
Human Brain Endothelial Cell-Derived Extracellular Vesicles Reduce Toxoplasma gondii Infection In Vitro in Human Brain and Umbilical Cord Vein Endothelial Cells
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introdution
2. Results
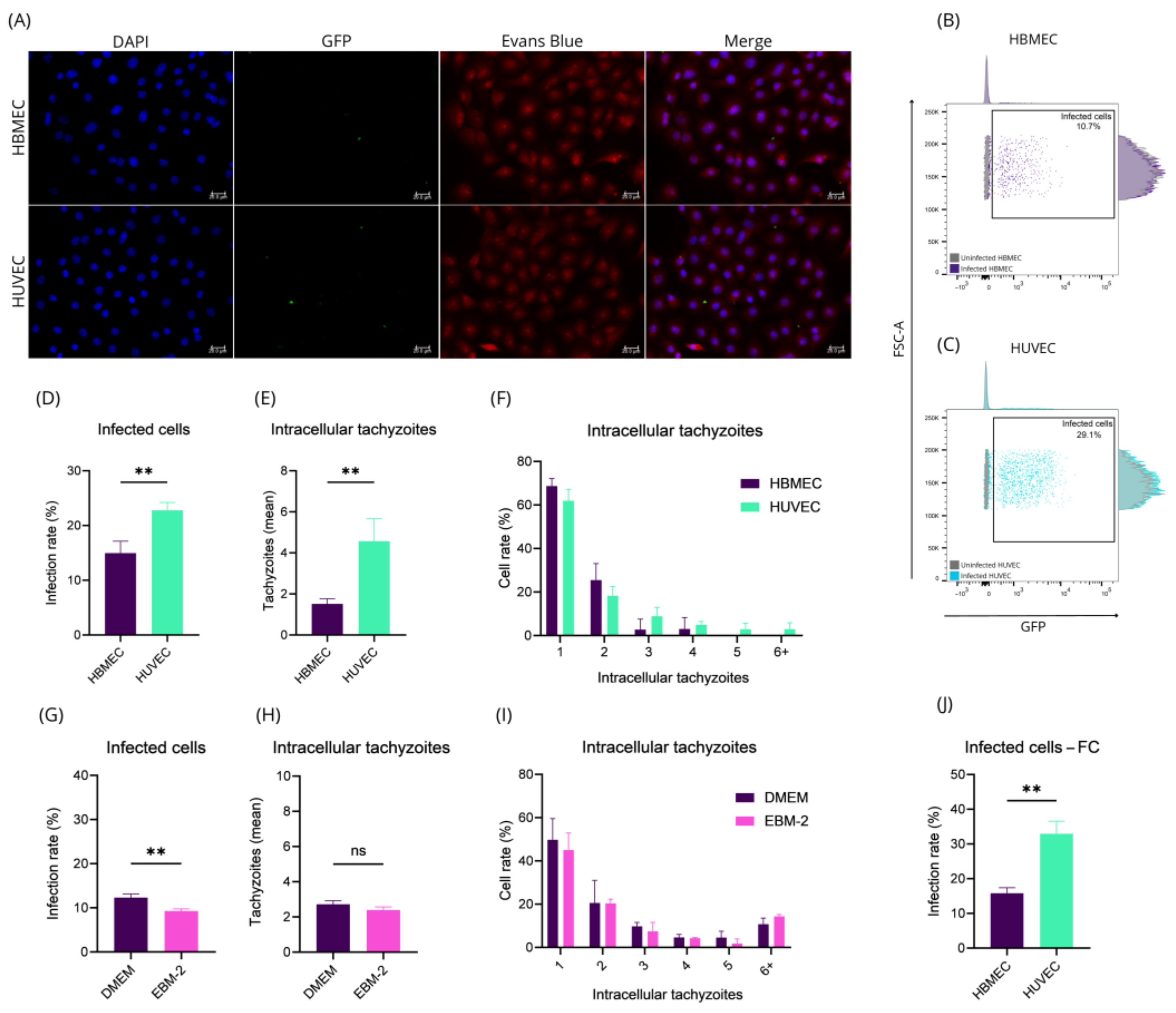
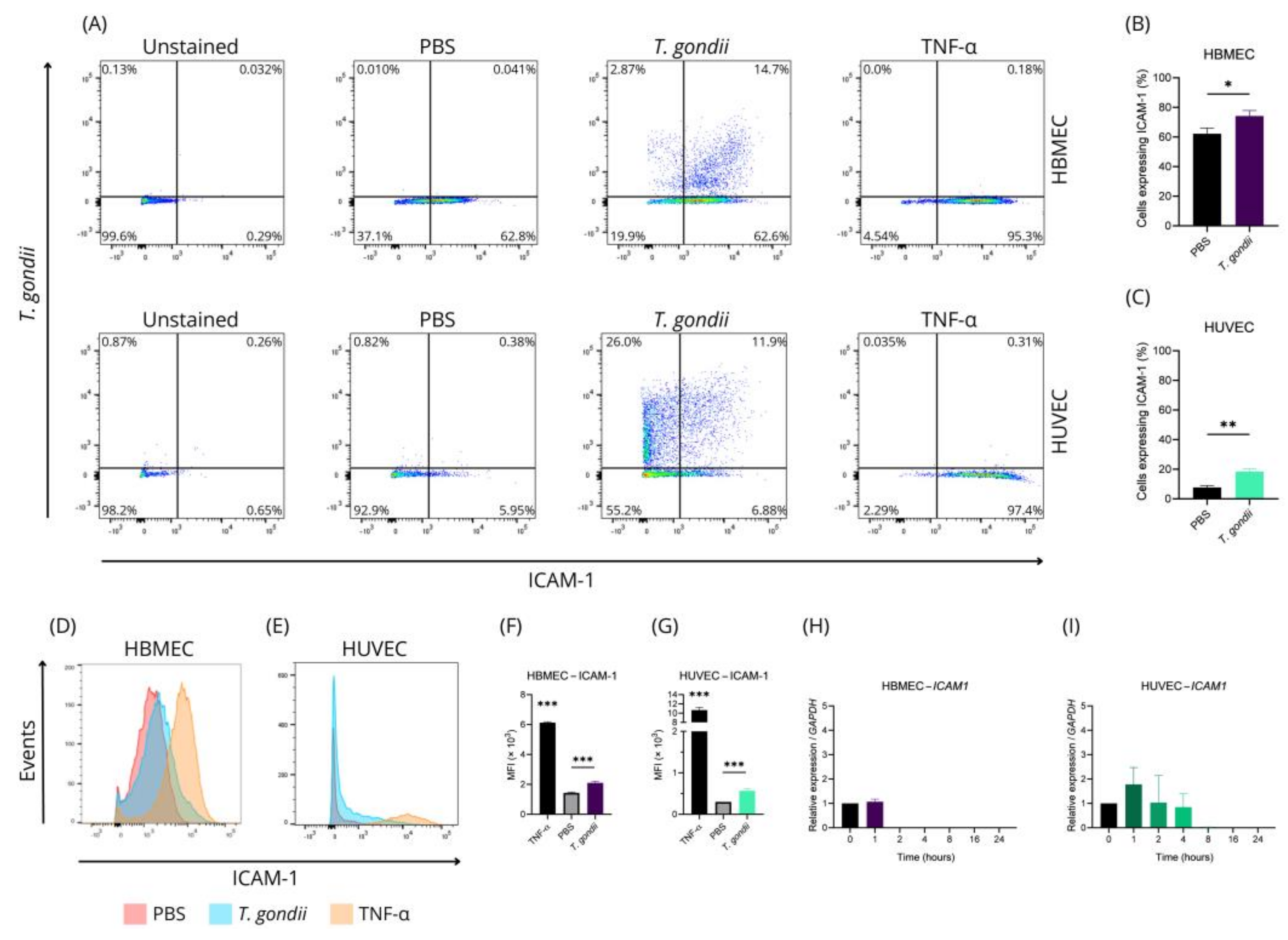
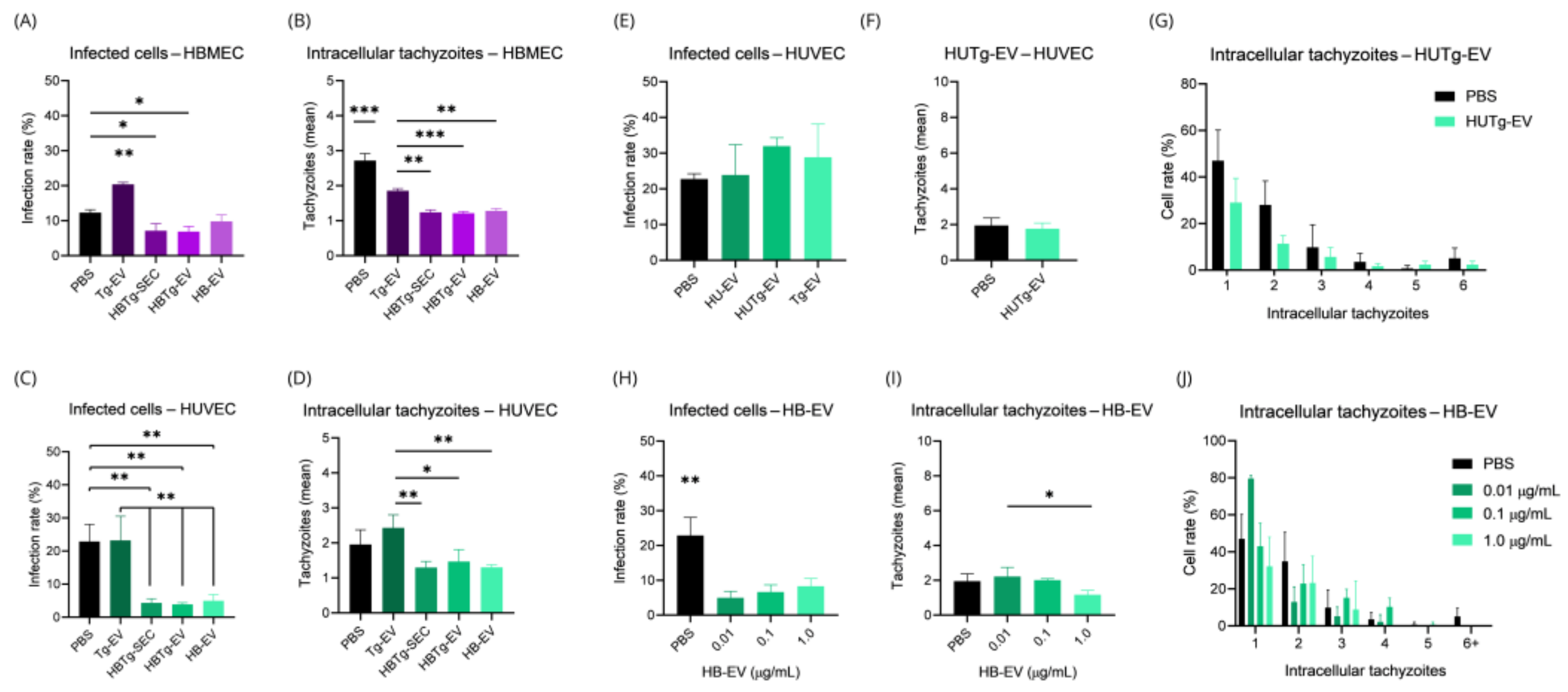
2.1. HBMEC and HUVEC Cells Respond Distinctly to Toxoplasma gondii Infection
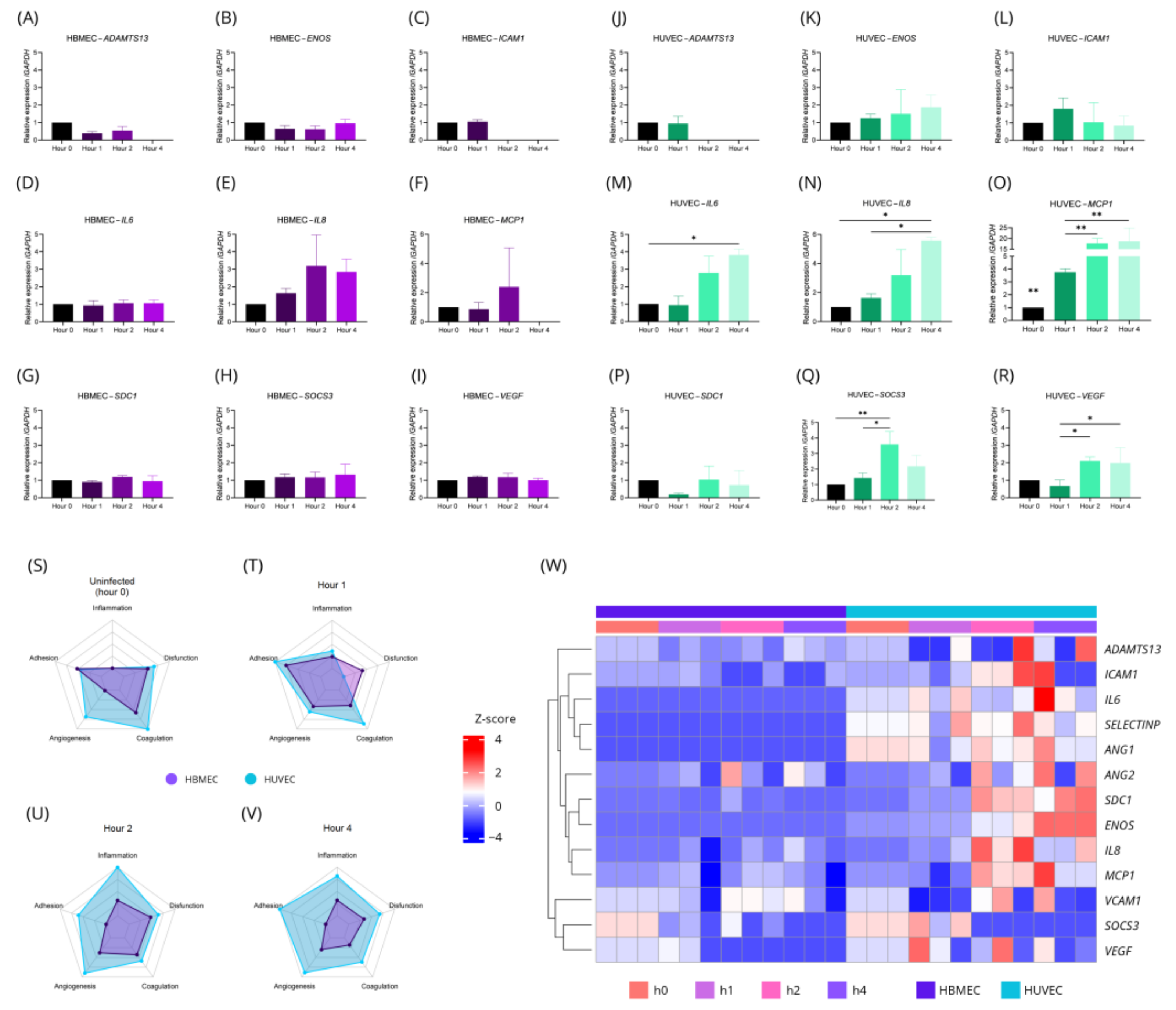
2.2. HBMEC and HUVEC Cells Have Characteristic Gene and Protein Changes in T. gondii Infection
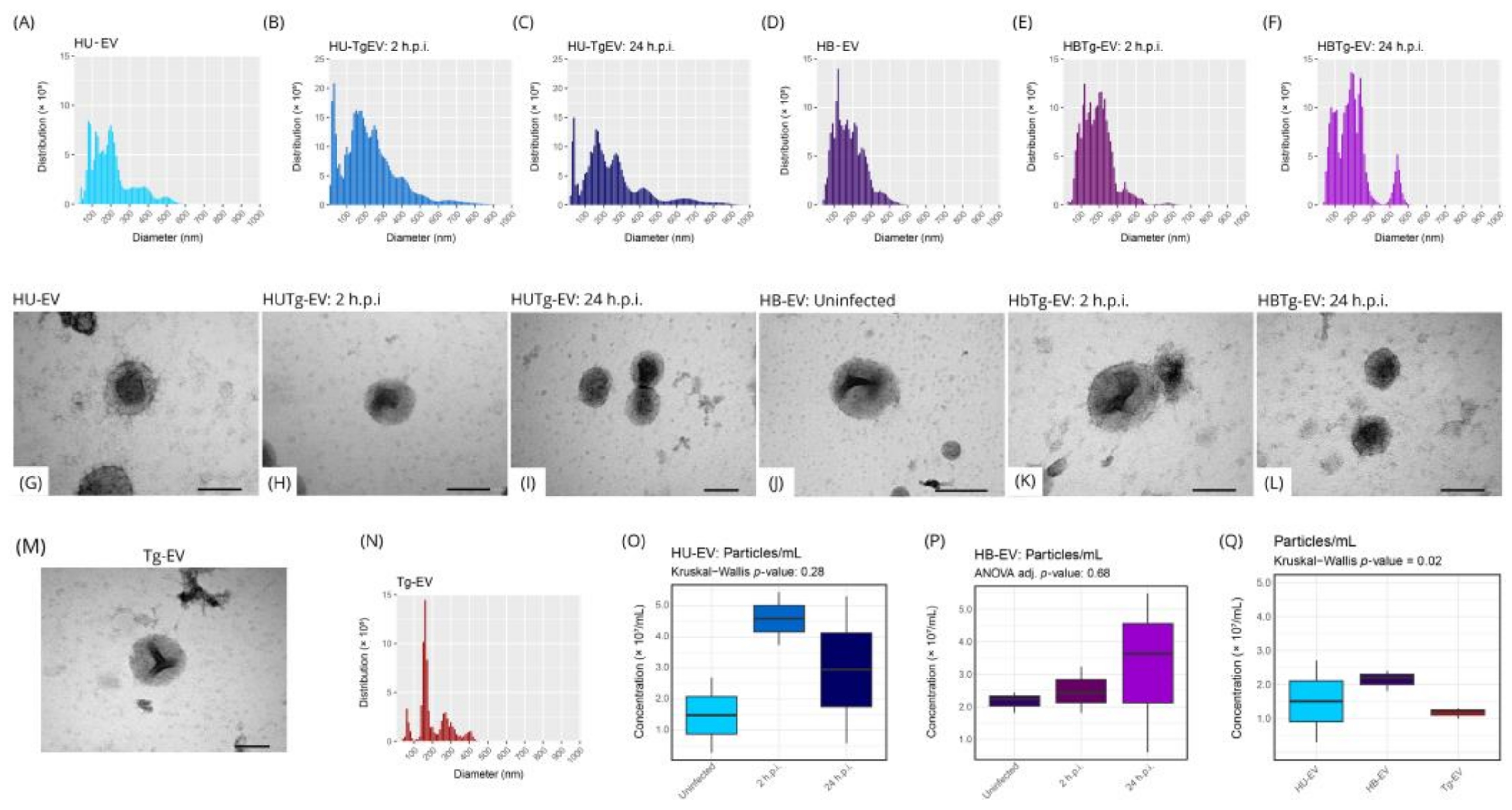
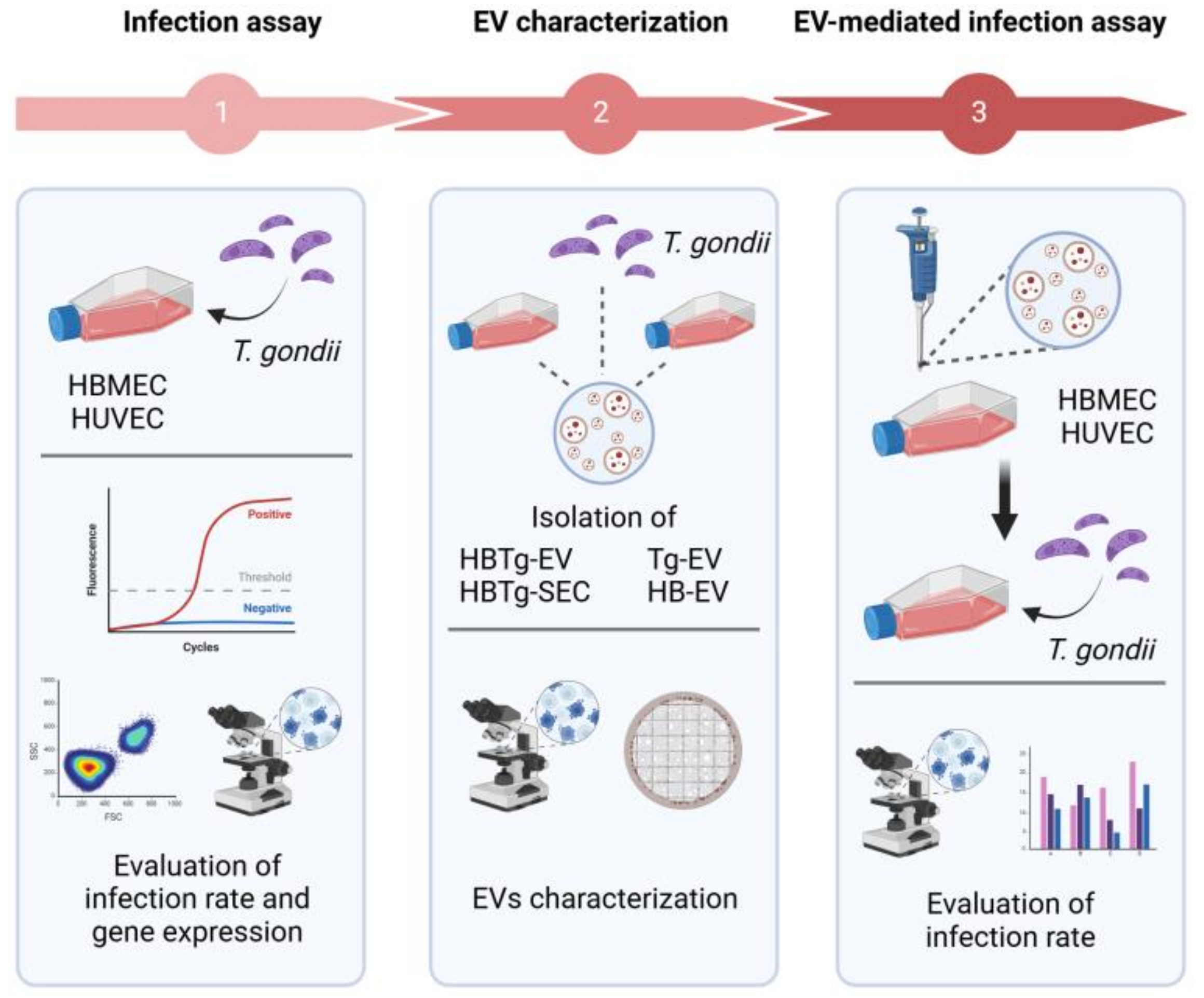
2.3. HBMEC HUVEC and T. gondii Secret EVs with Similar Size and Distribution
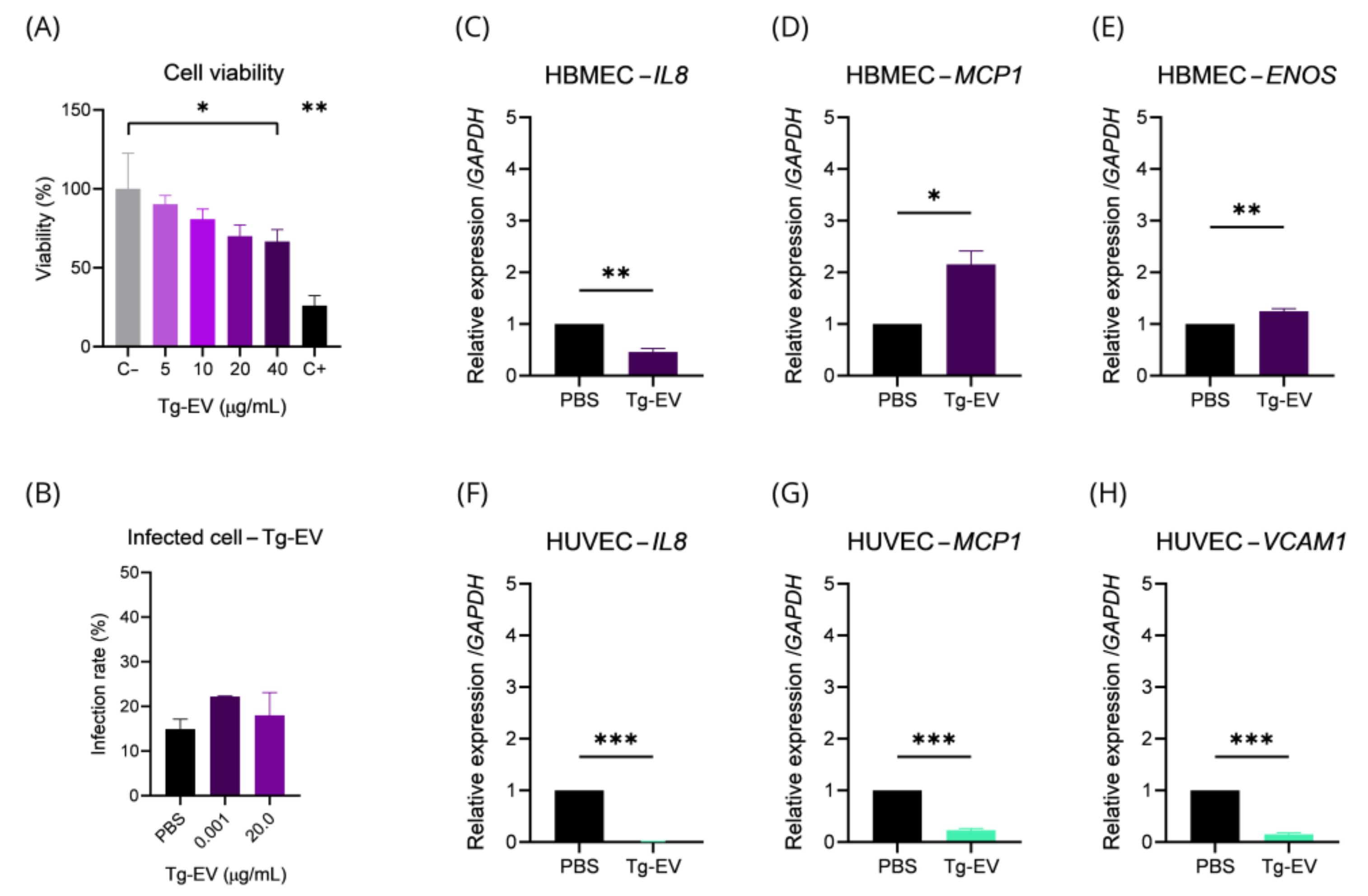
2.4. T. gondii EVs Can Affect Gene Transcription and Endothelial Cell Viability
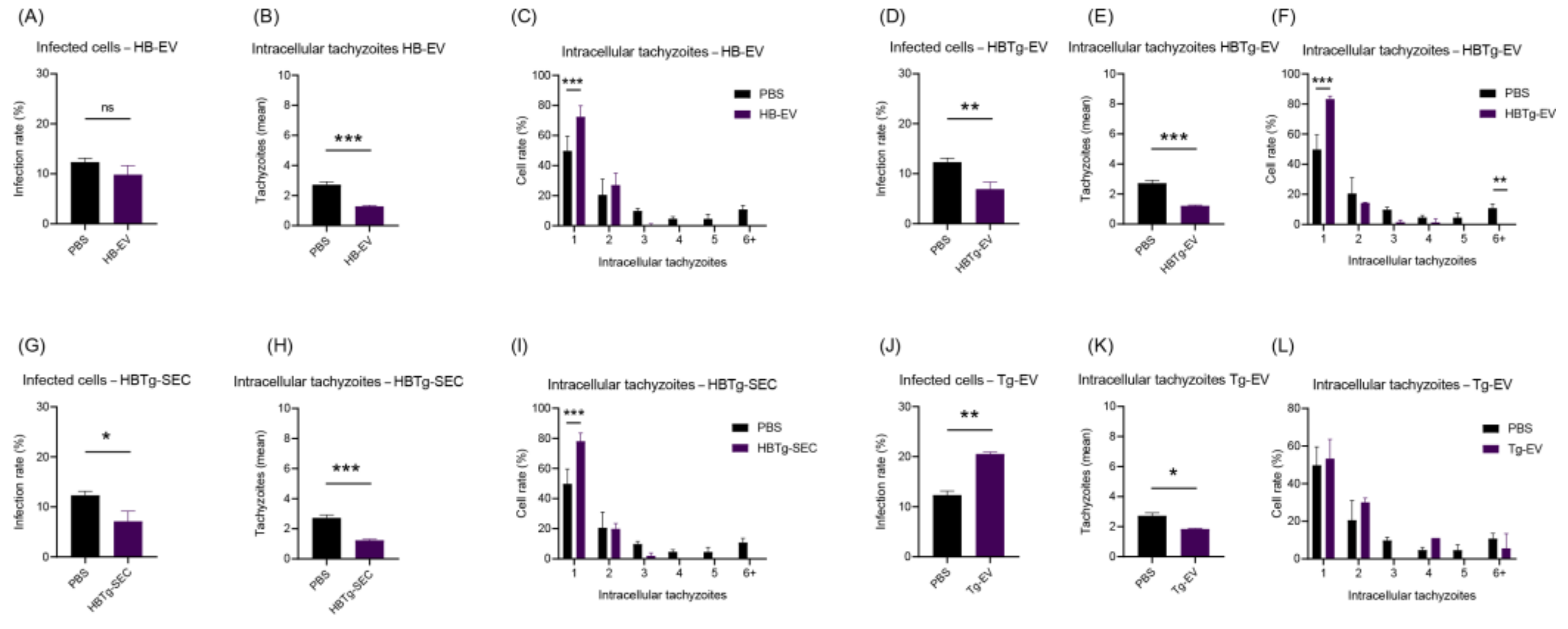
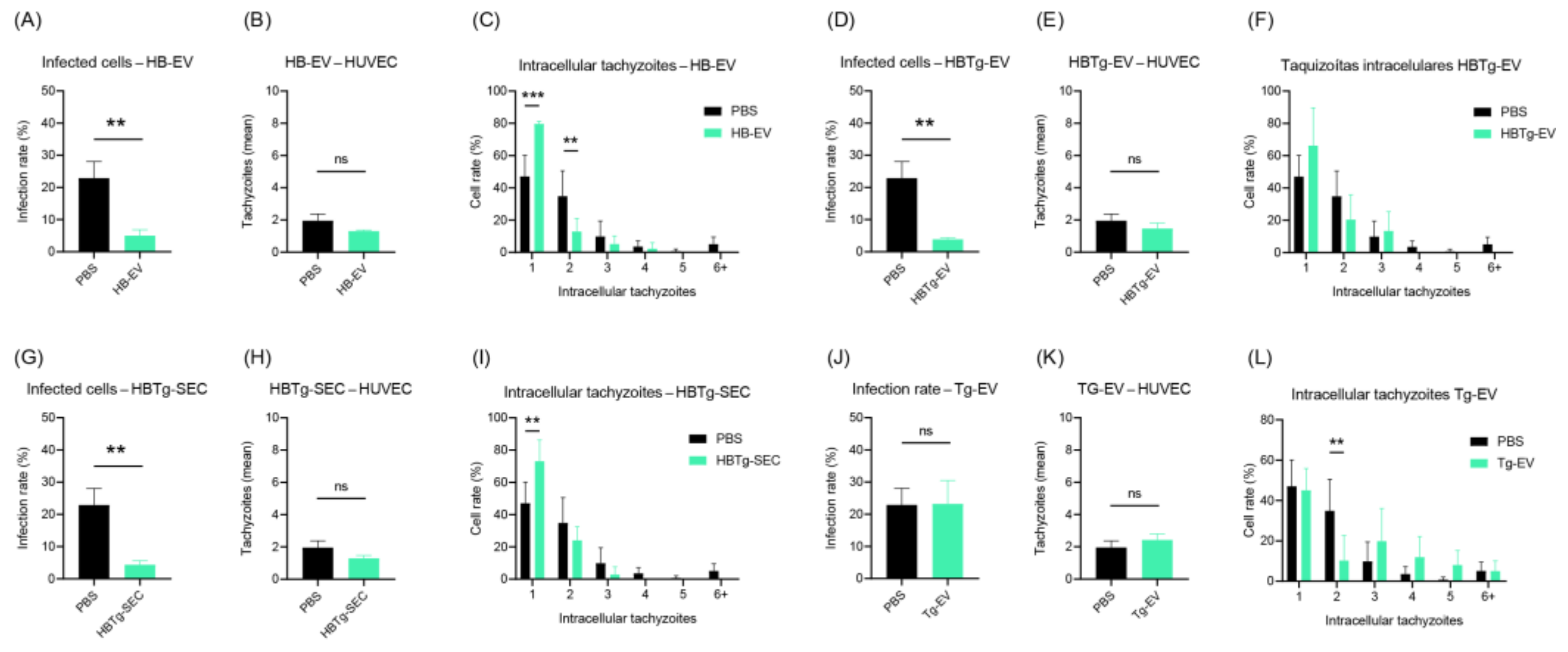
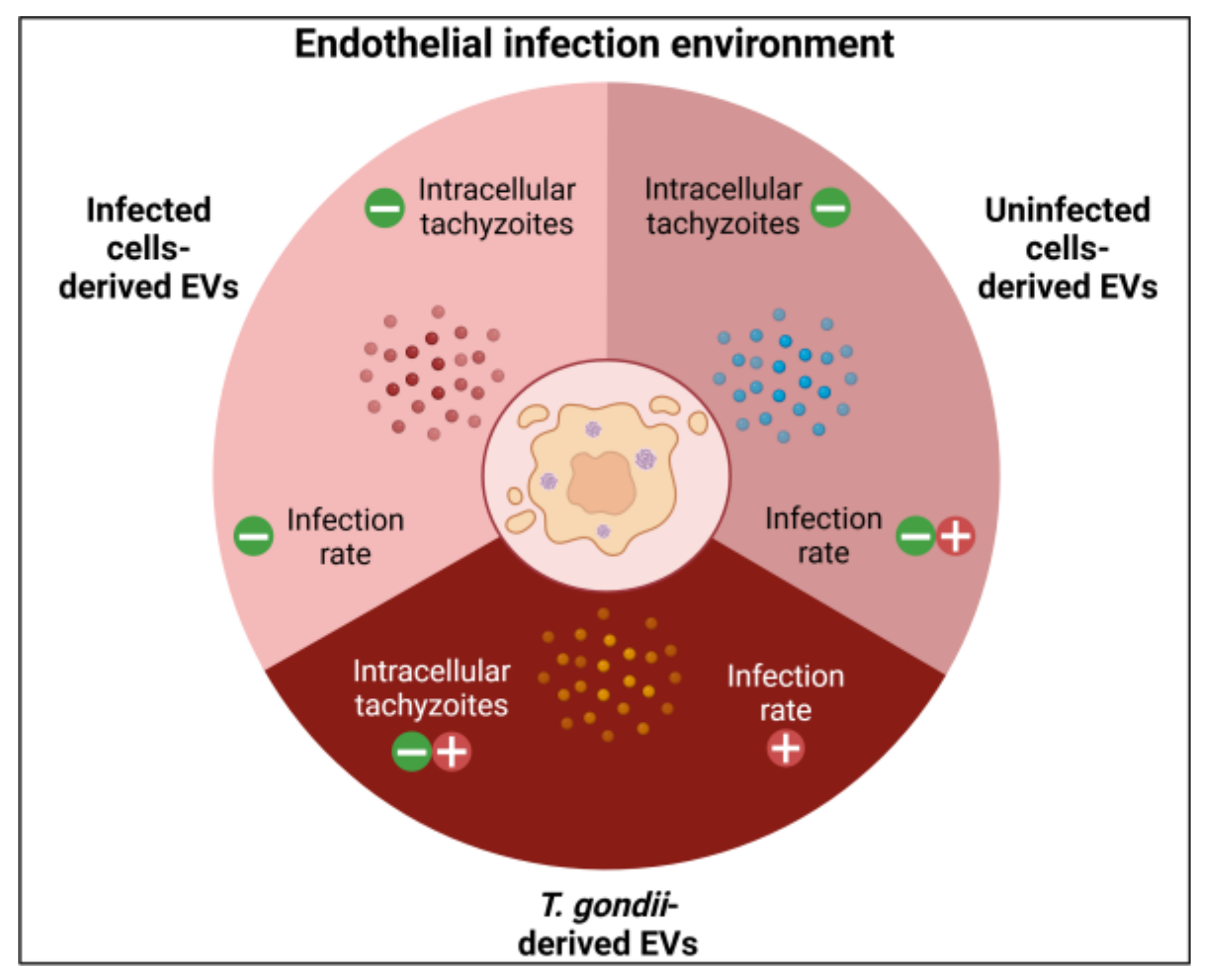
2.5. HBMECs Supernatant Contains EVs That Promote a Protective Effect in the Infectious Context on HBMEC and HUVEC
3. Discussion
4. Materials and Methods
4.1. Cells and Parasite Culture
4.2. Fetal Bovine Serum Depletion and Isolation of Extracellular Vesicles
4.3. Characterization of Extracellular Vesicles
4.4. Extracellular Vesicle Cytotoxicity Assay
4.5. Assessment of Gene Expression in Endothelial Cells Infected by T. gondii
4.6. Quantification of Transcripts by qPCR
4.7. Infection Assay
4.8. Assessment of T. gondii Infection and Adhesion Molecule Expression
4.9. Assessment of T. gondii by Electron Microscopy
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zovein, A.C.; Hofmann, J.J.; Lynch, M.; French, W.J.; Turlo, K.A.; Yang, Y.; Becker, M.S.; Zanetta, L.; Dejana, E.; Gasson, J.C.; et al. Article Fate Tracing Reveals the Endothelial Origin of Hematopoietic Stem Cells. Stem Cell 2008, 3, 625–636. [Google Scholar] [CrossRef]
- Pérez-Cremades, D.; Bueno-Betí, C.; García-Giménez, J.L.; Ibañez-Cabellos, J.S.; Hermenegildo, C.; Pallardó, F.V.; Novella, S. Extracellular histones disarrange vasoactive mediators release through a COX-NOS interaction in human endothelial cells. J. Cell. Mol. Med. 2017, 21, 1584–1592. [Google Scholar] [CrossRef]
- Schoergenhofer, C.; Schwameis, M.; Gelbenegger, G.; Buchtele, N.; Thaler, B.; Mussbacher, M.; Schabbauer, G.; Wojta, J.; Jilma-Stohlawetz, P.; Jilma, B. Inhibition of Protease-Activated Receptor (PAR1) Reduces Activation of the Endothelium, Coagulation, Fibrinolysis and Inflammation during Human Endotoxemia. Thromb. Haemost. 2018, 118, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Chia, P.Y.; Teo, A.; Yeo, T.W. Overview of the Assessment of Endothelial Function in Humans. Front. Med. 2020, 7, 542567. [Google Scholar] [CrossRef] [PubMed]
- Wilhelms, D.B.; Kirilov, M.; Mirrasekhian, E.; Eskilsson, A.; Kugelberg, U.O.; Klar, C.; Ridder, D.A.; Herschman, H.R.; Schwaninger, M.; Blomqvist, A.; et al. Deletion of Prostaglandin E2 Synthesizing Enzymes in Brain Endothelial Cells Attenuates Inflammatory Fever. J. Neurosci. 2014, 34, 11684–11690. [Google Scholar] [CrossRef]
- Hernanz, R.; Martínez-Revelles, S.; Palacios, R.; Martín, A.; Cachofeiro, V.; Aguado, A.; García-Redondo, L.; Barrús, M.T.; De Batista, P.R.; Briones, A.M.; et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br. J. Pharmacol. 2015, 172, 3159–3176. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Ledbetter, S.R.; Claffey, K.P.; Papadopoulos-Sergiou, A.; Perruzzi, C.A.; Detmar, M. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the α(v)β3 integrin, osteopontin, and thrombin. Am. J. Pathol. 1996, 149, 293–305. [Google Scholar]
- Ku, Y.H.; Cho, B.J.; Kim, M.J.; Lim, S.; Park, Y.J.; Jang, H.C.; Choi, S.H. Rosiglitazone increases endothelial cell migration and vascular permeability through Akt phosphorylation. BMC Pharmacol. Toxicol. 2017, 18, 62. [Google Scholar] [CrossRef]
- Hellenthal, K.E.M.; Brabenec, L.; Wagner, N.M. Regulation and Dysregulation of Endothelial Permeability during Systemic Inflammation. Cells 2022, 11, 1935. [Google Scholar] [CrossRef]
- Wang, S.; Wettschureck, N.; Offermanns, S.; Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef]
- Trimm, E.; Red-Horse, K. Vascular endothelial cell development and diversity. Nat. Rev. Cardiol. 2023, 20, 197–210. [Google Scholar] [CrossRef]
- Iwabayashi, M.; Taniyama, Y.; Sanada, F.; Azuma, J.; Iekushi, K. Role of serotonin in angiogenesis: Induction of angiogenesis by sarpogrelate via endothelial 5-HT1B/Akt/eNOS pathway in diabetic mice. Atherosclerosis 2012, 220, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Nobe, K.; Miyatake, M.; Sone, T.; Honda, K. High-Glucose-Altered Endothelial Cell Function Involves Both Disruption of Cell-to-Cell Connection and Enhancement of Force Development. J. Pharmacol. Exp. Ther. 2006, 318, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Masse, E.M.; Herzberg, K.T.; Meyers, M.S.; Yeo, K.T.; Yeo, T.K.; Sioussat, T.M.; Dvorak, H.F. Pathogenesis of Ascites Tumor Growth: Vascular Permeability Factor, Vascular Hyperpermeability, and Ascites Fluid Accumulation. Cancer Res. 1995, 55, 360–368. [Google Scholar] [PubMed]
- Herr, D.; Sallmann, A.; Bekes, I.; Konrad, R.; Holzheu, I.; Kreienberg, R.; Wulff, C. VEGF induces ascites in ovarian cancer patients via increasing peritoneal permeability by downregulation of Claudin 5. Gynecol. Oncol. 2012, 127, 210–216. [Google Scholar] [CrossRef]
- De Assis, M.C.; Da Costa, A.O.; Barja-Fidalgo, T.C.; Plotkowski, M.C. Human endothelial cells are activated by interferon-γ plus tumour necrosis factor-α to kill intracellular Pseudomonas aeruginosa. Immunology 2000, 101, 271–278. [Google Scholar] [CrossRef]
- Calvert, J.K.; Helbig, K.J.; Dimasi, D.; Cockshell, M.; Beard, M.R.; Pitson, S.M.; Bonder, C.S.; Carr, J.M. Dengue Virus Infection of Primary Endothelial Cells Induces Innate Immune Responses, Changes in Endothelial Cells Function and Is Restricted by Interferon-Stimulated Responses. J. Interf. Cytokine Res. 2015, 35, 654–665. [Google Scholar] [CrossRef]
- Liu, M.; Spellberg, B.; Phan, Q.T.; Fu, Y.; Fu, Y.; Lee, A.S.; Edwards, J.E.; Filler, S.G.; Ibrahim, A.S. The endothelial cell receptor GRP78 is required for mucormycosis pathogenesis in diabetic mice. J. Clin. Investig. 2010, 120, 1914–1924. [Google Scholar] [CrossRef]
- Debrah, A.Y.; Mand, S.; Toliat, M.R.; Marfo-Debrekyei, Y.; Batsa, L.; Nürnberg, P.; Lawson, B.; Adjei, O.; Hoerauf, A.; Pfarr, K. Plasma vascular endothelial growth factor-A (VEGF-A) and VEGF-A gene polymorphism are associated with hydrocele development in lymphatic filariasis. Am. J. Trop. Med. Hyg. 2007, 77, 601–608. [Google Scholar] [CrossRef]
- Knight, B.C.; Brunton, C.L.; Modi, N.C.; Wallace, G.R.; Stanford, M.R. The effect of Toxoplasma gondii infection on expression of chemokines by rat retinal vascular endothelial cells. J. Neuroimmunol. 2005, 160, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mao, Y.; Ramirez, S.H.; Tuma, R.F.; Chabrashvili, T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 2010, 171, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Riden, M. Retinal endothelial cells are more susceptible to oxidative stress and increased permeability than brain-derived endothelial cells. Microvasc. Res. 2003, 65, 18–23. [Google Scholar] [CrossRef]
- Konradt, C.; Ueno, N.; Christian, D.A.; Delong, J.H.; Pritchard, G.H.; Herz, J.; Bzik, D.J.; Koshy, A.A.; McGavern, D.B.; Lodoen, M.B.; et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 2016, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lachenmaier, S.M.; Deli, M.A.; Meissner, M.; Liesenfeld, O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J. Neuroimmunol. 2011, 232, 119–130. [Google Scholar] [CrossRef]
- Daneman, R.; Rescigno, M. The Gut Immune Barrier and the Blood-Brain Barrier: Are They So Different? Immunity 2009, 31, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Remington, J.S. Clinical Practice: Management of Toxoplasma gondii Infection during Pregnancy. Clin. Infect. Dis. 2008, 47, 554–566. [Google Scholar] [CrossRef]
- Ueno, N.; Harker, K.S.; Clarke, E.V.; McWhorter, F.Y.; Liu, W.F.; Tenner, A.J.; Lodoen, M.B. Real-time imaging of Toxoplasma-infected human monocytes under fluidic shear stress reveals rapid translocation of intracellular parasites across endothelial barriers. Cell Microbiol. 2014, 16, 580–595. [Google Scholar] [CrossRef]
- Harker, K.S.; Ueno, N.; Wang, T.; Bonhomme, C.; Liu, W.; Lodoen, M.B. Toxoplasma gondii modulates the dynamics of human monocyte adhesion to vascular endothelium under fluidic shear stress. J. Leukoc. Biol. 2013, 93, 789–800. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Bannai, H.; Xuan, X.; Nishikawa, Y. Toxoplasma gondii cyclophilin 18-mediated production of nitric oxide induces bradyzoite conversion in a CCR5-dependent manner. Infect. Immun. 2009, 77, 3686–3695. [Google Scholar] [CrossRef]
- Scharton-Kersten, T.M.; Yap, G.; Magram, J.; Sher, A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 1997, 185, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Franklin-Murray, A.L.; Mallya, S.; Jankeel, A.; Sureshchandra, S.; Messaoudi, I.; Lodoen, M., B. Toxoplasma gondii Dysregulates Barrier Function and Mechanotransduction Signaling in Human Endothelial Cells. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Harun, M.S.R.; Marsh, V.; Elsaied, N.A.; Webb, K.F.; Elsheikha, H.M. Effects of Toxoplasma gondii infection on the function and integrity of human cerebrovascular endothelial cells and the influence of verapamil treatment in vitro. Brain Res. 2020, 1746, 147002. [Google Scholar] [CrossRef]
- Dincel, G.C.; Atmaca, H.T. Nitric oxide production increases during Toxoplasma gondii encephalitis in mice. Exp. Parasitol. 2015, 156, 104–112. [Google Scholar] [CrossRef]
- Palviainen, M.; Saraswat, M.; Varga, Z.; Kitka, D.; Neuvonen, M.; Puhka, M.; Joenväärä, S.; Renkonen, R.; Nieuwland, R.; Takatalo, M.; et al. Extracellular vesicles from human plasma and serum are carriers of extravesicular cargo—Implications for biomarker discovery. PLoS ONE 2020, 15, e0236439. [Google Scholar] [CrossRef] [PubMed]
- Tessandier, N.; Melki, I.; Cloutier, N.; Allaeys, I.; Miszta, A.; Tan, S.; Milasan, A.; Michel, S.; Benmoussa, A.; Lévesque, T.; et al. Platelets Disseminate Extracellular Vesicles in Lymph in Rheumatoid Arthritis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 929–942. [Google Scholar] [CrossRef]
- Jimenez, L.; Yu, H.; McKenzie, A.J.; Franklin, J.L.; Patton, J.G.; Liu, Q.; Weaver, A.M. Quantitative Proteomic Analysis of Small and Large Extracellular Vesicles (EVs) Reveals Enrichment of Adhesion Proteins in Small EVs. J. Proteome Res. 2019, 18, 947–959. [Google Scholar] [CrossRef]
- Liang, K.; Liu, F.; Fan, J.; Sun, D.; Liu, C.; Lyon, C.J.; Bernard, D.W.; Li, Y.; Yokoi, K.; Katz, M.H.; et al. Nanoplasmonic quantification of tumour-derived extracellular vesicles in plasma microsamples for diagnosis and treatment monitoring. Nat. Biomed. Eng. 2017, 1, 1–11. [Google Scholar] [CrossRef]
- Lázaro-Ibáñez, E.; Sanz-Garcia, A.; Visakorpi, T.; Escobedo-Lucea, C.; Siljander, P.; Ayuso-Sacido, Á.; Yliperttula, M. Different gDNA Content in the Subpopulations of Prostate Cancer Extracellular Vesicles: Apoptotic Bodies, Microvesicles, and Exosomes. Prostate 2014, 74, 1379–1390. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nakayasu, E.S.; Oliveira, D.L.; Nimrichter, L.; Nosanchuk, J.D.; Almeida, I.C.; Casadevall, A. Extracellular Vesicles Produced by Cryptococcus neoformans Contain Protein Components Associated with Virulence. Eukaryot. Cell 2008, 7, 58–67. [Google Scholar] [CrossRef]
- Tedford, E.; Badya, N.B.; Laing, C.; Asaoka, N.; Kaneko, S.; Filippi, B.M.; McConkey, G.A. Infection-induced extracellular vesicles evoke neuronal transcriptional and epigenetic changes. Sci. Rep. 2023, 13, 1–16. [Google Scholar] [CrossRef]
- Silva, V.O.; Maia, M.M.; Torrecilhas, A.C.; Taniwaki, N.N.; Namiyama, G.M.; Oliveira, K.C.; Ribeiro, K.S.; Toledo, M.d.S.; Xander, P.; Pereira-Chioccola, V.L. Extracellular vesicles isolated from Toxoplasma gondii induce host immune response. Parasite Immunol. 2018, 40, e12571. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Xiu, F.; Wang, J.; Cong, H.; He, S.; Shi, Y.; Wang, X.; Li, X.; Zhou, H. Characterization of exosomes derived from Toxoplasma gondii and their functions in modulating immune responses. Int. J. Nanomed. 2018, 13, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Lozano-Amado, D.; Chowdhury, D.; Singh, U. Extracellular Vesicles and Their Impact on the Biology of Protozoan Parasites. Trop. Med. Infect. Dis. 2023, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Maia, M.M.; da Cruz, A.B.; Taniwaki, N.N.; Namiyama, G.M.; Gava, R.; Gomes, A.H.S.; Kanamura, C.T.; Barbo, M.L.P.; Pereira-Chioccola, V.L. Immunization with extracellular vesicles excreted by Toxoplasma gondii confers protection in murine infection, activating cellular and humoral responses. Int. J. Parasitol. 2021, 51, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Wowk, P.F.; Zardo, M.L.; Miot, H.T.; Goldenberg, S.; Carvalho, P.C.; Mörking, P.A. Proteomic profiling of extracellular vesicles secreted from Toxoplasma gondii. Proteomics 2017, 17, 15–16. [Google Scholar] [CrossRef]
- Quiarim, T.M.; Maia, M.M.; da Cruz, A.B.; Taniwaki, N.N.; Namiyama, G.M.; Pereira-Chioccola, V.L. Characterization of extracellular vesicles isolated from types I, II and III strains of Toxoplasma gondii. Acta Trop. 2021, 219, 105915. [Google Scholar] [CrossRef]
- Gendrin, C.; Mercier, C.; Braun, L.; Musset, K.; Dubremetz, J.F.; Cesbron-Delauw, M.F. Toxoplasma gondii uses unusual sorting mechanisms to deliver transmembrane proteins into the host-cell vacuole. Traffic 2008, 9, 1665–1680. [Google Scholar] [CrossRef]
- Furtado, J.M.; Bharadwaj, A.S.; Chipps, T.J.; Pan, Y.; Ashander, L.M.; Smith, J.R. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol. Cell Biol. 2012, 90, 912–915. [Google Scholar] [CrossRef]
- Bruce, A.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Lucas, G., Lewis Granum, S., Zayatz, E., Eds.; Garland Science, Taylor & Francis Group, LLC: New York, NY, USA, 2015; 1464p. [Google Scholar]
- Dubey, J.P.; Su, C. Population biology of Toxoplasma gondii: What’s out and where did they come from. Mem. Inst. Oswaldo Cruz 2009, 104, 190–195. [Google Scholar] [CrossRef]
- Baba, M.; Batanova, T.; Kitoh, K.; Takashima, Y. Adhesion of Toxoplasma gondii tachyzoite-infected vehicle leukocytes to capillary endothelial cells triggers timely parasite egression. Sci. Rep. 2017, 7, 5675. [Google Scholar] [CrossRef] [PubMed]
- Cañedo-solares, I.; Ortiz-alegría, L.B.; Ortiz-muñiz, A.R.; Correa, D. Endothelial cell invasion by Toxoplasma gondii: Differences between cell types and parasite strains. Parasitol. Res. 2013, 112, 3029–3033. [Google Scholar] [CrossRef]
- Lavine, M.D.; Arrizabalaga, G. Induction of mitotic S-phase of host and neighboring cells by Toxoplasma gondii enhances parasite invasion. Mol. Biochem. Parasitol. 2009, 164, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Jung, B.K.; Cho, J.; Song, H.; Pyo, K.H.; Lee, J.M.; Kim, M.K.; Chai, J.Y. Exosomes secreted by Toxoplasma gondii-infected L6 cells: Their effects on host cell proliferation and cell cycle changes. Korean J. Parasitol. 2016, 54, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Makó, V.; Czúcz, J.; Weiszhár, Z.; Herczenik, E.; Matkó, J.; Prohászka, Z.; Cervenak, L. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1β, TNF-α, and LPS. Cytometry A 2010, 77, 962–970. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Shang, H.; Xu, Y.; Qian, M. Monocyte chemoattractant protein-1 induces endothelial cell apoptosis in vitro through a p53-dependent mitochondrial pathway. Acta Biochim. Biophys. Sin. 2011, 43, 787–795. [Google Scholar] [CrossRef]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.-C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [CrossRef]
- Brenier-pinchart, M.; Villena, I.; Mercier, C.; Durand, F.; Simon, J.; Cesbron-delauw, M.; Pelloux, H. The Toxoplasma surface protein SAG1 triggers efficient in vitro secretion of chemokine ligand 2 (CCL2) from human fibroblasts. Microbes Infect. 2006, 8, 254–261. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Zou, Z.; Zhu, B.; Hu, Z.; Zeng, P.; Wu, L.; Xiong, J. Hepatitis C Virus Infection Induces Elevation of CXCL10 in Human Brain Microvascular Endothelial Cells. J. Med. Virol. 2016, 88, 1596–1603. [Google Scholar] [CrossRef]
- Casiraghi, C.; Dorovini-zis, K.; Horwitz, M.S. Epstein-Barr virus infection of human brain microvessel endothelial cells: A novel role in multiple sclerosis. J. Neuroimmunol. 2011, 230, 173–177. [Google Scholar] [CrossRef]
- Jong, A.; Wu, C.; Zhou, W.; Chen, H.; Huang, S. Infectomic Analysis of Gene Expression Profiles of Human Brain Microvascular Endothelial Cells Infected with Cryptococcus neoformans. J. Biomed. Biotechnol. 2008, 2008, 7. [Google Scholar] [CrossRef]
- Sawyer, A.J.; Tian, W.; Saucier-Sawyer, J.K.; Rizk, P.J.; Saltzman, W.M.; Bellamkonda, R.V.; Kyriakides, T.R. The effect of inflammatory cell-derived MCP-1 loss on neuronal survival during chronic neuroinflammation. Biomaterials 2014, 35, 6698–6706. [Google Scholar] [CrossRef]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of Dendritic Cells, MCP-1, and Activated Microglia/Macrophages in Amyotrophic Lateral Sclerosis Spinal Cord Tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Dyer, G.; Calderon, T.M.; Berman, J.W. HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent echanism: Possible role in NeuroAIDS. Glia 2005, 49, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wu, H.L.; Dong, B.H. Relationship between TLR4 and CCL2 expression and recurrent spontaneous abortion. Genet. Mol. Res. 2016, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Woodman, J.P.; Dimier, I.H.; Bout, D.T. Human endothelial cells are activated by IFN-gamma to inhibit Toxoplasma gondii replication. Inhibition is due to a different mechanism from that existing in mouse macrophages and human fibroblasts. J. Immunol. 1991, 147, 2019–2023. [Google Scholar] [CrossRef]
- Fujita, K.; Matsumiya, T.; Fujimoto, K.; Yoshida, H.; Kudoh, S.; Kudoh, D.; Mori, K.; Satoh, K.; Imaizumi, T.; Itaya, H. Expression of Tumor Necrosis Factor-α in Cultured Human Endothelial Cells Stimulated With Lipopolysaccharide or Interleukin-1α. Arterioscler. Thromb. Vasc. Biol. 2011, 20, 410–415. [Google Scholar] [CrossRef]
- Bennouna, S.; Sukhumavasi, W.; Denkers, E.Y. Toxoplasma gondii Inhibits Toll-Like Receptor 4 Ligand-Induced Mobilization of Intracellular Tumor Necrosis Factor Alpha to the Surface of Mouse Peritoneal Neutrophils. Infect. Immun. 2006, 74, 4274–4281. [Google Scholar] [CrossRef]
- Albrecht, E.W.J.A.; Stegeman, C.A.; Heeringa, P.; Henning, R.H.; van Goor, H. Protective role of endothelial nitric oxide synthase. J. Pathol. 2003, 199, 8–17. [Google Scholar] [CrossRef]
- Pfaff, A.W.; Mousli, M.; Sénégas, A.; Marcellin, L.; Takikawa, O.; Klein, J.P.; Candolfi, E. Impact of foetus and mother on IFN-γ-induced indoleamine 2,3-dioxygenase and inducible nitric oxide synthase expression in murine placenta following Toxoplasma gondii infection. Int. J. Parasitol. 2008, 38, 249–258. [Google Scholar] [CrossRef]
- Dincel, G.C.; Atmaca, H.T. Increased expressions of ADAMTS-13 and apoptosis contribute to neuropathology during Toxoplasma gondii encephalitis in mice. Neuropathology 2016, 36, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Vomund, A.N.; Majerus, E.M. ADAMTS13 Bound to Endothelial Cells Exhibits Enhanced Cleavage of von Willebrand Factor. J. Biol. Chem. 2009, 284, 30925–30932. [Google Scholar] [CrossRef] [PubMed]
- Breier, G. Angiogenesis in embryonic development—A review. Placenta 2000, 21, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Al-Mohaimen, N.A.; Ahmed, A.M. Vegf expression in trophoblastic tissue of women with spontaneous miscarriage and infected by toxoplasma gondii using insitu hybridization. Iraqi J. Biotechnol. 2012, 11, 494–502. [Google Scholar]
- Hwang, J.Y.; Park, J.W.; Hong, S.Y.; Park, H.S. Reduced expression of angiopoietin-1 in Hantaan virus-infected human umbilical vein endothelial cells increases their permeability. Acta Virol. 2009, 53, 7–13. [Google Scholar] [CrossRef]
- Conti, F.; Bartolucci, F. Angiopoietin-1 Reduces VEGF-Stimulated Leukocyte Adhesion to Endothelial Cells by Reducing ICAM-1, VCAM-1, and E-Selectin Expression. Geobot. Stud. 2015, 17–18. [Google Scholar] [CrossRef]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef]
- Tan, M.; Zhang, L.; Wang, W.; Sheng, C.; Jiang, C.; Sun, H.; Jing, Z.; Zhou, J. Hydrocortisone-conjugated glycine inhibits TNF-α induced inflammatory response through glucocorticoids receptor independent mechanism in human umbilical vein endothelial cells. Int. J. Clin. Exp. Med. 2017, 10, 9047–9055. [Google Scholar]
- Muro, S.; Schuchman, E.H.; Muzykantov, V.R. Lysosomal Enzyme Delivery by ICAM-1-Targeted Nanocarriers Bypassing Glycosylation- and Clathrin-Dependent Endocytosis. Mol. Ther. 2006, 13, 135–141. [Google Scholar] [CrossRef]
- Däubener, W.; Spors, B.; Hucke, C.; Adam, R.; Stins, M.; Kwang Sik Kim; Schroten, H. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect. Immun. 2001, 69, 6527–6531. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Miinick, C.R. Culture of Human Endothelial Cells Derived from Umbilical Veins. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.W. Salt Intake, Endothelial Cell Signaling, and Progression of Kidney Disease. Hypertension 2004, 43, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.J.O.; Giatromanolaki, A.; Cox, G.; Turley, H.; Steward, W.P.; Gatter, K.; Harris, A.L. Vascular endothelial growth factor, platelet-derived endothelial cell growth factor and angiogenesis in non-small-cell lung cancer. Br. J. Cancer 2000, 82, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Rust, N.M.; Papa, M.P.; Scovino, A.M.; da Silva, M.M.C.; Calzavara-Silva, C.E.; Marques, E.T. de A.; Peçanha, L.M.T.; Scharfstein, J.; Arruda, L.B. Bradykinin enhances Sindbis virus infection in human brain microvascular endothelial cells. Virology 2012, 422, 81–91. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sa, Q.; Gehman, M.; Ochiai, E. Interferon-gamma- and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev. Mol. Med. 2011, 13, 1–18. [Google Scholar] [CrossRef]
- Cossetti, C.; Iraci, N.; Mercer, T.R.; Leonardi, T.; Alpi, E.; Drago, D.; Alfaro-Cervello, C.; Saini, H.K.; Davis, M.P.; Schaeffer, J.; et al. Extracellular vesicles from neural stem cells transfer IFN-γ via Ifngr1 to activate Stat1 signaling in target cells. Mol. Cell 2014, 56, 193–204. [Google Scholar] [CrossRef]
- Mukhtar, F.; Guarnieri, A.; Brancazio, N.; Falcone, M.; Di Naro, M.; Azeem, M.; Zubair, M.; Nicolosi, D.; Di Marco, R.; Petronio Petronio, G. The role of Mycobacterium tuberculosis exosomal miRNAs in host pathogen cross-talk as diagnostic and therapeutic biomarkers. Front. Microbiol. 2024, 15, 1441781. [Google Scholar] [CrossRef]
- Hosseinkhani, B.; van den Akker, N.M.S.; Molin, D.G.M.; Michiels, L. (Sub)populations of extracellular vesicles released by TNF-α –triggered human endothelial cells promote vascular inflammation and monocyte migration. J. Extracell. Vesicles 2020, 9. [Google Scholar] [CrossRef]
- Bessieres, M.-H.; Swierczynski, B.; Miedouge, M.; Olle, P.; Segulela, J.; Pipy, B. Role of IFN-γ, TNF-α, IL4 and IL 10 in the regulation of experimental Toxoplasma gondii infection. J. Eukaryot. Microbiol. 1997, 44, 1997. [Google Scholar] [CrossRef]
- Giese, A.; Stuhlsatz, S.; Däubener, W.; MacKenzie, C.R. Inhibition of the Growth of Toxoplasma gondii in Immature Human Dendritic Cells Is Dependent on the Expression of TNF-α Receptor 2. J. Immunol. 2004, 173, 3366–3374. [Google Scholar] [CrossRef]
- Bonsergent, E.; Grisard, E.; Buchrieser, J.; Schwartz, O.; Théry, C.; Lavieu, G. Quantitative characterization of extracellular vesicle uptake and content delivery within mammalian cells. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, C.; Shah, N.; Ishii, M.; Annamalai, B.; Brandon, C.; Rodgers, J.; Nowling, T.; Rohrer, B. Mechanisms of extracellular vesicle uptake in stressed retinal pigment epithelial cell monolayers. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165608. [Google Scholar] [CrossRef] [PubMed]
- Aline, F.; Bout, D.; Amigorena, S.; Roingeard, P.; Dimier-Poisson, I. Toxoplasma gondii antigen-pulsed-dendritic cell-derived exosomes induce a protective immune response against T. gondii infection. Infect. Immun. 2004, 72, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Rana, S.; Baumann, A.; McAlear, J.; Hellwig, A.; Trendelenburg, M.; Lochnit, G.; Preissner, K.T.; Zöller, M. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. 2010, 70, 1668–1678. [Google Scholar] [CrossRef]
- Li, Y.Y.; Xiu, F.; Mou, Z.; Xue, Z.; Du, H.; Zhou, C.; Li, Y.Y.; Shi, Y.; He, S.; Zhou, H. Exosomes derived from Toxoplasma gondii stimulate an inflammatory response through JNK signaling pathway. Nanomedicine 2018, 13, 1157–1168. [Google Scholar] [CrossRef]
- Garcia, L.F.C.; Wowk, P.F.; Albrecht, L. Unraveling the impact of extracellular vesicle-depleted serum on endothelial cell characteristics over time. Int. J. Mol. Sci. 2024, 25, 4761. [Google Scholar] [CrossRef]
- Li, Y.; Yang, N.; Dong, B.; Yang, J.; Kou, L.; Qin, Q. MicroRNA-122 promotes endothelial cell apoptosis by targeting XIAP: Therapeutic implication for atherosclerosis. Life Sci. 2019, 232, 116590. [Google Scholar] [CrossRef]
- Liao, Z.; Muth, D.C.; Eitan, E.; Travers, M.; Learman, L.N.; Lehrmann, E.; Witwer, K.W. Serum extracellular vesicle depletion processes affect release and infectivity of HIV-1 in culture. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef]
- Van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. In Cancer Cell Culture: Methods and Protocols, 2nd ed.; Cree, I.A., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 237–245. [Google Scholar]
- Frentzou, G.A.; Bradford, C.; Harkness, K.A.; Haddock, G.; Woodroofe, M.N.; Cross, A.K. IL-1β down-regulates ADAMTS-13 mRNA expression in cells of the central nervous system. J. Mol. Neurosci. 2012, 46, 343–351. [Google Scholar] [CrossRef]
- Li, Y.; Viscidi, R.P.; Kannan, G.; McFarland, R.; Pletnikov, M.V.; Severance, E.G.; Yolken, R.H.; Xiao, J. Chronic Toxoplasma gondii Infection Induces Anti-N-Methyl-DAspartate Receptor Autoantibodies and Associated Behavioral Changes and Neuropathology. Infect. Immun. 2018, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Yao, H.; Hu, G.; Chen, X.M.; Lund, A.K.; Buch, S. HIV Tat Induces Expression of ICAM-1 in HUVECs: Implications for miR-221/-222 in HIV-Associated Cardiomyopathy. PLoS ONE 2013, 8, e60170. [Google Scholar] [CrossRef] [PubMed]
- Melo, E.J.L.; Attias, M.; De Souza, W. The single mitochondrion of tachyzoites of Toxoplasma gondii. J. Struct. Biol. 2000, 130, 27–33. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, L.F.C.; Cavalari, V.C.; Wowk, P.F.; Albrecht, L. Human Brain Endothelial Cell-Derived Extracellular Vesicles Reduce Toxoplasma gondii Infection In Vitro in Human Brain and Umbilical Cord Vein Endothelial Cells. Int. J. Mol. Sci. 2025, 26, 2640. https://doi.org/10.3390/ijms26062640
Garcia LFC, Cavalari VC, Wowk PF, Albrecht L. Human Brain Endothelial Cell-Derived Extracellular Vesicles Reduce Toxoplasma gondii Infection In Vitro in Human Brain and Umbilical Cord Vein Endothelial Cells. International Journal of Molecular Sciences. 2025; 26(6):2640. https://doi.org/10.3390/ijms26062640
Chicago/Turabian StyleGarcia, Luiz Fernando Cardoso, Victoria Cruz Cavalari, Pryscilla Fanini Wowk, and Letusa Albrecht. 2025. "Human Brain Endothelial Cell-Derived Extracellular Vesicles Reduce Toxoplasma gondii Infection In Vitro in Human Brain and Umbilical Cord Vein Endothelial Cells" International Journal of Molecular Sciences 26, no. 6: 2640. https://doi.org/10.3390/ijms26062640
APA StyleGarcia, L. F. C., Cavalari, V. C., Wowk, P. F., & Albrecht, L. (2025). Human Brain Endothelial Cell-Derived Extracellular Vesicles Reduce Toxoplasma gondii Infection In Vitro in Human Brain and Umbilical Cord Vein Endothelial Cells. International Journal of Molecular Sciences, 26(6), 2640. https://doi.org/10.3390/ijms26062640