

Models of Early Resistance to CDK4/6 Inhibitors Unveil Potential Therapeutic Treatment Sequencing
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Early Resistance to CDK4/6 Inhibitor Plus Tamoxifen
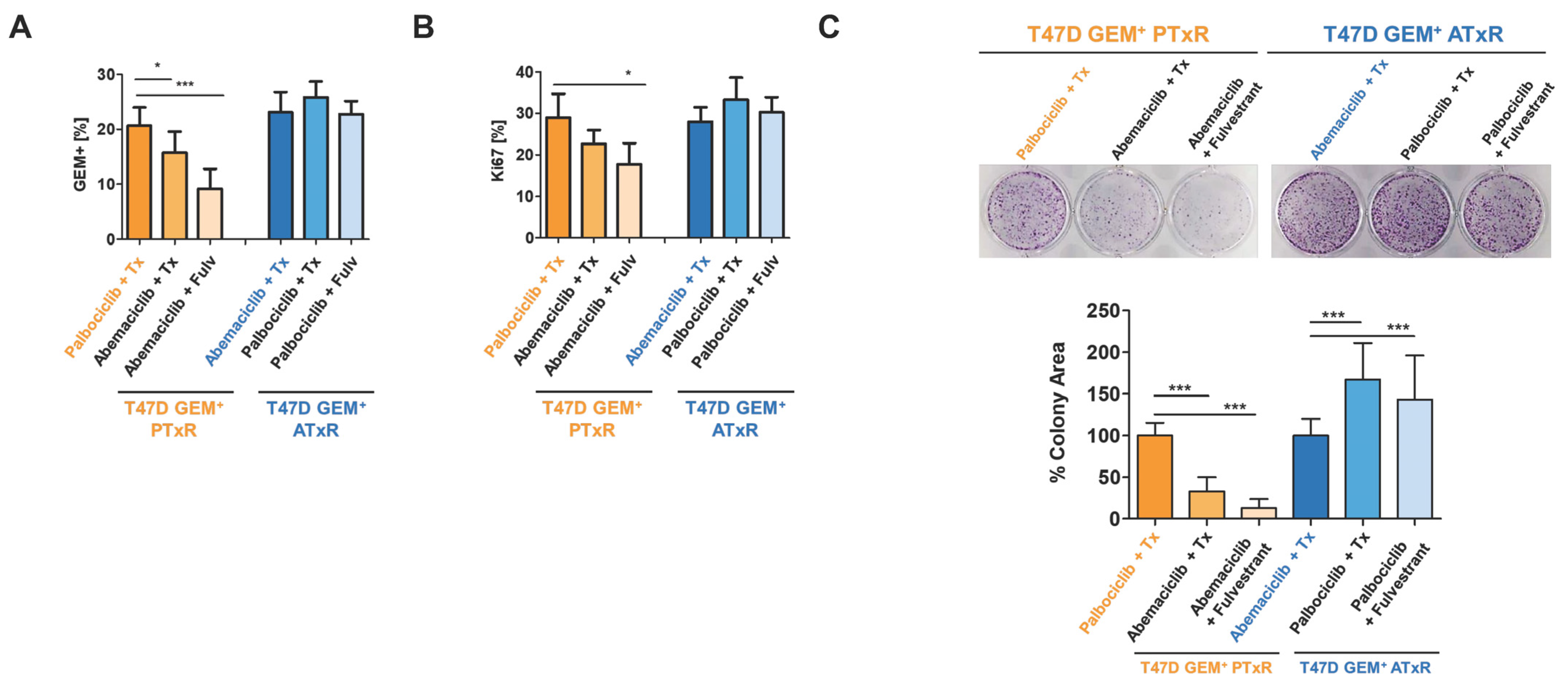
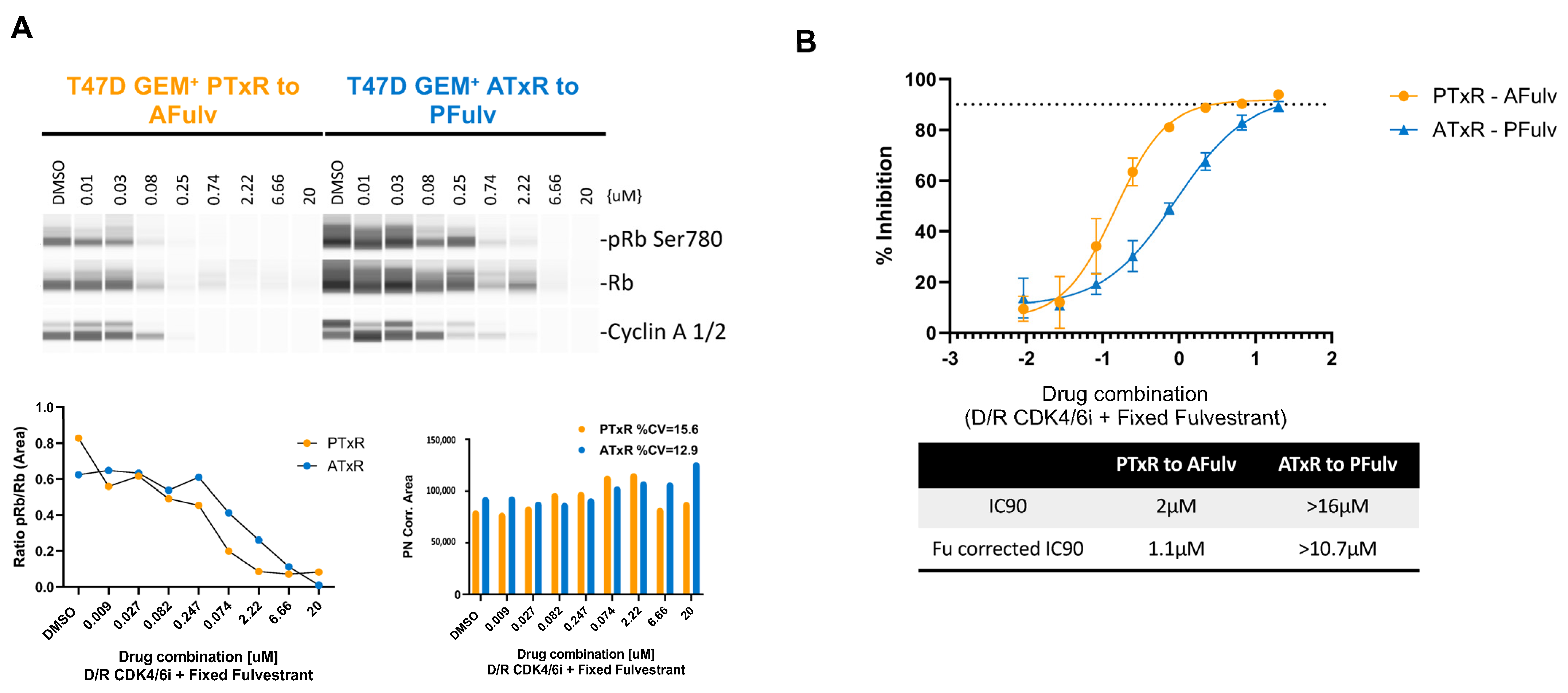
2.2. Switch to Abemaciclib + ET Resensitizes Cells to CDK4/6i Following Progression on Palbociclib + Tamoxifen
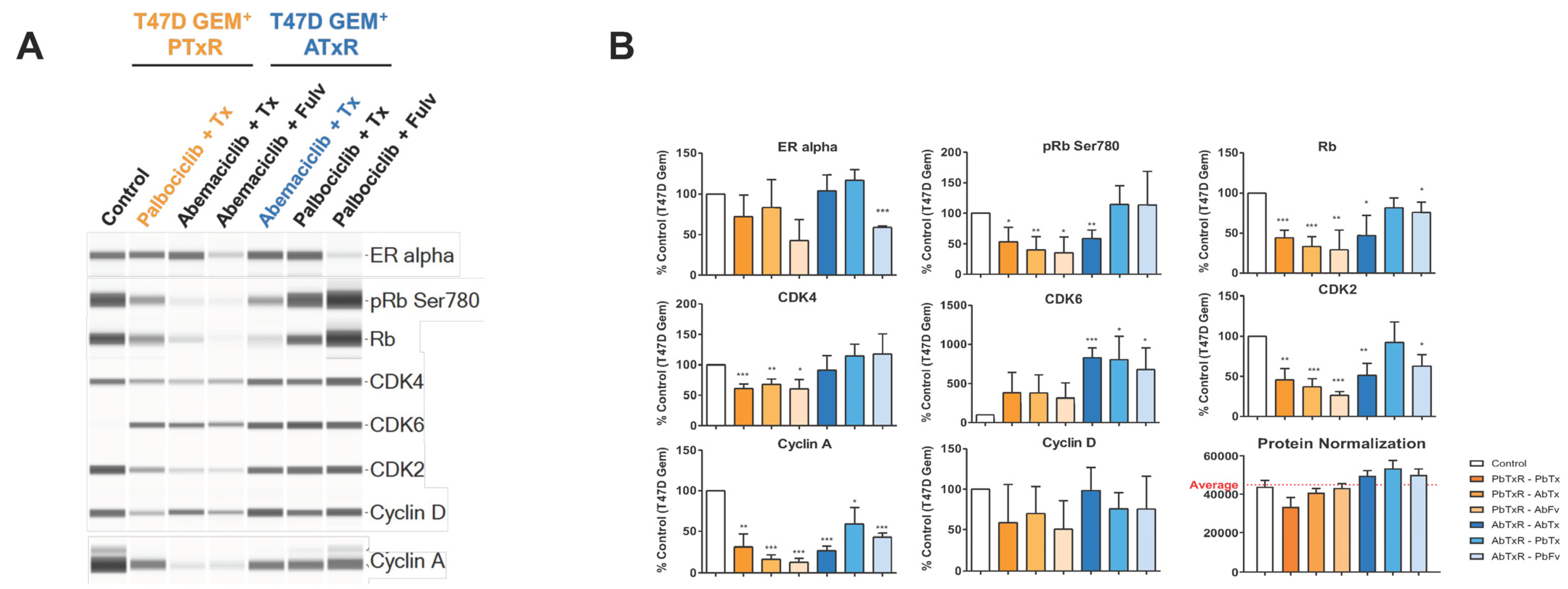
2.3. Biomarker Modulation
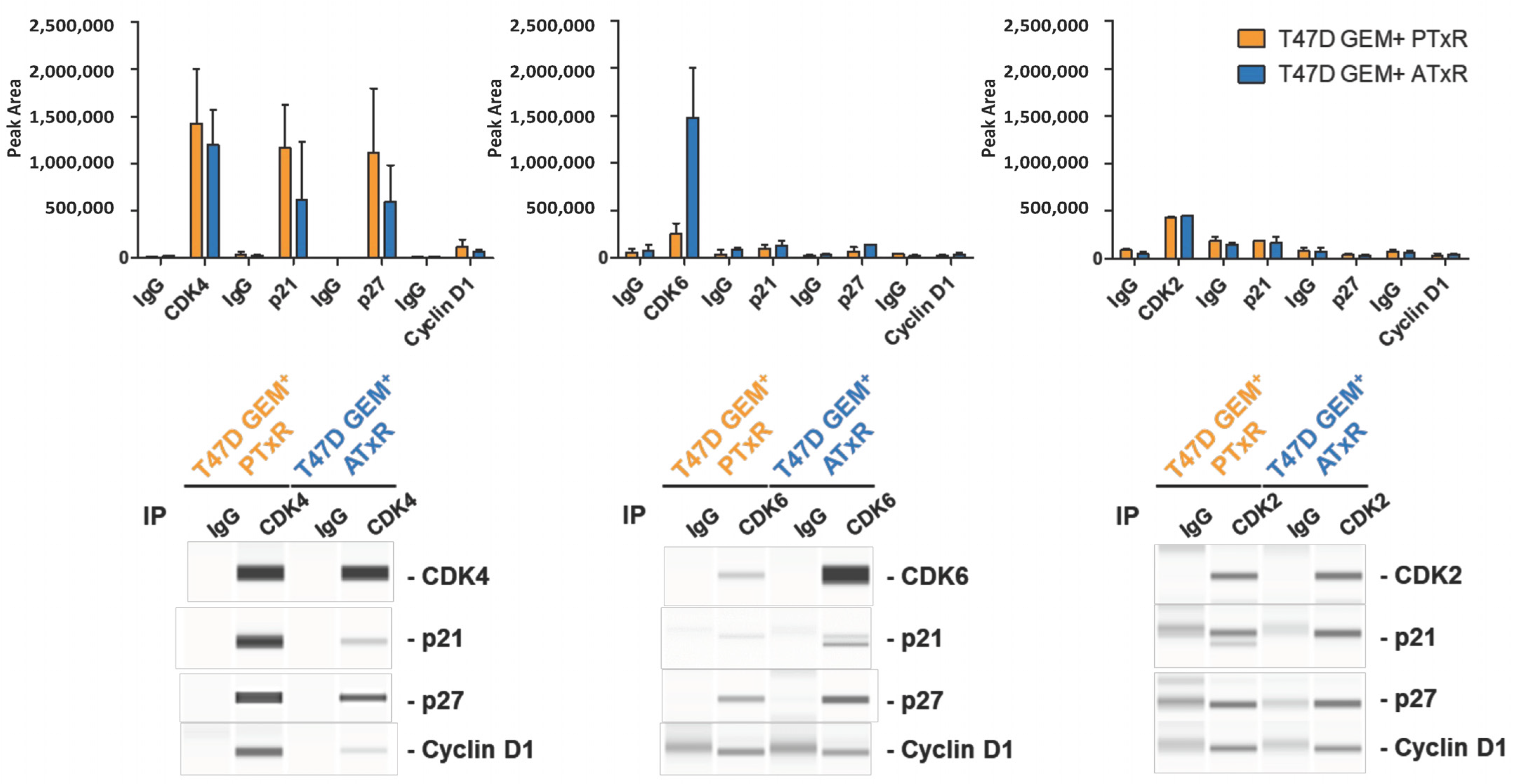
2.4. coIPs and Jess Analysis
2.5. Molecular Characterization of Resistant Cell Lines Following Therapeutic Switch
3. Discussion
4. Materials and Methods
4.1. Cell Line Generation and Culture Conditions
4.2. Chemicals
4.3. Determination of Clinically Relevant Drug Concentrations
4.4. Flow Cytometry Assays
4.4.1. Geminin Analysis
4.4.2. Ki-67 Analysis
4.4.3. Annexin V-DAPI Analysis
4.5. Colony Formation Assay
4.6. Dose–Response Assay
4.7. Western Blot and Jess Simple WesternTM Assays
4.8. Co-Immunoprecipitation
4.9. RNAseq
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen response |
| ATxR | Abemaciclib + tamoxifen resistant |
| CDK4/6 | Cyclin-dependent kinase 4 and 6 |
| CDK4/6i | CDK4/6 inhibitor |
| ET | Endocrine therapy (tamoxifen, fulvestrant) |
| GEM+ | Geminin positive |
| HER2 | Human epidermal growth factor receptor 2 negative |
| HR+ | Hormone receptor positive |
| PTxR | Palbociclib + tamoxifen resistant |
| Tamoxifen | 4-hydroxytamoxifen; 4-OH-tamoxifen |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; Andre, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 116–124. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Kalinsky, K.; Layman, R.M.; Kaufman, P.A.; Graff, S.L.; Bianchini, G.; Martin, M.; Zhou, Y.; Knoderer, H.; Litchfield, L.; Wander, S.A. postMONARCH: A phase 3 study of abemaciclib plus fulvestrant versus placebo plus fulvestrant in patients with HR+, HER2−, metastatic breast cancer following progression on a CDK4 & 6 inhibitor and endocrine therapy. J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS1117. [Google Scholar] [CrossRef]
- Kalinsky, K.; Bianchini, G.; Hamilton, E.; Graff, S.L.; Park, K.H.; Jeselsohn, R.; Demirci, U.; Martin, M.; Layman, R.M.; Hurvitz, S.A.; et al. Abemaciclib Plus Fulvestrant in Advanced Breast Cancer After Progression on CDK4/6 Inhibition: Results from the Phase III postMONARCH Trial. J. Clin. Oncol. 2024, JCO2402086. [Google Scholar] [CrossRef]
- Yeo, B.; Turner, N.C.; Jones, A. An update on the medical management of breast cancer. BMJ 2014, 348, g3608. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Han, H.S.; Zangardi, M.L.; Niemierko, A.; Mariotti, V.; Kim, L.S.L.; Xi, J.; Pandey, A.; Dunne, S.; Nasrazadani, A.; et al. Clinical Outcomes with Abemaciclib After Prior CDK4/6 Inhibitor Progression in Breast Cancer: A Multicenter Experience. J. Natl. Compr. Cancer Netw. 2021, 19, 149–156. [Google Scholar] [CrossRef]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.M.; Llombart-Cussac, A.; Soliman, H.; Wang, H.; Wijayawardana, S.; Jansen, V.M.; et al. Clinical Significance of PIK3CA and ESR1 Mutations in circulating tumor DNA: Analysis from the MONARCH 2 Study of Abemaciclib Plus Fulvestrant. Clin. Cancer Res. 2022, 28, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, X.; Xiong, Y.; Li, R.; Ito, T.; Ahmed, T.A.; Karoulia, Z.; Adamopoulos, C.; Wang, H.; Wang, L.; et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat. Cancer 2021, 2, 429–443. [Google Scholar] [CrossRef]
- Wang, B.; Li, R.; Wu, S.; Liu, X.; Ren, J.; Li, J.; Bi, K.; Wang, Y.; Jia, H. Breast Cancer Resistance to Cyclin-Dependent Kinases 4/6 Inhibitors: Intricacy of the Molecular Mechanisms. Front. Oncol. 2021, 11, 651541. [Google Scholar] [CrossRef]
- Pack, L.R.; Daigh, L.H.; Chung, M.; Meyer, T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat. Commun. 2021, 12, 3356. [Google Scholar] [CrossRef]
- Freeman-Cook, K.; Hoffman, R.L.; Miller, N.; Almaden, J.; Chionis, J.; Zhang, Q.; Eisele, K.; Liu, C.; Zhang, C.; Huser, N.; et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 2021, 39, 1404–1421.e1411. [Google Scholar] [CrossRef]
- Navarro-Yepes, J.; Kettner, N.M.; Rao, X.; Bishop, C.S.; Bui, T.N.; Wingate, H.F.; Singareeka Raghavendra, A.; Wang, Y.; Wang, J.; Sahin, A.A.; et al. Abemaciclib Is Effective in Palbociclib-Resistant Hormone Receptor-Positive Metastatic Breast Cancers. Cancer Res. 2023, 83, 3264–3283. [Google Scholar] [CrossRef]
- Yano, S.; Tazawa, H.; Kagawa, S.; Fujiwara, T.; Hoffman, R.M. FUCCI Real-Time Cell-Cycle Imaging as a Guide for Designing Improved Cancer Therapy: A Review of Innovative Strategies to Target Quiescent Chemo-Resistant Cancer Cells. Cancers 2020, 12, 2655. [Google Scholar] [CrossRef] [PubMed]
- Bagegni, N.; Thomas, S.; Liu, N.; Luo, J.; Hoog, J.; Northfelt, D.W.; Goetz, M.P.; Forero, A.; Bergqvist, M.; Karen, J.; et al. Serum thymidine kinase 1 activity as a pharmacodynamic marker of cyclin-dependent kinase 4/6 inhibition in patients with early-stage breast cancer receiving neoadjuvant palbociclib. Breast Cancer Res. 2017, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Groenland, S.L.; Martinez-Chavez, A.; van Dongen, M.G.J.; Beijnen, J.H.; Schinkel, A.H.; Huitema, A.D.R.; Steeghs, N. Clinical Pharmacokinetics and Pharmacodynamics of the Cyclin-Dependent Kinase 4 and 6 Inhibitors Palbociclib, Ribociclib, and Abemaciclib. Clin. Pharmacokinet. 2020, 59, 1501–1520. [Google Scholar] [CrossRef]
- Harbeck, N.; Rastogi, P.; Martin, M.; Tolaney, S.M.; Shao, Z.M.; Fasching, P.A.; Huang, C.S.; Jaliffe, G.G.; Tryakin, A.; Goetz, M.P.; et al. Adjuvant abemaciclib combined with endocrine therapy for high-risk early breast cancer: Updated efficacy and Ki-67 analysis from the monarchE study. Ann. Oncol. 2021, 32, 1571–1581. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef]
- Kumarasamy, V.; Vail, P.; Nambiar, R.; Witkiewicz, A.K.; Knudsen, E.S. Functional Determinants of Cell Cycle Plasticity and Sensitivity to CDK4/6 Inhibition. Cancer Res. 2021, 81, 1347–1360. [Google Scholar] [CrossRef]
- Shrestha, M.; Wang, D.Y.; Ben-David, Y.; Zacksenhaus, E. CDK4/6 inhibitors and the pRB-E2F1 axis suppress PVR and PD-L1 expression in triple-negative breast cancer. Oncogenesis 2023, 12, 29. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem. Biol. 2019, 26, 1067–1080.e1068. [Google Scholar] [CrossRef]
- Gharbi, S.I.; Pelletier, L.A.; Espada, A.; Gutierrez, J.; Sanfeliciano, S.M.G.; Rauch, C.T.; Ganado, M.P.; Baquero, C.; Zapatero, E.; Zhang, A.; et al. Crystal structure of active CDK4-cyclin D and mechanistic basis for abemaciclib efficacy. NPJ Breast Cancer 2022, 8, 126. [Google Scholar] [CrossRef]
- Hickey, T.E.; Selth, L.A.; Chia, K.M.; Laven-Law, G.; Milioli, H.H.; Roden, D.; Jindal, S.; Hui, M.; Finlay-Schultz, J.; Ebrahimie, E.; et al. The androgen receptor is a tumor suppressor in estrogen receptor-positive breast cancer. Nat. Med. 2021, 27, 310–320. [Google Scholar] [CrossRef]
- Srivastava, T.P.; Ajmeriya, S.; Goel, I.; Talukdar, J.; Srivastava, A.; Parshad, R.; Deo, S.V.S.; Mathur, S.R.; Gogia, A.; Rai, A.; et al. Prognostic role of Androgen Receptor splice variant 7 (AR-V7) in the pathogenesis of breast cancer. BMC Cancer 2024, 24, 1398. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, D.; Kang, J.; Luo, H.; Tian, Y.; Liu, X.; Shao, C. ARv7 promotes the escape of prostate cancer cells from androgen deprivation therapy-induced senescence by mediating the SKP2/p27 axis. BMC Biol. 2025, 23, 66. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kim, C.Y.; Oh, J.H.; Kim, M.H. NR4A1 Regulates Tamoxifen Resistance by Suppressing ERK Signaling in ER-Positive Breast Cancer. Cells 2021, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, N.; Tang, J.; Wang, W.; Fang, F.; Sood, R.; O’Hagan, H.M.; Miller, K.D.; Clarke, R.; Nephew, K.P. Poly-ADP-Ribosylation of Estrogen Receptor-Alpha by PARP1 Mediates Antiestrogen Resistance in Human Breast Cancer Cells. Cancers 2019, 11, 43. [Google Scholar] [CrossRef]
- Fu, H.; Wu, Z.X.; Lei, Z.N.; Teng, Q.X.; Yang, Y.; Ashby, C.R.; Lei, Y.; Lian, Y.; Chen, Z.S. The Resistance of Cancer Cells to Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, is Mediated by the ABCB1 Transporter. Front. Pharmacol. 2022, 13, 861642. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef]
- Dhakal, A.; Falkson, C.; O’Regan, R.M. Adjuvant cyclin-dependent kinase 4/6 inhibition in hormone receptor-positive breast cancer: One Monarch to rule them all? Cancer 2021, 127, 3302–3309. [Google Scholar] [CrossRef]
- Hamilton, E.; Cortes, J.; Ozyilkan, O.; Chen, S.C.; Petrakova, K.; Manikhas, A.; Jerusalem, G.; Hegg, R.; Huober, J.; Chapman, S.C.; et al. nextMONARCH: Abemaciclib Monotherapy or Combined With Tamoxifen for Metastatic Breast Cancer. Clin. Breast Cancer 2021, 21, 181–190.e182. [Google Scholar] [CrossRef]
- Hamilton, E.; Cortes, J.; Ozyilkan, O.; Chen, S.C.; Petrakova, K.; Manikhas, A.; Jerusalem, G.; Hegg, R.; Huober, J.; Zhang, W.; et al. nextMONARCH Phase 2 randomized clinical trial: Overall survival analysis of abemaciclib monotherapy or in combination with tamoxifen in patients with endocrine-refractory HR+, HER2− metastatic breast cancer. Breast Cancer Res. Treat. 2022, 195, 55–64. [Google Scholar] [CrossRef]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Kalinsky, K.; Bianchini, G.; Hamilton, E.P.; Graff, S.L.; Park, K.H.; Jeselsohn, R.; Demirci, U.; Martin, M.; Layman, R.M.; Hurvitz, S.A.; et al. Abemaciclib plus fulvestrant vs fulvestrant alone for HR+, HER2− advanced breast cancer following progression on a prior CDK4/6 inhibitor plus endocrine therapy: Primary outcome of the phase 3 postMONARCH trial. J. Clin. Oncol. 2024, 42 (Suppl. S17), LBA1001. [Google Scholar] [CrossRef]
- Kalinsky, K.; Accordino, M.K.; Chiuzan, C.; Mundi, P.S.; Sakach, E.; Sathe, C.; Ahn, H.; Trivedi, M.S.; Novik, Y.; Tiersten, A.; et al. Randomized Phase II Trial of Endocrine Therapy with or Without Ribociclib After Progression on Cyclin-Dependent Kinase 4/6 Inhibition in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: MAINTAIN Trial. J. Clin. Oncol. 2023, 41, 4004–4013. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Chen, S. The development, application and limitations of breast cancer cell lines to study tamoxifen and aromatase inhibitor resistance. J. Steroid. Biochem. Mol. Biol. 2012, 131, 83–92. [Google Scholar] [CrossRef]
- Abstract CT048: Population pharmacokinetics and pharmacodynamics for an oral Notch inhibitor, LY3039478, in the first-in-man study. Cancer Res. 2016, 76. [CrossRef]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef]
- Pesch, A.M.; Hirsh, N.H.; Michmerhuizen, A.R.; Jungles, K.M.; Wilder-Romans, K.; Chandler, B.C.; Liu, M.; Lerner, L.M.; Nino, C.A.; Ward, C.; et al. RB expression confers sensitivity to CDK4/6 inhibitor-mediated radiosensitization across breast cancer subtypes. JCI Insight 2022, 7, e154402. [Google Scholar] [CrossRef]
- Xu, X.Q.; Pan, X.H.; Wang, T.T.; Wang, J.; Yang, B.; He, Q.J.; Ding, L. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol. Sin. 2021, 42, 171–178. [Google Scholar] [CrossRef]
- Torres-Guzman, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef] [PubMed]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Spring, L.M.; Bardia, A.; Wander, S.A. Mechanisms of Resistance to CDK4/6 Blockade in Advanced Hormone Receptor-positive, HER2−negative Breast Cancer and Emerging Therapeutic Opportunities. Clin. Cancer Res. 2022, 28, 821–830. [Google Scholar] [CrossRef]
- Belli, S.; Esposito, D.; Ascione, C.M.; Messina, F.; Napolitano, F.; Servetto, A.; De Angelis, C.; Bianco, R.; Formisano, L. EGFR and HER2 hyper-activation mediates resistance to endocrine therapy and CDK4/6 inhibitors in ER+ breast cancer. Cancer Lett. 2024, 593, 216968. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.; Chenoweth, A.M.; Johansson, A.; Laddach, R.; Guppy, N.; Trendell, J.; Esapa, B.; Mavousian, A.; Navarro-Llinas, B.; Haider, S.; et al. Anti-EGFR Antibody-Drug Conjugate Carrying an Inhibitor Targeting CDK Restricts Triple-Negative Breast Cancer Growth. Clin. Cancer Res. 2024, 30, 3298–3315. [Google Scholar] [CrossRef]
- Puppe, J.; Opdam, M.; Schouten, P.C.; Jozwiak, K.; Lips, E.; Severson, T.; van de Ven, M.; Brambillasca, C.; Bouwman, P.; van Tellingen, O.; et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin. Cancer Res. 2019, 25, 4351–4362. [Google Scholar] [CrossRef]
- Menendez, J.A.; Papadimitropoulou, A.; Vander Steen, T.; Cuyas, E.; Oza-Gajera, B.P.; Verdura, S.; Espinoza, I.; Vellon, L.; Mehmi, I.; Lupu, R. Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer. Cancers 2021, 13, 1132. [Google Scholar] [CrossRef]
- Vafeiadou, V.; Hany, D.; Picard, D. Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERalpha-Positive Breast Cancer. Cancers 2022, 14, 954. [Google Scholar] [CrossRef]
- Sakaue-Sawano, A.; Kobayashi, T.; Ohtawa, K.; Miyawaki, A. Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biol. 2011, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Volsky, D.J. PAGE: Parametric analysis of gene set enrichment. BMC Bioinform. 2005, 6, 144. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Turner, K.; Chappell, J.; Kulanthaivel, P.; Ng, W.T.; Royalty, J. Food effect on the pharmacokinetics of 200-mg abemaciclib in healthy subject [abstract]. In Proceedings of the 107th Annual Meeting of the American Association for Cancer Research, New Orleans, LA, USA, 16–20 April 2016. Abstract no. CT152.. [Google Scholar]
- Flaherty, K.T.; LoRusso, P.M.; DeMichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, Dose-Escalation Trial of the Oral Cyclin-Dependent Kinase 4/6 Inhibitor PD 0332991, Administered Using a 21-Day Schedule in Patients with Advanced Cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical Pharmacology Review Ribociclib. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209092Orig1s000ChemR.pdf (accessed on 22 February 2024).
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef]
- Austin, R.P.; Barton, P.; Cockroft, S.L.; Wenlock, M.C.; Riley, R.J. The Influence of Nonspecific Microsomal Binding on Apparent Intrinsic Clearance, and Its Prediction from Physicochemical Properties. Drug Metab. Dispos. 2002, 30, 1497–1503. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapatero-Solana, E.; Ding, Y.; Pulliam, N.; de Dios, A.; Ortiz-Ruiz, M.J.; Lallena, M.J. Models of Early Resistance to CDK4/6 Inhibitors Unveil Potential Therapeutic Treatment Sequencing. Int. J. Mol. Sci. 2025, 26, 2643. https://doi.org/10.3390/ijms26062643
Zapatero-Solana E, Ding Y, Pulliam N, de Dios A, Ortiz-Ruiz MJ, Lallena MJ. Models of Early Resistance to CDK4/6 Inhibitors Unveil Potential Therapeutic Treatment Sequencing. International Journal of Molecular Sciences. 2025; 26(6):2643. https://doi.org/10.3390/ijms26062643
Chicago/Turabian StyleZapatero-Solana, Elisabet, Yan Ding, Nicholas Pulliam, Alfonso de Dios, Maria Jesus Ortiz-Ruiz, and María José Lallena. 2025. "Models of Early Resistance to CDK4/6 Inhibitors Unveil Potential Therapeutic Treatment Sequencing" International Journal of Molecular Sciences 26, no. 6: 2643. https://doi.org/10.3390/ijms26062643
APA StyleZapatero-Solana, E., Ding, Y., Pulliam, N., de Dios, A., Ortiz-Ruiz, M. J., & Lallena, M. J. (2025). Models of Early Resistance to CDK4/6 Inhibitors Unveil Potential Therapeutic Treatment Sequencing. International Journal of Molecular Sciences, 26(6), 2643. https://doi.org/10.3390/ijms26062643