
Antitumor Activity of Radiation Therapy Combined with Checkpoint Kinase Inhibition in SHH/p53-Mutated Human Medulloblastoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
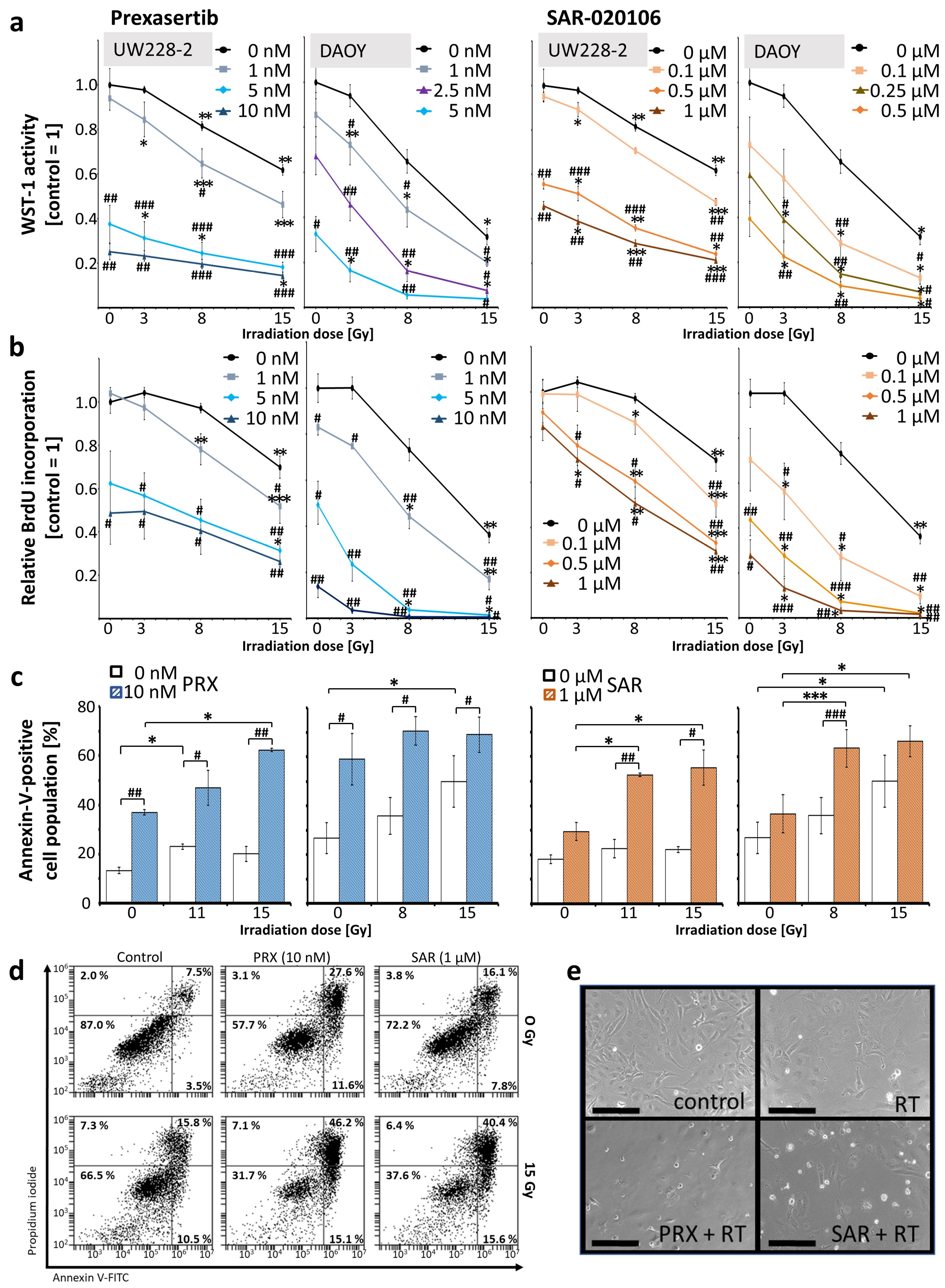
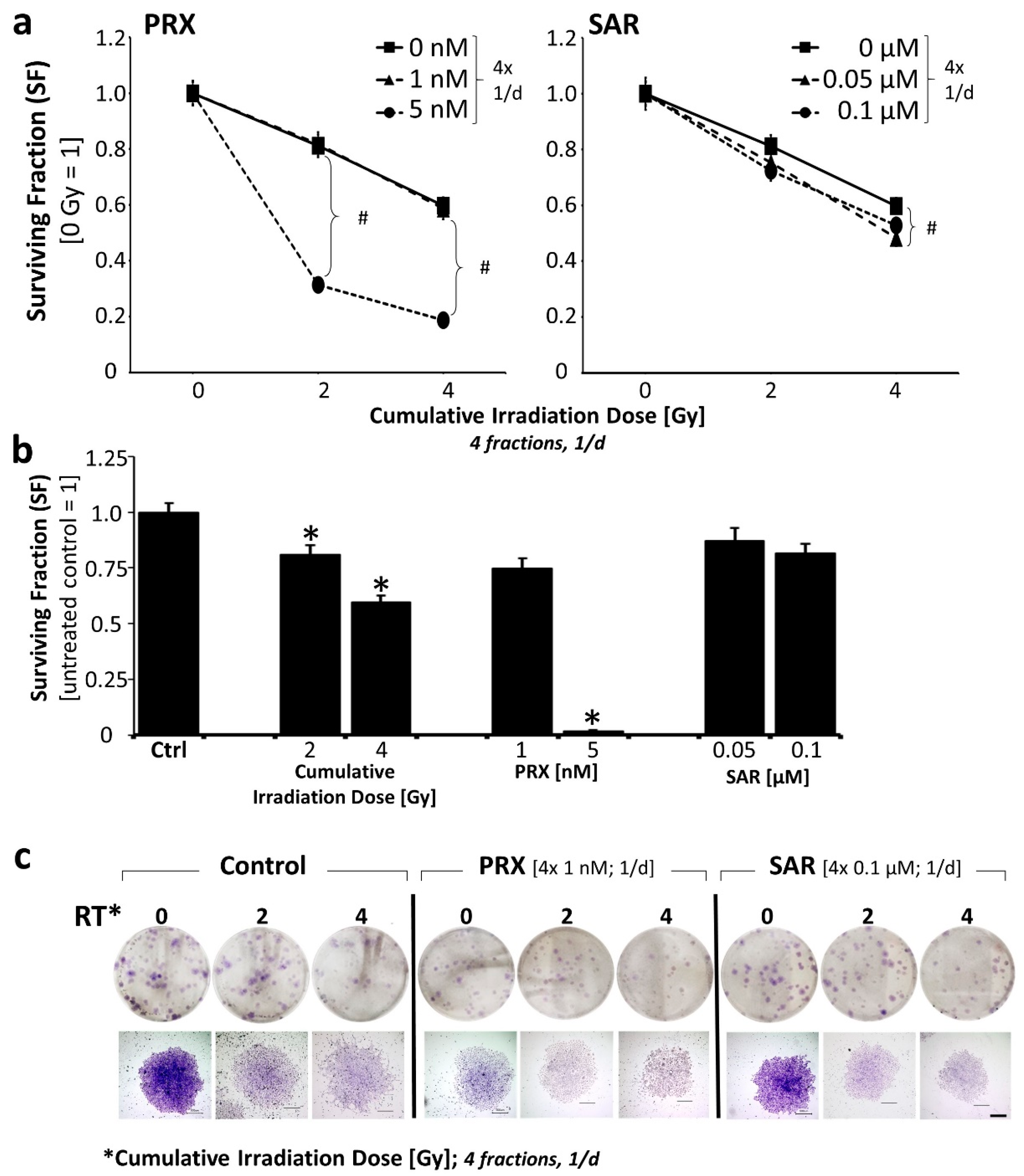
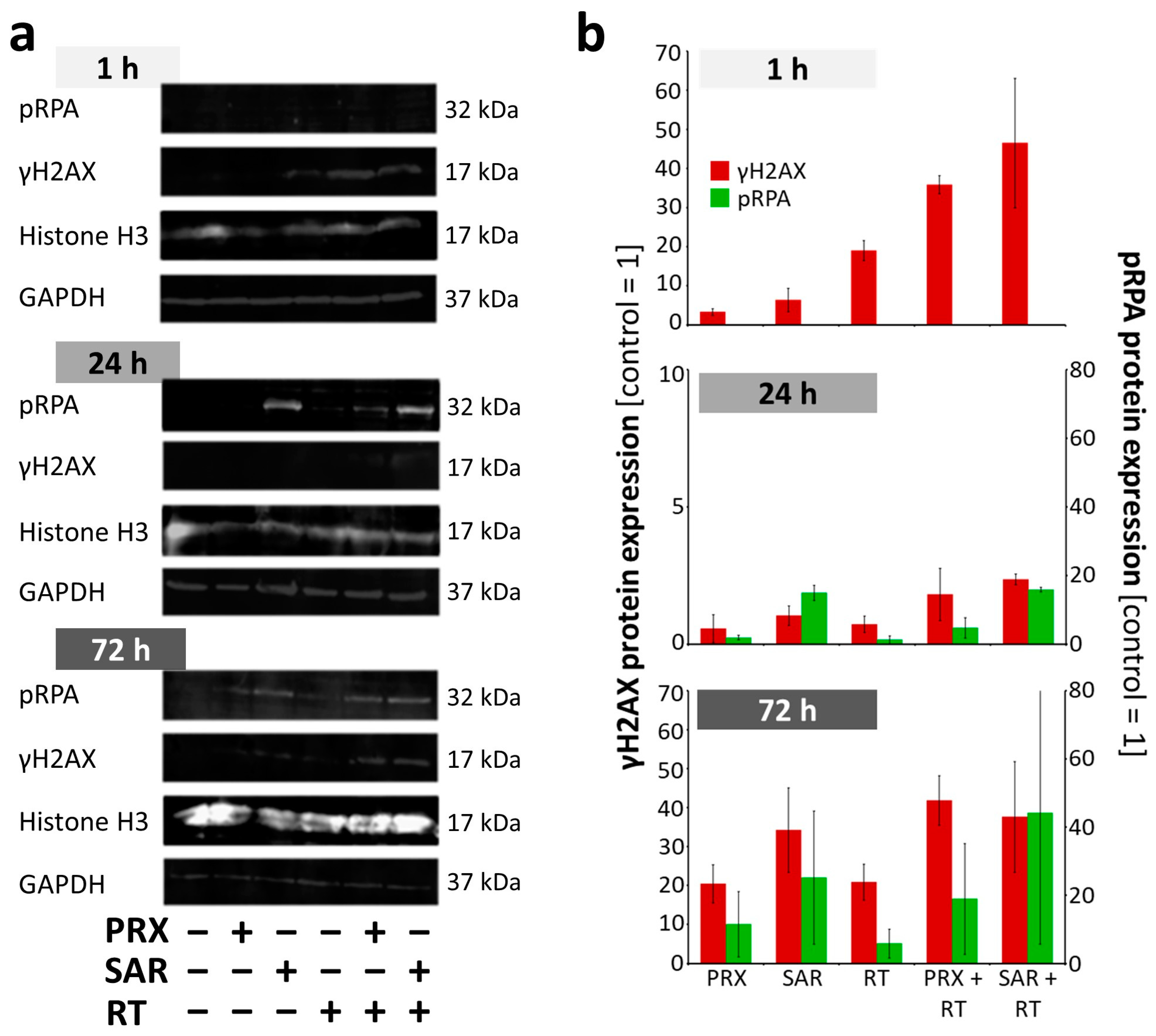
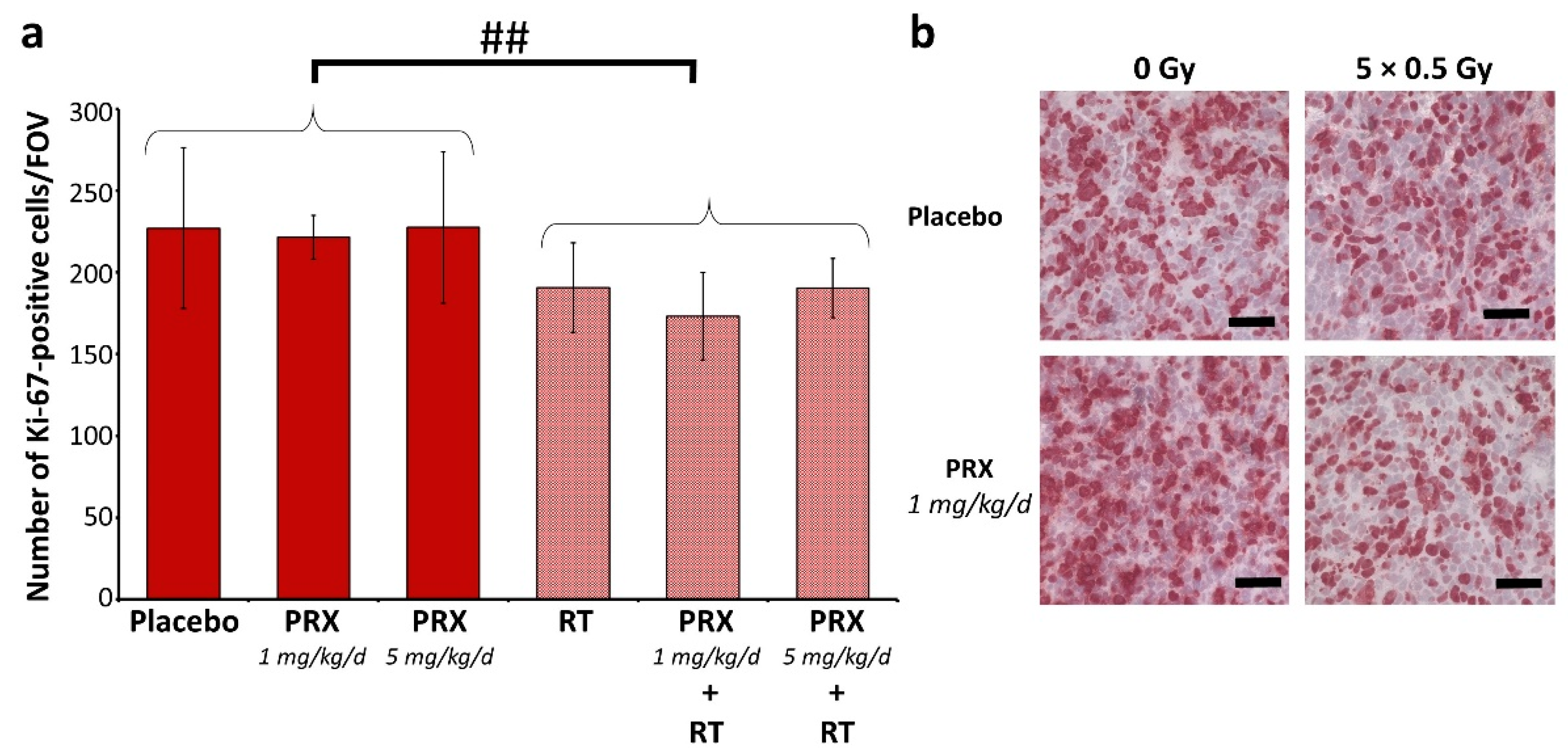
2. Results and Discussion
3. Materials and Methods
3.1. Cell Lines
3.2. Drugs
3.3. Cell Culture Treatment and Assays
3.4. Fluorescence–Microscopic Analyses of DNA Damage in S Phase Cells
3.5. Western Blot of DNA Damage Proteins
3.6. Mouse Model, Treatment, and Imaging
3.7. Tissue Preparation and Staining
3.8. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATR | Ataxia telangiectasia and Rad3-related protein |
| BLI | Bioluminescence imaging |
| BW | Body weight |
| Chk | Checkpoint kinase |
| Chk-i | Checkpoint kinase inhibitor |
| HNSCC | Head and neck squamous cell carcinoma |
| MB | Medulloblastoma |
| NSG mouse | Nod-SCID gamma mouse |
| OS | Overall survival |
| PDX | Patient-derived xenograft |
| PE | Plating efficacy |
| PRX | Prexasertib |
| RT | Irradiation; radiation therapy |
| SAR | SAR-020106 |
| SF | Surviving fraction |
| SHH/p53-mut | SHH-activated and TP53-mutated |
| WB | Western blot |
References
- Ostrom, Q.T.; Price, M.; Ryan, K.; Edelson, J.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Pediatric Brain Tumor Foundation Childhood and Adolescent Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2022, 24, iii1–iii38. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Robinson, G.W.; Smith, K.S.; Lin, T.; Merchant, T.E.; Chintagumpala, M.; Mahajan, A.; Su, J.; Bouffet, E.; Bartels, U.; et al. Outcomes by Clinical and Molecular Features in Children with Medulloblastoma Treated with Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J. Clin. Oncol. 2021, 39, 822–835. [Google Scholar] [CrossRef]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Doz, F.; Milde, T. An International Prospective Trial on Medulloblastoma (MB) in Children Older than 3 to 5 Years with Wnt Biological Profile (Pnet 5 MB-LR and Pnet 5 MB-WNT-HR), Average-Risk Biological Profile (PNET 5 MB-SR), or TP53 Mutation, and Registry for MB Occurring in the Context of Genetic Predisposition: NCT02066220, Siop Pnet 5 MB. Available online: https://clinicaltrials.gov/ct2/show/NCT02066220 (accessed on 14 June 2023).
- Chevignard, M.; Câmara-Costa, H.; Doz, F.; Dellatolas, G. Core deficits and quality of survival after childhood medulloblastoma: A review. Neurooncol. Pract. 2017, 4, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Castellino, S.M.; Ullrich, N.J.; Whelen, M.J.; Lange, B.J. Developing interventions for cancer-related cognitive dysfunction in childhood cancer survivors. J. Natl. Cancer Inst. 2014, 106, 186. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Zeng, L.; Beggs, R.R.; Cooper, T.S.; Weaver, A.N.; Yang, E.S. Combining Chk1/2 Inhibition with Cetuximab and Radiation Enhances In Vitro and In Vivo Cytotoxicity in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2017, 16, 591–600. [Google Scholar] [CrossRef]
- Prince, E.W.; Balakrishnan, I.; Shah, M.; Mulcahy Levy, J.M.; Griesinger, A.M.; Alimova, I.; Harris, P.S.; Birks, D.K.; Donson, A.M.; Davidson, N.; et al. Checkpoint kinase 1 expression is an adverse prognostic marker and therapeutic target in MYC-driven medulloblastoma. Oncotarget 2016, 7, 53881–53894. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Ditano, J.P.; Eastman, A. Comparative Activity and Off-Target Effects in Cells of the CHK1 Inhibitors MK-8776, SRA737, and LY2606368. ACS Pharmacol. Transl. Sci. 2021, 4, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.D.; VanWye, A.B.; Dowless, M.; Blosser, W.; Falcon, B.L.; Stewart, J.; Stephens, J.; Beckmann, R.P.; Lin, A.B.; Stancato, L.F. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin. Cancer Res. 2017, 23, 4354–4363. [Google Scholar] [CrossRef]
- Zeng, L.; Nikolaev, A.; Xing, C.; Della Manna, D.L.; Yang, E.S. CHK1/2 Inhibitor Prexasertib Suppresses NOTCH Signaling and Enhances Cytotoxicity of Cisplatin and Radiation in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2020, 19, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Endersby, R.; Whitehouse, J.; Pribnow, A.; Kuchibhotla, M.; Hii, H.; Carline, B.; Gande, S.; Stripay, J.; Ancliffe, M.; Howlett, M.; et al. Small-molecule screen reveals synergy of cell cycle checkpoint kinase inhibitors with DNA-damaging chemotherapies in medulloblastoma. Sci. Transl. Med. 2021, 13, eaba7401. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W. Evaluation of LY2606368 Therapy in Combination with Cyclophosphamide or Gemcitabine for Children and Adolescents with Refractory or Recurrent Group 3/Group 4 or SHH Medulloblastoma Brain Tumors: NCT04023669. Available online: https://clinicaltrials.gov/study/NCT04023669 (accessed on 8 January 2025).
- Walton, M.I.; Eve, P.D.; Hayes, A.; Valenti, M.; Haven Brandon, A.D.; Box, G.; Boxall, K.J.; Aherne, G.W.; Eccles, S.A.; Raynaud, F.I.; et al. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol. Cancer Ther. 2010, 9, 89–100. [Google Scholar] [CrossRef]
- Reader, J.C.; Matthews, T.P.; Klair, S.; Cheung, K.-M.J.; Scanlon, J.; Proisy, N.; Addison, G.; Ellard, J.; Piton, N.; Taylor, S.; et al. Structure-guided evolution of potent and selective CHK1 inhibitors through scaffold morphing. J. Med. Chem. 2011, 54, 8328–8342. [Google Scholar] [CrossRef]
- Borst, G.R.; McLaughlin, M.; Kyula, J.N.; Neijenhuis, S.; Khan, A.; Good, J.; Zaidi, S.; Powell, N.G.; Meier, P.; Collins, I.; et al. Targeted radiosensitization by the Chk1 inhibitor SAR-020106. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1110–1118. [Google Scholar] [CrossRef]
- Patties, I.; Kallendrusch, S.; Böhme, L.; Kendzia, E.; Oppermann, H.; Gaunitz, F.; Kortmann, R.-D.; Glasow, A. The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide, and to decitabine treatment. J. Exp. Clin. Cancer Res. 2019, 38, 420. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.-J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin. Cancer Res. 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Chaudhary, R.; Slebos, R.J.C.; Song, F.; McCleary-Sharpe, K.P.; Masannat, J.; Tan, A.C.; Wang, X.; Amaladas, N.; Wu, W.; Hall, G.E.; et al. Effects of checkpoint kinase 1 inhibition by prexasertib on the tumor immune microenvironment of head and neck squamous cell carcinoma. Mol. Carcinog. 2021, 60, 138–150. [Google Scholar] [CrossRef]
- Manic, G.; Signore, M.; Sistigu, A.; Russo, G.; Corradi, F.; Siteni, S.; Musella, M.; Vitale, S.; Angelis, M.L.d.; Pallocca, M.; et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 2018, 67, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Nair, J.; Huang, T.-T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.-M. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Wan, Y.; Wang, H.; Liu, J.; Yang, J.; Sun, R.; Meng, H.; Ma, X.; Jiang, Y.; Cheng, W. CXCL2-mediated ATR/CHK1 signaling pathway and platinum resistance in epithelial ovarian cancer. J. Ovarian Res. 2021, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Moureau, S.; Luessing, J.; Harte, E.C.; Voisin, M.; Lowndes, N.F. A role for the p53 tumour suppressor in regulating the balance between homologous recombination and non-homologous end joining. Open Biol. 2016, 6, 160225. [Google Scholar] [CrossRef]
- Hsu, W.-H.; Zhao, X.; Zhu, J.; Kim, I.-K.; Rao, G.; McCutcheon, J.; Hsu, S.-T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Checkpoint Kinase 1 Inhibition Enhances Cisplatin Cytotoxicity and Overcomes Cisplatin Resistance in SCLC by Promoting Mitotic Cell Death. J. Thorac. Oncol. 2019, 14, 1032–1045. [Google Scholar] [CrossRef]
- Campagne, O.; Davis, A.; Maharaj, A.R.; Zhong, B.; Stripay, J.; Farmer, D.; Roussel, M.F.; Stewart, C.F. CNS penetration and pharmacodynamics of the CHK1 inhibitor prexasertib in a mouse Group 3 medulloblastoma model. Eur. J. Pharm. Sci. 2020, 142, 105106. [Google Scholar] [CrossRef]
- Yang, E.S.; Deutsch, E.; Mehmet, A.; Fayette, J.; Tao, Y.; Nabell, L.; Spencer, S.A.; Wang, X.A.; Spoljoric, E.A.; Zhang, W.; et al. A Phase 1b trial of prexasertib in combination with chemoradiation in patients with locally advanced head and neck squamous cell carcinoma. Radiother. Oncol. 2021, 157, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Hong, D.S.; Patel, M.R.; Pant, S.; Ulahannan, S.V.; Jones, S.; Meric-Bernstam, F.; Wang, J.S.; Aljumaily, R.; Hamilton, E.P.; et al. A Phase 1b Trial of Prexasertib in Combination with Standard-of-Care Agents in Advanced or Metastatic Cancer. Target. Oncol. 2021, 16, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Lampert, E.J.; Cimino-Mathews, A.; Lee, J.S.; Nair, J.; Lee, M.-J.; Yuno, A.; An, D.; Trepel, J.B.; Ruppin, E.; Lee, J.-M. Clinical outcomes of prexasertib monotherapy in recurrent BRCA wild-type high-grade serous ovarian cancer involve innate and adaptive immune responses. J. Immunother. Cancer 2020, 8, e000516. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Yamamoto, N.; Shitara, K.; Tamura, K.; Matsubara, N.; Tajimi, M.; Lin, A.B.; Asou, H.; Cai, Z.; Inoue, K.; et al. Dose-finding study of the checkpoint kinase 1 inhibitor, prexasertib, in Japanese patients with advanced solid tumors. Cancer Sci. 2018, 109, 3216–3223. [Google Scholar] [CrossRef]
- Hong, D.S.; Moore, K.; Patel, M.; Grant, S.C.; Burris, H.A.; William, W.N.; Jones, S.; Meric-Bernstam, F.; Infante, J.; Golden, L.; et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3263–3272. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients with Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.-J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013–e1824. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lee, J.-M.; Gao, B.; Miller, R.; Lee, J.-Y.; Colombo, N.; Vergote, I.; Credille, K.M.; Young, S.R.; McNeely, S.; et al. A Phase 2 study of prexasertib (LY2606368) in platinum resistant or refractory recurrent ovarian cancer. Gynecol. Oncol. 2022, 167, 213–225. [Google Scholar] [CrossRef]
- Cash, T.; Fox, E.; Liu, X.; Minard, C.G.; Reid, J.M.; Scheck, A.C.; Weigel, B.J.; Wetmore, C. A phase 1 study of prexasertib (LY2606368), a CHK1/2 inhibitor, in pediatric patients with recurrent or refractory solid tumors, including CNS tumors: A report from the Children’s Oncology Group Pediatric Early Phase Clinical Trials Network (ADVL1515). Pediatr. Blood Cancer 2021, 68, e29065. [Google Scholar] [CrossRef]
- Di Giulio, S.; Colicchia, V.; Pastorino, F.; Pedretti, F.; Fabretti, F.; Di Nicolis Robilant, V.; Ramponi, V.; Scafetta, G.; Moretti, M.; Licursi, V.; et al. A combination of PARP and CHK1 inhibitors efficiently antagonizes MYCN-driven tumors. Oncogene 2021, 40, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Patties, I.; Kortmann, R.-D.; Glasow, A. Inhibitory effects of epigenetic modulators and differentiation inducers on human medulloblastoma cell lines. J. Exp. Clin. Cancer Res. 2013, 32, 27. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, H.; Purcz, K.; Birkemeyer, C.; Baran-Schmidt, R.; Meixensberger, J.; Gaunitz, F. Carnosine’s inhibitory effect on glioblastoma cell growth is independent of its cleavage. Amino Acids 2019, 51, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Gringmuth, M.; Walther, J.; Greiser, S.; Toussaint, M.; Schwalm, B.; Kool, M.; Kortmann, R.-D.; Glasow, A.; Patties, I. Enhanced Survival of High-Risk Medulloblastoma-Bearing Mice after Multimodal Treatment with Radiotherapy, Decitabine, and Abacavir. Int. J. Mol. Sci. 2022, 23, 3815. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuchařová, Z.; Glasow, A.; Kortmann, R.-D.; Patties, I. Antitumor Activity of Radiation Therapy Combined with Checkpoint Kinase Inhibition in SHH/p53-Mutated Human Medulloblastoma. Int. J. Mol. Sci. 2025, 26, 2577. https://doi.org/10.3390/ijms26062577
Kuchařová Z, Glasow A, Kortmann R-D, Patties I. Antitumor Activity of Radiation Therapy Combined with Checkpoint Kinase Inhibition in SHH/p53-Mutated Human Medulloblastoma. International Journal of Molecular Sciences. 2025; 26(6):2577. https://doi.org/10.3390/ijms26062577
Chicago/Turabian StyleKuchařová, Zuzana, Annegret Glasow, Rolf-Dieter Kortmann, and Ina Patties. 2025. "Antitumor Activity of Radiation Therapy Combined with Checkpoint Kinase Inhibition in SHH/p53-Mutated Human Medulloblastoma" International Journal of Molecular Sciences 26, no. 6: 2577. https://doi.org/10.3390/ijms26062577
APA StyleKuchařová, Z., Glasow, A., Kortmann, R.-D., & Patties, I. (2025). Antitumor Activity of Radiation Therapy Combined with Checkpoint Kinase Inhibition in SHH/p53-Mutated Human Medulloblastoma. International Journal of Molecular Sciences, 26(6), 2577. https://doi.org/10.3390/ijms26062577