
Study on the Mechanism of Formononetin Against Hepatocellular Carcinoma: Regulating Metabolic Pathways of Ferroptosis and Cell Cycle


Abstract

1. Introduction
2. Results
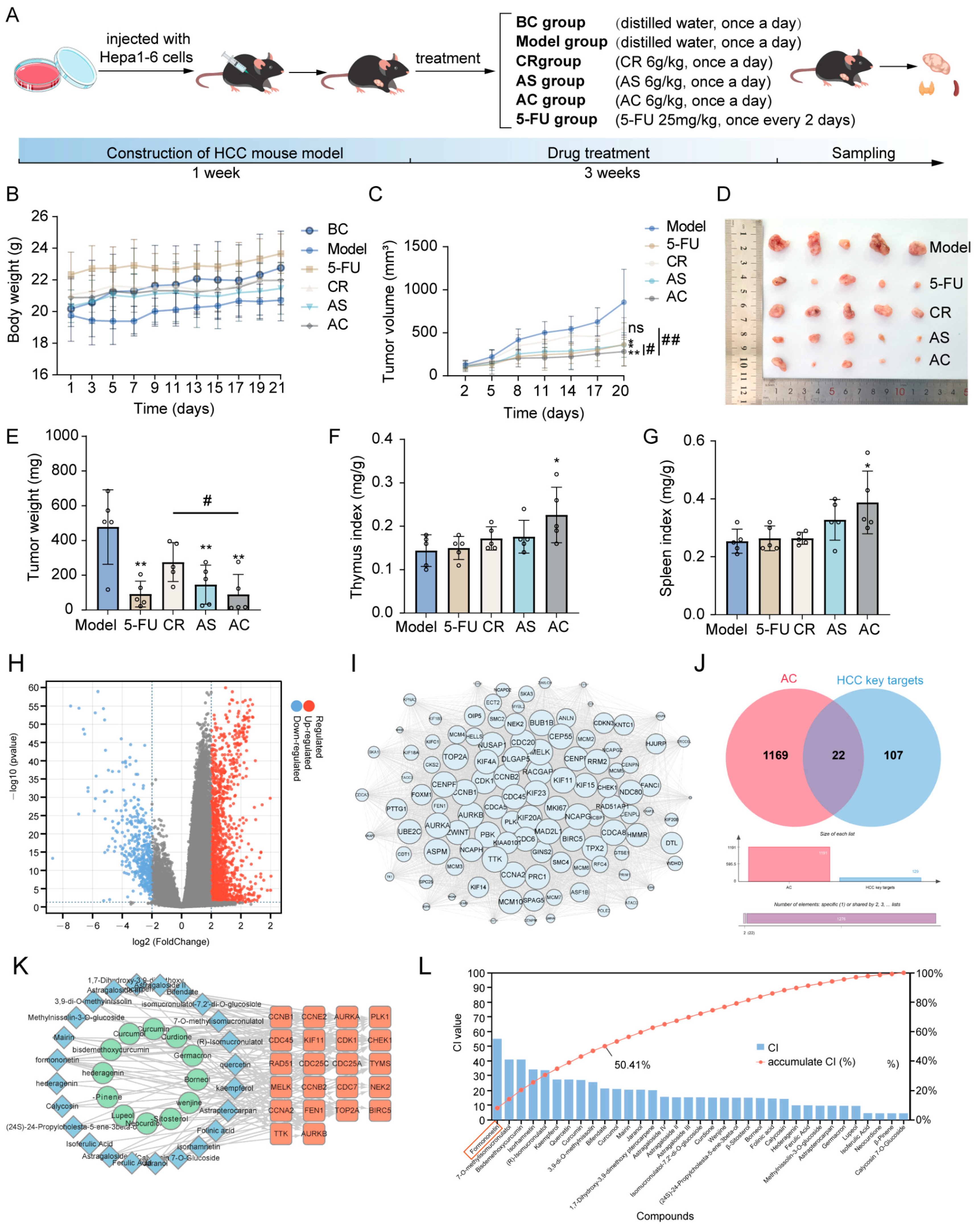
2.1. Network Pharmacology Prediction and Core Component Identification
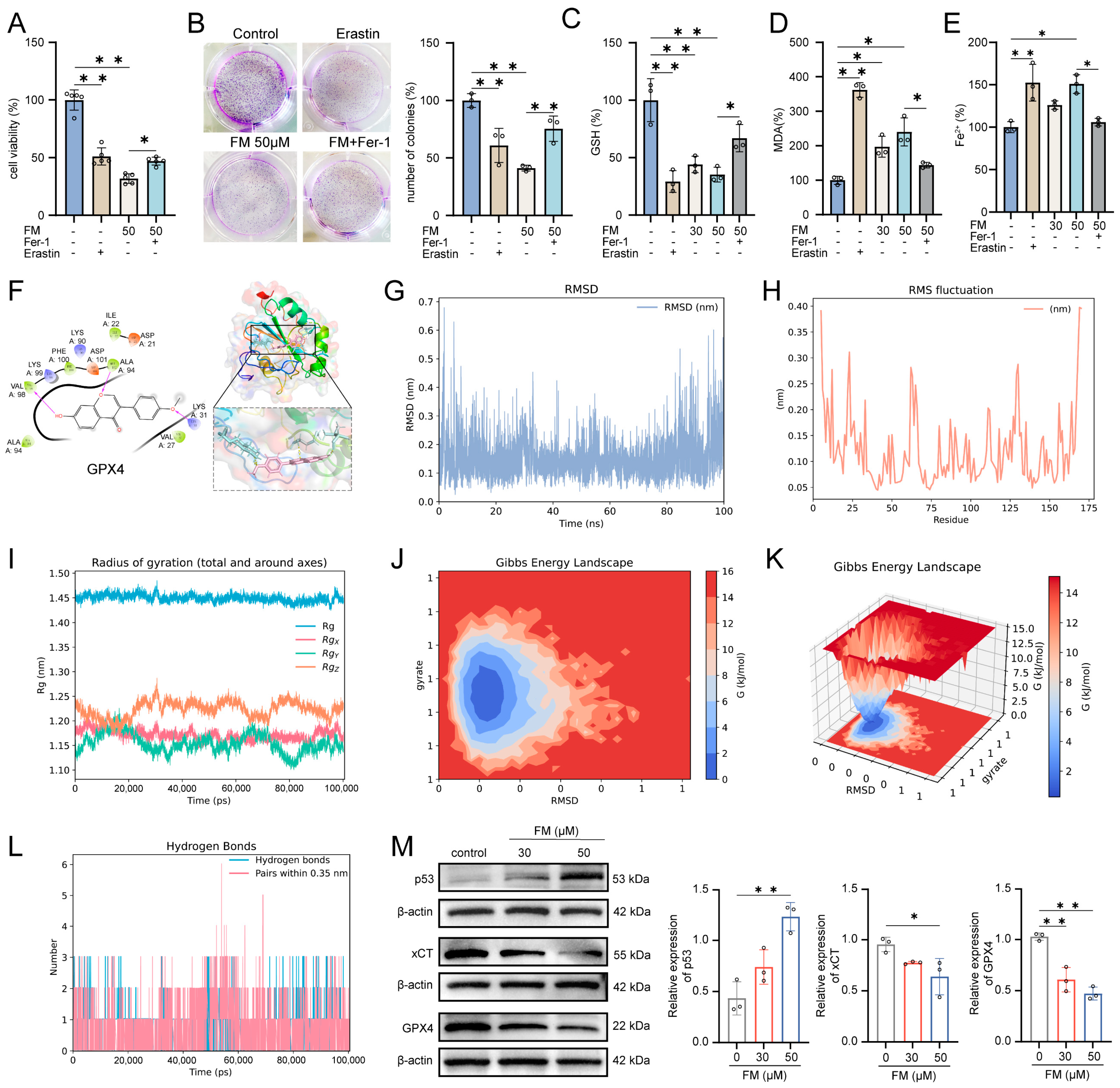
2.2. Bioinformatics Analysis and Molecular Docking Analysis
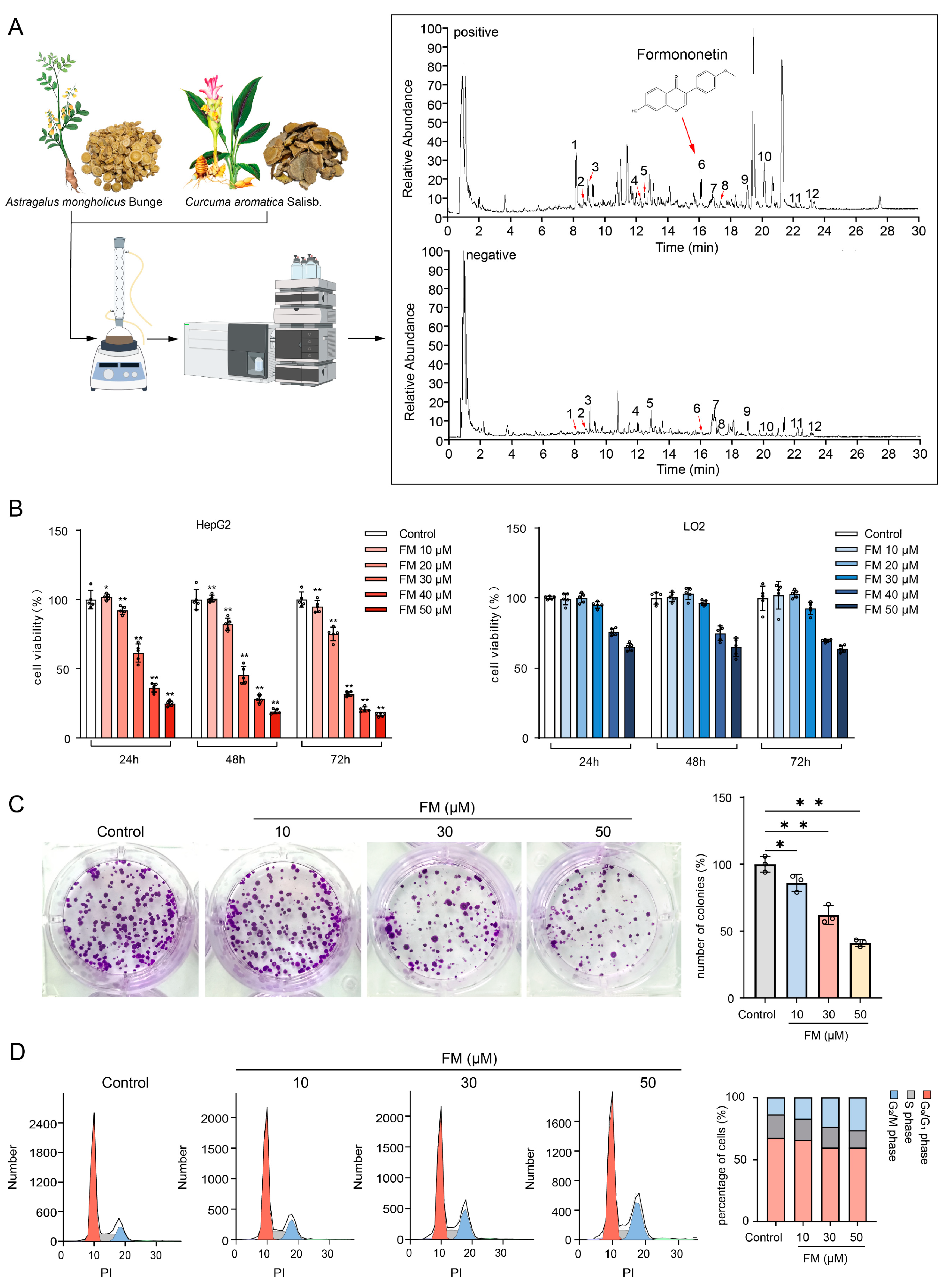
2.3. FM Inhibits the Proliferation of HepG2 Cells
2.4. FM Induces DNA Damage by Accumulating ROS
2.5. FM Regulates Cellular Metabolism
2.6. FM Induces Ferroptosis in HepG2 Cells
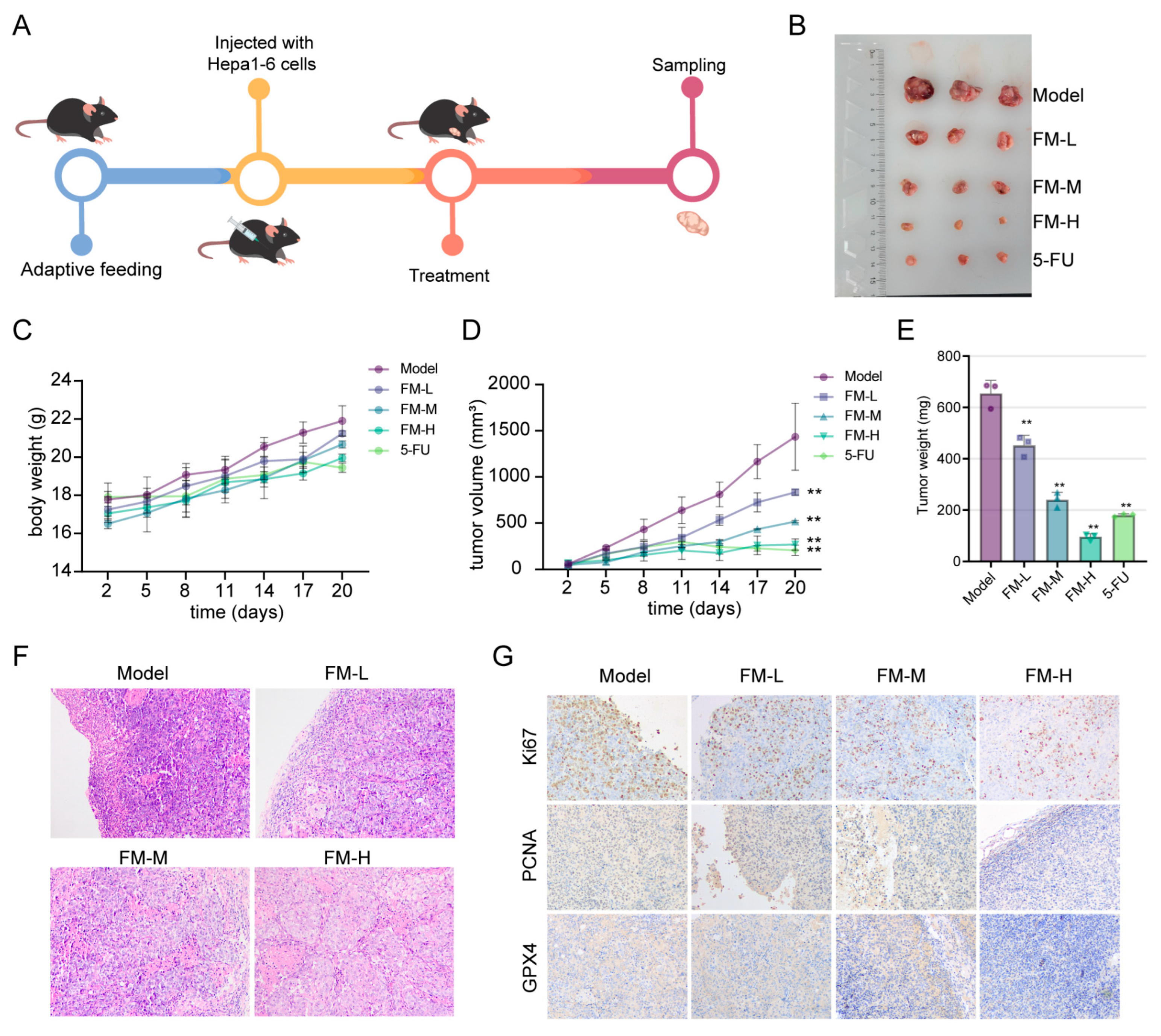
2.7. FM Inhibits Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Network Pharmacology Analysis and Bioinformatics Analysis
4.1.1. Data Sources and DEGs Analysis
4.1.2. Identification of Key Gene Expression Modules
4.1.3. Collection of Active Components and Targets
4.1.4. Construction of Component–Target (C-T) Network
4.1.5. Core Component Identification
4.1.6. Bioinformatics Analysis of Core Component
4.1.7. Molecular Docking Studies
4.1.8. Molecular Dynamics Simulation
4.2. Experimental Validation
4.2.1. Reagents
4.2.2. Identification of Main Components
4.2.3. Animal Experiments
4.2.4. Hematoxylin and Eosin (HE) Staining and Immunohistochemical (IHC) Experiment
4.2.5. Cell Viability and Proliferation Assays
4.2.6. Cell Cycle Assay
4.2.7. Analysis of MMP and ROS
4.2.8. Metabolomics Analysis
4.2.9. Analysis of MDA, Fe2+, and GSH
4.2.10. Western Blotting Assay
4.2.11. Statistics and Reproducibility
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| TCM | traditional Chinese medicine |
| AC | Astragalus mongholicus Bunge-Curcuma aromatica Salisb. |
| FM | formononetin |
| CDKs | cyclin-dependent kinases |
| CCNB | cyclin B |
| PPI | protein–protein interaction |
| MTT | methyl thiazolyl tetrazolium |
| MDA | malondialdehyde |
| DEGs | differential expression genes |
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Talib, W.H.; Baban, M.M.; Bulbul, M.F.; Al-Zaidaneen, E.; Allan, A.; Al-Rousan, E.W.; Ahmad, R.H.Y.; Alshaeri, H.K.; Alasmari, M.M.; Law, D. Natural Products and Altered Metabolism in Cancer: Therapeutic Targets and Mechanisms of Action. Int. J. Mol. Sci. 2024, 25, 9593. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Gu, Y.Y.; Nie, W.Y.; Zhu, X.M.; Qi, M.J.; Zhao, R.M.; Zhu, W.Z.; Zhang, X.L. Formononetin Induces Ferroptosis in Activated Hepatic Stellate Cells to Attenuate Liver Fibrosis by Targeting NADPH Oxidase 4. Phytother. Res. 2024, 38, 5988–6003. [Google Scholar] [CrossRef]
- Tay, K.C.; Tan, L.T.; Chan, C.K.; Hong, S.L.; Chan, K.-G.; Yap, W.H.; Pusparajah, P.; Lee, L.-H.; Goh, B.-H. Formononetin: A Review of Its Anticancer Potentials and Mechanisms. Front. Pharmacol. 2019, 10, 820. [Google Scholar] [CrossRef]
- Chen, J. Essential role of medicine and food homology in health and wellness. Chin. Herb. Med. 2023, 15, 347–348. [Google Scholar] [CrossRef]
- Korde, L.A.; Wu, A.H.; Fears, T.; Nomura, A.M.; West, D.W.; Kolonel, L.N.; Pike, M.C.; Hoover, R.N.; Ziegler, R.G. Childhood soy intake and breast cancer risk in Asian American women. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1050–1059. [Google Scholar] [CrossRef]
- Aliya, S.; Alhammadi, M.; Park, U.; Tiwari, J.N.; Lee, J.-H.; Han, Y.-K.; Huh, Y.S. The potential role of formononetin in cancer treatment: An updated review. Biomed. Pharmacother. 2023, 168, 115811. [Google Scholar] [CrossRef]
- Kim, C.; Lee, S.G.; Yang, W.M.; Arfuso, F.; Um, J.-Y.; Kumar, A.P.; Bian, J.; Sethi, G.; Ahn, K.S. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018, 431, 123–141. [Google Scholar] [CrossRef]
- He, M.; Shi, J.; Wu, C.; Xu, Y.-J.; Liu, Y. Integrating Lipidomics, Metabolomics, and Network Pharmacology to Reveal the Mechanism of Cannabidiol against Inflammation in High-Fat, High-Cholesterol Diet-Induced Mice. J. Agric. Food Chem. 2024, 72, 19246–19256. [Google Scholar] [CrossRef]
- Cheung, M.K.; Yue, G.G.; Gomes, A.J.; Wong, E.C.; Lee, J.K.; Kwok, F.H.; Chiu, P.W.; Lau, C.B. Network pharmacology reveals potential functional components and underlying molecular mechanisms of Andrographis paniculata in esophageal cancer treatment. Phytother. Res. 2022, 36, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kang, J.; Guo, W.; Wang, F.; Guo, M.; Feng, S.; Zhou, W.; Li, J.; Tahir, A.T.; Wang, S.; et al. An optimal medicinal and edible Chinese herbal formula attenuates particulate matter-induced lung injury through its anti-oxidative, anti-inflammatory and anti-apoptosis activities. Chin. Herb. Med. 2023, 15, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, J.; Qi, K.; Li, Y.; Chen, Y. Combined analysis of transcriptomics and metabolomics revealed complex metabolic genes for diterpenoids biosynthesis in different organs of Anoectochilus roxburghii. Chin. Herb. Med. 2023, 15, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, X.; Wang, Y.; Song, Y.; Pan, B.; Fan, B.; Wang, F.; Chen, X.; Tu, P.; Han, J.; et al. Neuroprotective effects of Longxue Tongluo Capsule on ischemic stroke rats revealed by LC-MS/MS-based metabolomics approach. Chin. Herb. Med. 2023, 15, 430–438. [Google Scholar] [CrossRef]
- Jäger, N. Bioinformatics workflows for clinical applications in precision oncology. Semin. Cancer Biol. 2022, 84, 103–112. [Google Scholar] [CrossRef]
- Sun, Y.; Tao, Q.; Cao, Y.; Yang, T.; Zhang, L.; Luo, Y.; Wang, L. Kaempferol has potential anti-coronavirus disease 2019 (COVID-19) targets based on bioinformatics analyses and pharmacological effects on endotoxin-induced cytokine storm. Phytother. Res. 2023, 37, 2290–2304. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, W.; Abdul Razak, S.R.; Han, T.; Ahmad, N.H.; Li, X. Ferroptosis as a potential target for cancer therapy. Cell Death Dis. 2023, 14, 460. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Tang, D.; Kroemer, G.; Ren, J. Ferroptosis in hepatocellular carcinoma: Mechanisms and targeted therapy. Br. J. Cancer 2023, 128, 190–205. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022, 29, 467–480. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Zhang, J.; Wu, D.; Zhang, J.; Zhou, H.; Rong, Y.; Liu, F.; Wei, B.; Xu, X. Ponicidin suppresses pancreatic cancer growth by inducing ferroptosis: Insight gained by mass spectrometry-based metabolomics. Phytomedicine 2022, 98, 153943. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ding, M.; Feng, M.; Fan, X.; Li, Z. Polyunsaturated Fatty Acids in Quinoa Induce Ferroptosis of Colon Cancer by Suppressing Stemness. J. Agric. Food Chem. 2024, 72, 16152–16162. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan BW, Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Xu, C.; Jiang, Z.B.; Shao, L.; Zhao, Z.-M.; Fan, X.-X.; Sui, X.; Yu, L.-L.; Wang, X.-R.; Zhang, R.-N.; Wang, W.-J.; et al. β-Elemene enhances erlotinib sensitivity through induction of ferroptosis by upregulating lncRNA H19 in EGFR-mutant non-small cell lung cancer. Pharmacol. Res. 2023, 191, 106739. [Google Scholar] [CrossRef]
- Li, H.; Yu, Y.; Liu, Y.; Luo, Z.; Law, B.Y.K.; Zheng, Y.; Huang, X.; Li, W. Ursolic acid enhances the antitumor effects of sorafenib associated with Mcl-1-related apoptosis and SLC7A11-dependent ferroptosis in human cancer. Pharmacol. Res. 2022, 182, 106306. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, J.Y.; Oh, M.; Lee, E.-W. An integrated view of lipid metabolism in ferroptosis revisited via lipidomic analysis. Exp. Mol. Med. 2023, 55, 1620–1631. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, S.; Yang, X.; Li, T.; Xia, Y.; Meng, X. Metabolomic study of the intervention effects of Shuihonghuazi Formula, a Traditional Chinese Medicinal formulae, on hepatocellular carcinoma (HCC) rats using performance HPLC/ESI-TOF-MS. J. Ethnopharmacol. 2017, 198, 468–478. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Bao, N.; Chen, Z.; Liu, M.; Zhao, C.; Li, X.; Zhang, Z. Mechanisms of Astragali Radix-Curcumae Rhi-zoma herb pair for anti-hepatocellular carcinoma by integrating bioinformatics and experimental validation. Chin. Tradit. Herb. Drugs 2024, 55, 114–126. (In Chinese) [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ni, F.; Qiu, T.; Wolff, J.T.; Tsai, S.C.; Luo, R. Molecular Basis for Polyketide Ketoreductase-Substrate Interactions. Int. J. Mol. Sci. 2020, 21, 7562. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Nguyen, H.; Roe, D.R.; Simmerling, C. Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput. 2013, 9, 2020–2034. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Liang, Z.Q.; Bian, Y.; Gu, J.F.; Yin, G.; Sun, R.L.; Liang, Y.; Wan, L.L.; Yin, Q.H.; Wang, X.; Gao, J.; et al. Exploring the anti-metastatic effects of Astragalus mongholicus Bunge-Curcuma aromatica Salisb. on colorectal cancer: A network-based metabolomics and pharmacology approach. Phytomedicine 2023, 114, 154772. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Jiang, M.W.; Li, M.Y.; Zhang, Z.H.; Xing, Y.; Ri, M.; Jin, C.H.; Xu, G.H.; Piao, L.X.; Jin, H.L.; et al. Formononetin represses cervical tumorigenesis by interfering with the activation of PD-L1 through MYC and STAT3 downregulation. J. Nutr. Biochem. 2022, 100, 108899. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qin, X.; Tian, J.; Gao, X.; Wu, X.; Du, G.; Zhou, Y. Liquiritin protects PC12 cells from corticosterone-induced neurotoxicity via regulation of metabolic disorders, attenuation ERK1/2-NF-κB pathway, activation Nrf2-Keap1 pathway, and inhibition mitochondrial apoptosis pathway. Food Chem. Toxicol. 2020, 146, 111801. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | PDB ID | Resolution | Docking Score | Amino Acid Residue |
|---|---|---|---|---|
| CHK1 | 5oq7 | 2.10 Å | −7.015 | ASP-148 |
| Cdc25C | 3op3 | 2.63 Å | −5.317 | TRP-414, TYR-485 |
| CDK1 | 4yc6 | 2.6 Å | −5.802 | ASP-146 |
| CCNB1 | 5lqf | 2.06 Å | −7.888 | LEU-83 |
| CCNB2 | O95067 | - | −5.012 | ASN-350, LYS-353 |
| CCNA2 | 7mkx | 3.08 Å | −5.858 | LYS-33 |
| KIF11 | 4a51 | 2.75 Å | −6.091 | TRP-127, ARG-221 |
| AURKB | 4af3 | 2.75 Å | −7.129 | LYS-106, ALA-157 |
| FEN1 | 5fv7 | 2.84 Å | −4.675 | ASP86 |
| BIRC5 | 2rax | 3.3 Å | −3.487 | ASP-71 |
| No. | Metabolites | Formula | m/z | RT [min] | HMDB | VIP | p | FC | Trend | Scan Mode |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | L-Lysine | C6H14N2O2 | 147.1128 | 1.103 | HMDB0000182 | 1.073 | 1.70 × 10−2 | 2.36 | ↑ | + |
| 2 | Choline | C5H14NO | 105.1075 | 1.285 | HMDB0000097 | 5.457 | 1.46 × 10−2 | 3.90 | ↑ | + |
| 3 | L-Serine | C3H7NO3 | 106.0926 | 1.287 | HMDB0000187 | 1.766 | 3.60 × 10−2 | 6.13 | ↑ | + |
| 4 | Citrulline | C6H13N3O3 | 176.1031 | 1.295 | HMDB0000904 | 1.629 | 3.93 × 10−3 | 127.97 | ↑ | + |
| 5 | L-Arginine | C6H14N4O2 | 175.1190 | 1.296 | HMDB0000517 | 1.308 | 1.12 × 10−3 | 0.06 | ↓ | + |
| 6 | L-Threonine | C4H9NO3 | 120.0658 | 1.304 | HMDB0000167 | 1.046 | 1.02 × 10−2 | 0.12 | ↓ | + |
| 7 | N-Acetylneuraminic acid | C11H19NO9 | 308.0993 | 1.319 | HMDB0000230 | 1.226 | 1.74 × 10−2 | 0.43 | ↓ | − |
| 8 | Asymmetric dimethylarginine | C8H18N4O2 | 203.1504 | 1.328 | HMDB0001539 | 1.134 | 5.37 × 10−3 | 18.90 | ↑ | + |
| 9 | Uridine diphosphate-N-acetylglucosamine | C17H27N3O17P2 | 606.0758 | 1.639 | HMDB0000290 | 1.183 | 4.92 × 10−5 | 0.05 | ↓ | − |
| 10 | L-Valine | C5H11NO2 | 118.0865 | 1.941 | HMDB0000883 | 6.734 | 9.46 × 10−4 | 2.76 | ↑ | + |
| 11 | dGDP | C10H15N5O10P2 | 426.0231 | 1.968 | HMDB0000960 | 2.681 | 1.33 × 10−3 | 0.03 | ↓ | − |
| 12 | L-Methionine | C5H11NO2S | 150.0584 | 2.023 | HMDB0000696 | 5.192 | 2.34 × 10−3 | 2.51 | ↑ | + |
| 13 | L-Isoleucine | C6H13NO2 | 132.1021 | 2.081 | HMDB0000172 | 1.996 | 2.61 × 10−2 | 0.32 | ↓ | + |
| 14 | Glutathione | C10H17N3O6S | 306.0770 | 2.084 | HMDB0062697 | 2.849 | 9.29 × 10−3 | 0.07 | ↓ | − |
| 15 | Oxoglutaric acid | C5H6O5 | 145.0981 | 2.102 | HMDB0000208 | 4.288 | 1.57 × 10−3 | 0.20 | ↓ | − |
| 16 | Hypoxanthine | C5H4N4O | 137.0459 | 2.181 | HMDB0000157 | 8.273 | 4.04 × 10−3 | 148.68 | ↑ | + |
| 17 | 5-Oxoproline | C5H7NO3 | 130.0501 | 2.254 | HMDB0000267 | 2.655 | 1.59 × 10−3 | 5.92 | ↑ | + |
| 18 | Acetaminophen | C8H9NO2 | 152.0707 | 2.287 | HMDB0001859 | 1.962 | 3.85 × 10−4 | 2.81 | ↑ | + |
| 19 | Niacinamide | C6H6N2O | 123.0556 | 2.338 | HMDB0001406 | 4.396 | 1.61 × 10−2 | 2.61 | ↑ | + |
| 20 | Xanthine | C5H4N4O2 | 153.0408 | 2.433 | HMDB0000292 | 2.371 | 4.51 × 10−3 | 184.22 | ↑ | + |
| 21 | N-Acetyl-L-aspartic acid | C6H9NO5 | 174.0399 | 2.524 | HMDB0000812 | 2.366 | 2.13 × 10−2 | 0.19 | ↓ | − |
| 22 | Isocitric acid | C6H8O7 | 191.0191 | 2.558 | HMDB0000193 | 1.904 | 4.84 × 10−3 | 0.13 | ↓ | − |
| 23 | L-Tyrosine | C9H11NO3 | 182.0812 | 2.590 | HMDB0000158 | 8.759 | 1.59 × 10−3 | 2.78 | ↑ | + |
| 24 | Spermine | C10H26N4 | 203.2231 | 2.601 | HMDB0001256 | 1.816 | 2.70 × 10−3 | 51.58 | ↑ | + |
| 25 | Uracil | C4H4N2O2 | 113.0350 | 2.624 | HMDB0000300 | 1.159 | 2.42 × 10−2 | 112.62 | ↑ | + |
| 26 | L-Glutamate | C5H9NO4 | 148.0605 | 2.756 | HMDB0000148 | 1.631 | 3.03 × 10−2 | 0.05 | ↓ | + |
| 27 | L-Glutamine | C5H10N2O3 | 147.0765 | 2.881 | HMDB0000641 | 1.761 | 3.05 × 10−2 | 7.46 | ↑ | + |
| 28 | L-Cysteine | C3H7NO2S | 120.1580 | 2.909 | HMDB0000574 | 1.099 | 2.99 × 10−2 | 0.01 | ↓ | − |
| 29 | L-Norleucine | C6H13NO2 | 132.1020 | 2.931 | HMDB0001645 | 14.419 | 3.20 × 10−3 | 2.52 | ↑ | + |
| 30 | Triethanolamine | C6H15NO3 | 150.1126 | 3.208 | HMDB0032538 | 1.102 | 1.05 × 10−4 | 2.10 | ↑ | + |
| 31 | L-Proline | C5H9NO2 | 116.0710 | 3.267 | HMDB0000162 | 2.820 | 2.38 × 10−2 | 2.39 | ↑ | + |
| 32 | Spermidine | C7H19N3 | 146.1652 | 4.531 | HMDB0001257 | 6.166 | 3.30 × 10−4 | 4.68 | ↑ | + |
| 33 | Oxidized glutathione | C20H32N6O12S2 | 611.1457 | 4.960 | HMDB0003337 | 4.990 | 1.79 × 10−3 | 0.14 | ↓ | − |
| 34 | L-Phenylalanine | C9H11NO2 | 166.0863 | 5.044 | HMDB0000159 | 10.928 | 6.97 × 10−3 | 2.15 | ↑ | + |
| 35 | N′-Formylkynurenine | C11H12N2O4 | 237.0871 | 5.046 | HMDB0001200 | 1.027 | 6.80 × 10−5 | 6.34 | ↑ | + |
| 36 | Adenine | C5H5N5 | 136.0619 | 5.128 | HMDB0000034 | 2.159 | 9.75 × 10−3 | 12.75 | ↑ | + |
| 37 | N-Formyl-L-methionine | C6H11NO3S | 176.0379 | 5.258 | HMDB0001015 | 1.437 | 8.47 × 10−4 | 2.80 | ↑ | − |
| 38 | Pantothenic acid | C9H17NO5 | 218.1032 | 5.333 | HMDB0000210 | 1.190 | 3.19 × 10−4 | 2.46 | ↑ | − |
| 39 | 8-Isoprostaglandin E2 | C20H32O5 | 351.2183 | 8.836 | HMDB0005844 | 1.806 | 4.87 × 10−2 | 3.11 | ↑ | − |
| 40 | Glycoursodeoxycholic acid | C26H43NO5 | 448.3076 | 11.325 | HMDB0000708 | 1.338 | 1.85 × 10−2 | 0.07 | ↓ | − |
| 41 | 8,9-DiHETrE | C20H34O4 | 339.4816 | 18.790 | HMDB0002311 | 1.830 | 5.31 × 10−3 | 0.26 | ↓ | + |
| 42 | L-Palmitoylcarnitine | C23H46NO4 | 399.3280 | 22.009 | HMDB0000222 | 1.753 | 9.76 × 10−5 | 0.21 | ↓ | − |
| 43 | Eicosapentaenoic acid | C20H30O2 | 301.2179 | 23.755 | HMDB0001999 | 1.468 | 3.24 × 10−2 | 14.10 | ↑ | − |
| 44 | Arachidonic acid | C20H32O2 | 303.2335 | 25.677 | HMDB0001043 | 3.689 | 1.12 × 10−2 | 12.90 | ↑ | − |
| 45 | Palmitic acid | C16H32O2 | 255.2331 | 27.686 | HMDB0000220 | 1.442 | 2.94 × 10−2 | 6.08 | ↑ | − |
| 46 | Oleic acid | C18H34O2 | 281.2491 | 28.297 | HMDB0000207 | 1.925 | 3.27 × 10−3 | 4.13 | ↑ | − |
| 47 | Stearic acid | C18H36O2 | 283.2647 | 30.628 | HMDB0000827 | 1.510 | 4.62 × 10−2 | 4.64 | ↑ | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, N.; Chen, Z.; Li, B.; Yang, H.; Li, X.; Zhang, Z. Study on the Mechanism of Formononetin Against Hepatocellular Carcinoma: Regulating Metabolic Pathways of Ferroptosis and Cell Cycle. Int. J. Mol. Sci. 2025, 26, 2578. https://doi.org/10.3390/ijms26062578
Bao N, Chen Z, Li B, Yang H, Li X, Zhang Z. Study on the Mechanism of Formononetin Against Hepatocellular Carcinoma: Regulating Metabolic Pathways of Ferroptosis and Cell Cycle. International Journal of Molecular Sciences. 2025; 26(6):2578. https://doi.org/10.3390/ijms26062578
Chicago/Turabian StyleBao, Ning, Zichao Chen, Baohong Li, Haolin Yang, Xiao Li, and Zhen Zhang. 2025. "Study on the Mechanism of Formononetin Against Hepatocellular Carcinoma: Regulating Metabolic Pathways of Ferroptosis and Cell Cycle" International Journal of Molecular Sciences 26, no. 6: 2578. https://doi.org/10.3390/ijms26062578
APA StyleBao, N., Chen, Z., Li, B., Yang, H., Li, X., & Zhang, Z. (2025). Study on the Mechanism of Formononetin Against Hepatocellular Carcinoma: Regulating Metabolic Pathways of Ferroptosis and Cell Cycle. International Journal of Molecular Sciences, 26(6), 2578. https://doi.org/10.3390/ijms26062578