.; Wellinger, L.C.; Marbach, D.; Griesser, V.; Pettazzoni, P.; et al. DUSP4 Protects BRAF- and NRAS-Mutant Melanoma from Oncogene Overdose through Modulation of MITF. Life Sci. Alliance 2022, 5, e202101235. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.P.; Feng, T.; Sigoillot, F.D.; Geyer, F.C.; Shirley, M.D.; Ruddy, D.A.; Rakiec, D.P.; Freeman, A.K.; Engelman, J.A.; Jaskelioff, M.; et al. Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-Mutant Melanoma. Mol. Cancer Res. 2019, 17, 199–211. [Google Scholar] [CrossRef]
- Dias, M.H.; Bernards, R. Playing Cancer at Its Own Game: Activating Mitogenic Signaling as a Paradoxical Intervention. Mol. Oncol. 2021, 15, 1975–7985. [Google Scholar] [CrossRef]
- Dias, M.H.; Friskes, A.; Wang, S.; Fernandes Neto, J.M.; van Gemert, F.; Mourragui, S.; Papagianni, C.; Kuiken, H.J.; Mainardi, S.; Alvarez-Villanueva, D.; et al. Paradoxical Activation of Oncogenic Signaling as a Cancer Treatment Strategy. Cancer Discov. 2024, 14, 1276–1301. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor Mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor That Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef]
- Koch, B.; Barugahare, A.A.; Lo, T.L.; Huang, C.; Schittenhelm, R.B.; Powell, D.R.; Beilharz, T.H.; Traven, A. A Metabolic Checkpoint for the Yeast-to-Hyphae Developmental Switch Regulated by Endogenous Nitric Oxide Signaling. Cell Rep. 2018, 25, 2244–2258.e7. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-Seq Analysis Identifies Different Transcriptomic Types and Developmental Trajectories of Primary Melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Nagel, T.E.; Stuart, D.D. Mechanism and Consequences of RAF Kinase Activation by Small-Molecule Inhibitors. Br. J. Cancer 2014, 111, 640–645. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Gibney, G.T.; Smalley, K.S.M. NRAS Mutant Melanoma: Biological Behavior and Future Strategies for Therapeutic Management. Oncogene 2013, 32, 3009–3018. [Google Scholar] [CrossRef]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling Vemurafenib Resistance in Melanoma Reveals a Strategy to Forestall Drug Resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Jung, N.Y.; Atari, A.; Rodriguez, D.J.; Kesar, D.; Song, T.-Y.; Rees, M.G.; Ronan, M.; Li, R.; Ruiz, P.; et al. Systematic Profiling of Conditional Pathway Activation Identifies Context-Dependent Synthetic Lethalities. Nat. Genet. 2023, 55, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Young, M.J.; Li, R.; Jain, S.; Wernitznig, A.; Krill-Burger, J.M.; Lemke, C.T.; Monducci, D.; Rodriguez, D.J.; Chang, L.; et al. Paralog Knockout Profiling Identifies DUSP4 and DUSP6 as a Digenic Dependence in MAPK Pathway-Driven Cancers. Nat. Genet. 2021, 53, 1664–1672. [Google Scholar] [CrossRef]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.-Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef]
- Noma, I.H.Y.; Carvalho, L.A.d.C.; Camarena, D.E.M.; Silva, R.O.; de Moraes Junior, M.O.; de Souza, S.T.; Newton-Bishop, J.; Nsengimana, J.; Maria-Engler, S.S. Peroxiredoxin-2 Represses NRAS-Mutated Melanoma Cells Invasion by Modulating EMT Markers. Biomed. Pharmacother. 2024, 177, 116953. [Google Scholar] [CrossRef]
- Carvalho, L.A.d.C.; Noma, I.H.Y.; Uehara, A.H.; Siena, Á.D.D.; Osaki, L.H.; Mori, M.P.; Pinto, N.C. de S.; Freitas, V.M.; Junior, W.A.S.; Smalley, K.S.M.; et al. Modeling Melanoma Heterogeneity In Vitro: Redox, Resistance and Pigmentation Profiles. Antioxidants 2024, 13, 555. [Google Scholar] [CrossRef]
- McGrail, K.; Granado-Martínez, P.; Esteve-Puig, R.; García-Ortega, S.; Ding, Y.; Sánchez-Redondo, S.; Ferrer, B.; Hernandez-Losa, J.; Canals, F.; Manzano, A.; et al. BRAF Activation by Metabolic Stress Promotes Glycolysis Sensitizing NRASQ61-Mutated Melanomas to Targeted Therapy. Nat. Commun. 2022, 13, 7113. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting Mitochondria Metabolism for Cancer Therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Arismendi-Morillo, G. Electron Microscopy Morphology of the Mitochondrial Network in Human Cancer. Int. J. Biochem. Cell Biol. 2009, 41, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuchner, J.; Kuznetsov, A.V.; Obexer, P.; Ausserlechner, M.J. BIRC5/Survivin Enhances Aerobic Glycolysis and Drug Resistance by Altered Regulation of the Mitochondrial Fusion/Fission Machinery. Oncogene 2013, 32, 4748–4757. [Google Scholar] [CrossRef] [PubMed]
- Inoue-Yamauchi, A.; Oda, H. Depletion of Mitochondrial Fission Factor DRP1 Causes Increased Apoptosis in Human Colon Cancer Cells. Biochem. Biophys. Res. Commun. 2012, 421, 81–85. [Google Scholar] [CrossRef]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of Mitochondrial Fission Prevents Cell Cycle Progression in Lung Cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2175–2186. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial Dynamics Regulates Migration and Invasion of Breast Cancer Cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial Division Is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef]
- Zacharioudakis, E.; Gavathiotis, E. Mitochondrial Dynamics Proteins as Emerging Drug Targets. Trends Pharmacol. Sci. 2023, 44, 112–127. [Google Scholar] [CrossRef]
- Chen, K.-H.; Dasgupta, A.; Ding, J.; Indig, F.E.; Ghosh, P.; Longo, D.L. Role of Mitofusin 2 (Mfn2) in Controlling Cellular Proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 382–394. [Google Scholar] [CrossRef]
- Zhang, G.-E.; Jin, H.-L.; Lin, X.-K.; Chen, C.; Liu, X.-S.; Zhang, Q.; Yu, J.-R. Anti-Tumor Effects of Mfn2 in Gastric Cancer. Int. J. Mol. Sci. 2013, 14, 13005–13021. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, Z.; Zhang, Y.; Zhang, P.; Zhu, X.; Huang, J.; Ma, T.; Lu, T.; Song, Q.; Li, Q.; et al. Adenovirus-Expressed Human Hyperplasia Suppressor Gene Induces Apoptosis in Cancer Cells. Mol. Cancer Ther. 2008, 7, 222–232. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, L.; Wang, L.; Wang, J.; Wang, D.; Jiang, J.; Zhang, J.; Zhou, Q. Mitochondrial Division Inhibitor (Mdivi-1) Inhibits Proliferation and Epithelial-Mesenchymal Transition via the NF-κB Pathway in Thyroid Cancer Cells. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2023, 88, 105552. [Google Scholar] [CrossRef]
- Fang, C.-T.; Kuo, H.-H.; Yuan, C.-J.; Yao, J.-S.; Yih, L.-H. Mdivi-1 Induces Spindle Abnormalities and Augments Taxol Cytotoxicity in MDA-MB-231 Cells. Cell Death Discov. 2021, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Min, K.; Kwon, T.K. Inhibition of Drp1 Sensitizes Cancer Cells to Cisplatin-Induced Apoptosis through Transcriptional Inhibition of c-FLIP Expression. Molecules 2020, 25, 5793. [Google Scholar] [CrossRef]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small Molecule Metabolites: Discovery of Biomarkers and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef]
- Jang, J.E.; Hwang, D.Y.; Eom, J.-I.; Cheong, J.-W.; Jeung, H.-K.; Cho, H.; Chung, H.; Kim, J.S.; Min, Y.H. DRP1 Inhibition Enhances Venetoclax-Induced Mitochondrial Apoptosis in TP53-Mutated Acute Myeloid Leukemia Cells through BAX/BAK Activation. Cancers 2023, 15, 745. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; de Luxán-Delgado, B.; Penin-Peyta, L.; Royo-García, A.; Parejo-Alonso, B.; Jagust, P.; Alcalá, S.; Rubiolo, J.A.; Sánchez, L.; Sainz, B.; et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers 2021, 13, 698. [Google Scholar] [CrossRef]
- Che, L.; Wu, J.-S.; Du, Z.-B.; He, Y.-Q.; Yang, L.; Lin, J.-X.; Lei, Z.; Chen, X.-X.; Guo, D.-B.; Li, W.-G.; et al. Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma. Cancers 2022, 14, 821. [Google Scholar] [CrossRef]
- Cheng, J.; Sha, Z.; Zhang, R.; Ge, J.; Chen, P.; Kuang, X.; Chang, J.; Ren, K.; Luo, X.; Chen, S.; et al. L22 Ribosomal Protein Is Involved in Dynamin-Related Protein 1-Mediated Gastric Carcinoma Progression. Bioengineered 2022, 13, 6650–6664. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: Beyond the Warburg Effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed]
- Petti, C.; Vegetti, C.; Molla, A.; Bersani, I.; Cleris, L.; Mustard, K.J.; Formelli, F.; Hardie, G.D.; Sensi, M.; Anichini, A. AMPK Activators Inhibit the Proliferation of Human Melanomas Bearing the Activated MAPK Pathway. Melanoma Res. 2012, 22, 341–350. [Google Scholar] [CrossRef]
- Woodard, J.; Platanias, L.C. AMP-Activated Kinase (AMPK)-Generated Signals in Malignant Melanoma Cell Growth and Survival. Biochem. Biophys. Res. Commun. 2010, 398, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Q.; Deng, G.; Kuang, S.; Lian, J.; Wang, M.; Zhu, H. AMPK Activation by GSK621 Inhibits Human Melanoma Cells in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2016, 480, 515–521. [Google Scholar] [CrossRef]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin Blocks Melanoma Invasion and Metastasis Development in AMPK/P53-Dependent Manner. Mol. Cancer Ther. 2013, 12, 1605–1615. [Google Scholar] [CrossRef]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.-H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin Enhances the Therapeutic Benefit of BRAF(V600E) Inhibition in Melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef]
- Schöckel, L.; Glasauer, A.; Basit, F.; Bitschar, K.; Truong, H.; Erdmann, G.; Algire, C.; Hägebarth, A.; Willems, P.H.; Kopitz, C.; et al. Targeting Mitochondrial Complex I Using BAY 87-2243 Reduces Melanoma Tumor Growth. Cancer Metab. 2015, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Figarola, J.L.; Singhal, J.; Singhal, S.; Kusari, J.; Riggs, A. Bioenergetic Modulation with the Mitochondria Uncouplers SR4 and Niclosamide Prevents Proliferation and Growth of Treatment-Naïve and Vemurafenib-Resistant Melanomas. Oncotarget 2018, 9, 36945–36965. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, L.S.; Watashi, C.M.; Colturato-Kido, C.; Pelegrino, M.T.; Paredes-Gamero, E.J.; Weller, R.B.; Seabra, A.B.; Rodrigues, T. Antitumor Potential of S-Nitrosothiol-Containing Polymeric Nanoparticles against Melanoma. Mol. Pharm. 2018, 15, 1160–1168. [Google Scholar] [CrossRef]
- Medeiros, H.C.D.; Colturato-Kido, C.; Ferraz, L.S.; Costa, C.A.; Moraes, V.W.R.; Paredes-Gamero, E.J.; Tersariol, I.L.S.; Rodrigues, T. AMPK Activation Induced by Promethazine Increases NOXA Expression and Beclin-1 Phosphorylation and Drives Autophagy-Associated Apoptosis in Chronic Myeloid Leukemia. Chem. Biol. Interact. 2020, 315, 108888. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinform. Oxf. Engl. 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Vemurafenib (µM) | Mdivi-1 (µM) | CI |
|---|---|---|---|
| 24 | 1.0 | 1.0 | 0.0219300 * |
| 48 | 1.0 | 1.0 | 0.0000216 * |
| 72 | 1.0 | 1.0 | 0.0569500 * |
| Time (h) | Vemurafenib (µM) | CCCP (nM) | CI |
|---|---|---|---|
| 4 | 1.0 | 25 | 0.08958 * |
| 48 | 1.0 | 25 | 0.00290 * |
| 72 | 1.0 | 25 | 0.20948 * |
| Time (h) | Vemurafenib (µM) | Antimycin A (nM) | CI |
| 24 | 1.0 | 25 | 3.5060700 * |
| 48 | 1.0 | 25 | 0.0000448 * |
| 72 | 1.0 | 25 | 0.1414900 * |
| Time (h) | Vemurafenib (µM) | Rotenone (nM) | CI |
| 24 | 1.0 | 25 | 6.87541 * |
| 48 | 1.0 | 25 | 0.46380 * |
| 72 | 1.0 | 25 | 0.81222 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado-Souza, L.F.L.d.; Ferraz, L.S.; Citrangulo Tortelli, T., Jr.; Ribeiro, C.A.J.; Amaral, D.T.d.; Arruda, D.C.; Oliveira, É.A.d.; Chammas, R.; Maria-Engler, S.S.; Rodrigues, T. Exploiting Paradoxical Activation of Oncogenic MAPK Signaling by Targeting Mitochondria to Sensitize NRAS Mutant-Melanoma to Vemurafenib. Int. J. Mol. Sci. 2025, 26, 2675. https://doi.org/10.3390/ijms26062675
Prado-Souza LFLd, Ferraz LS, Citrangulo Tortelli T Jr., Ribeiro CAJ, Amaral DTd, Arruda DC, Oliveira ÉAd, Chammas R, Maria-Engler SS, Rodrigues T. Exploiting Paradoxical Activation of Oncogenic MAPK Signaling by Targeting Mitochondria to Sensitize NRAS Mutant-Melanoma to Vemurafenib. International Journal of Molecular Sciences. 2025; 26(6):2675. https://doi.org/10.3390/ijms26062675
Chicago/Turabian StylePrado-Souza, Laura Francisca Leite do, Letícia Silva Ferraz, Tharcísio Citrangulo Tortelli, Jr., César Augusto João Ribeiro, Danilo Trabuco do Amaral, Denise Costa Arruda, Érica Aparecida de Oliveira, Roger Chammas, Silvya Stuchi Maria-Engler, and Tiago Rodrigues. 2025. "Exploiting Paradoxical Activation of Oncogenic MAPK Signaling by Targeting Mitochondria to Sensitize NRAS Mutant-Melanoma to Vemurafenib" International Journal of Molecular Sciences 26, no. 6: 2675. https://doi.org/10.3390/ijms26062675
APA StylePrado-Souza, L. F. L. d., Ferraz, L. S., Citrangulo Tortelli, T., Jr., Ribeiro, C. A. J., Amaral, D. T. d., Arruda, D. C., Oliveira, É. A. d., Chammas, R., Maria-Engler, S. S., & Rodrigues, T. (2025). Exploiting Paradoxical Activation of Oncogenic MAPK Signaling by Targeting Mitochondria to Sensitize NRAS Mutant-Melanoma to Vemurafenib. International Journal of Molecular Sciences, 26(6), 2675. https://doi.org/10.3390/ijms26062675