
Neuroimmune Interactions and Their Role in Immune Cell Trafficking in Cardiovascular Diseases and Cancer

 ,
,  ,
,
Abstract
1. Introduction
2. Sympathetic Innervation of Lymphoid Organs
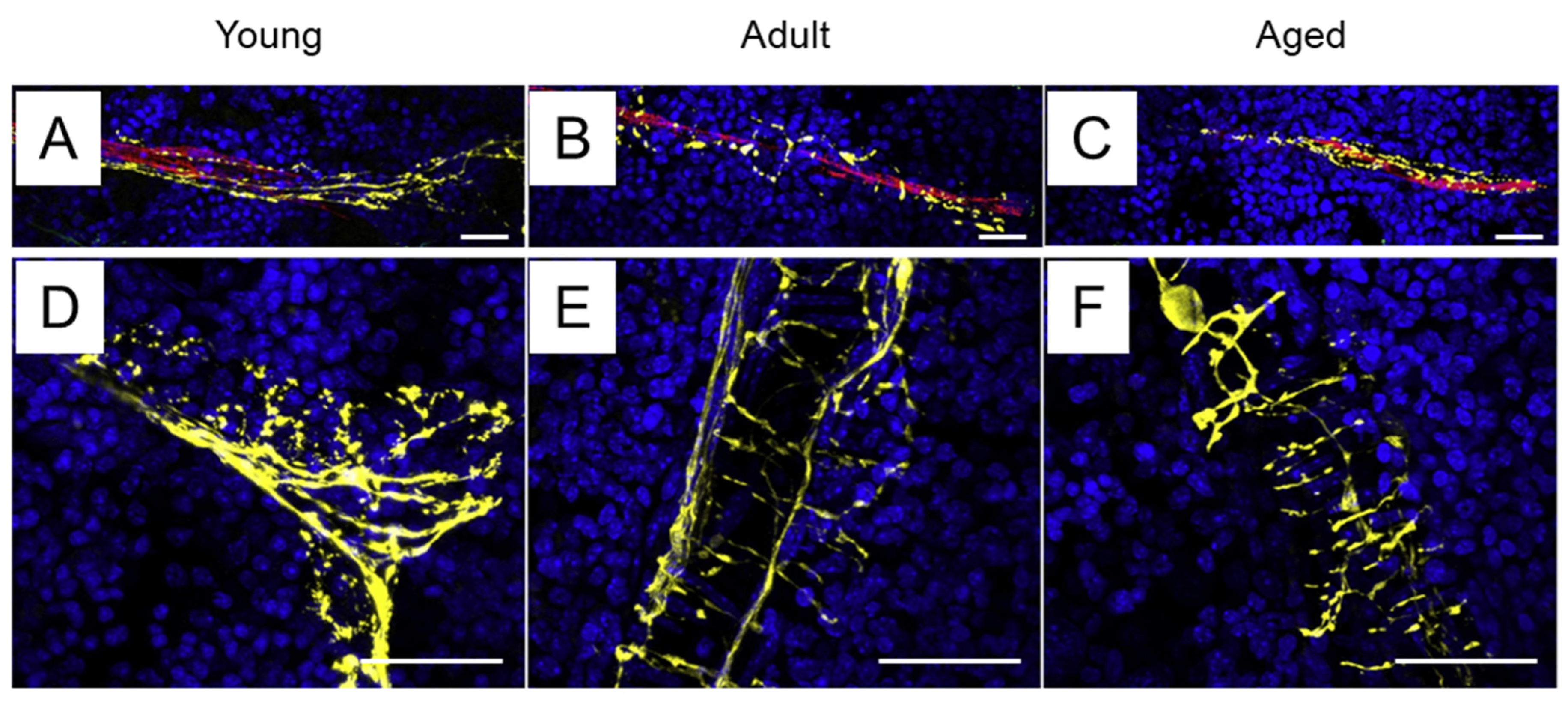
2.1. Sympathetic Innervation of the Bone Marrow
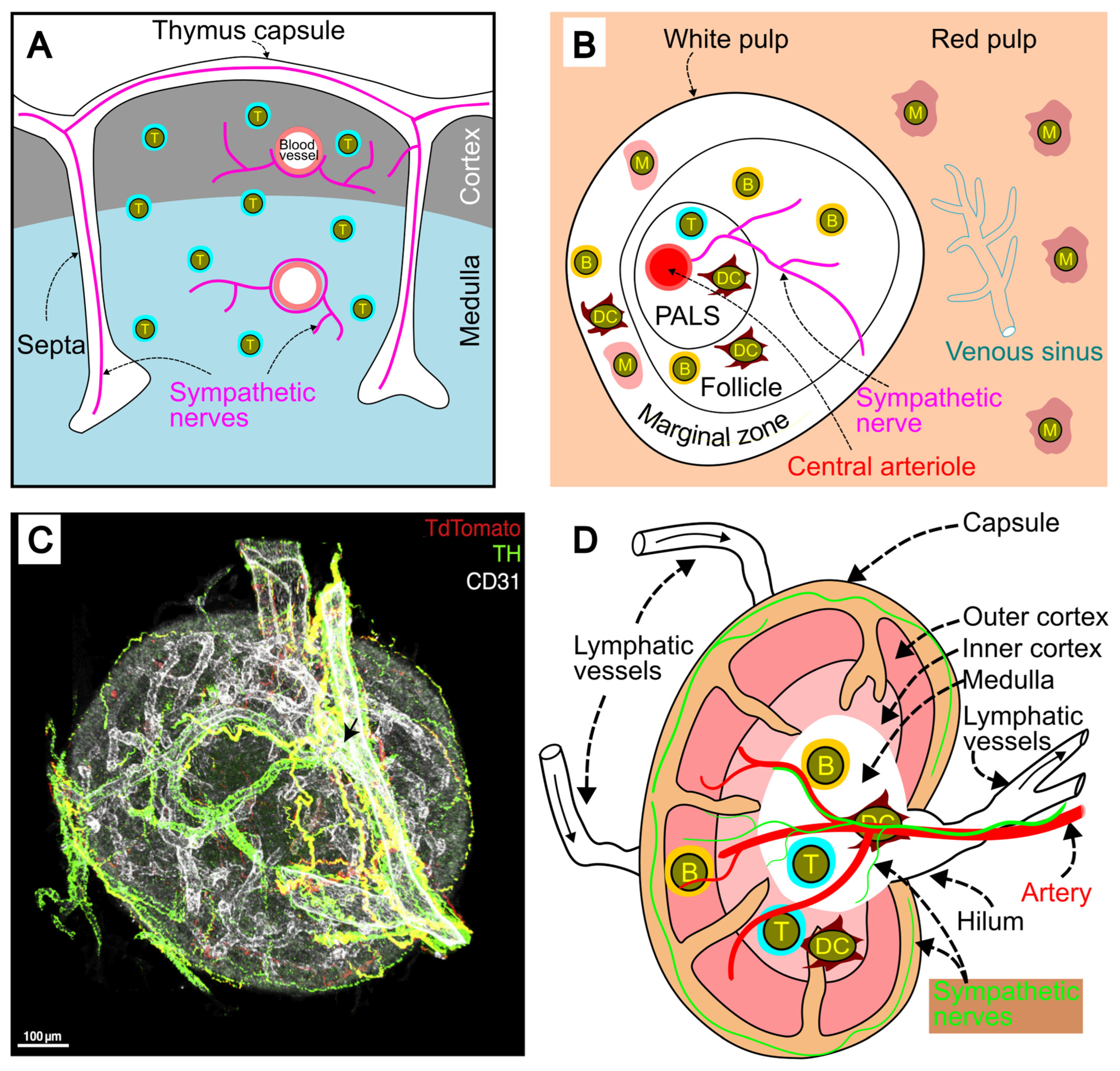
2.2. Sympathetic Innervation of the Thymus
2.3. Sympathetic Innervation of the Spleen
2.4. Sympathetic Innervation of Lymph Nodes
2.5. Sympathetic Innervation of Tertiary Lymphoid Organs
3. Expression of Adrenergic Receptors on Immune Cells
4. Adrenergic Signaling Pathways
4.1. α1-Adrenergic Signaling Pathway
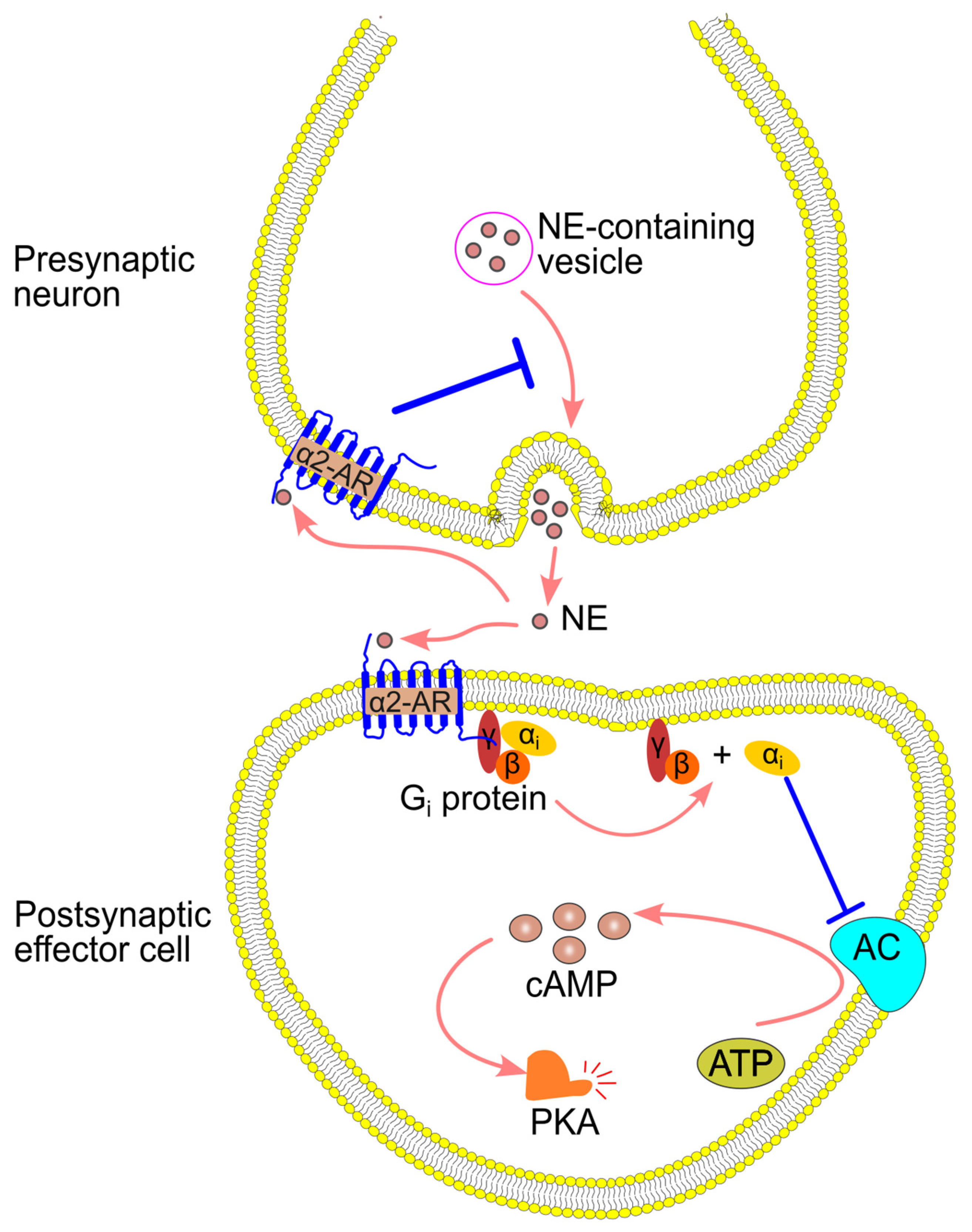
4.2. α2-Adrenergic Signaling Pathway
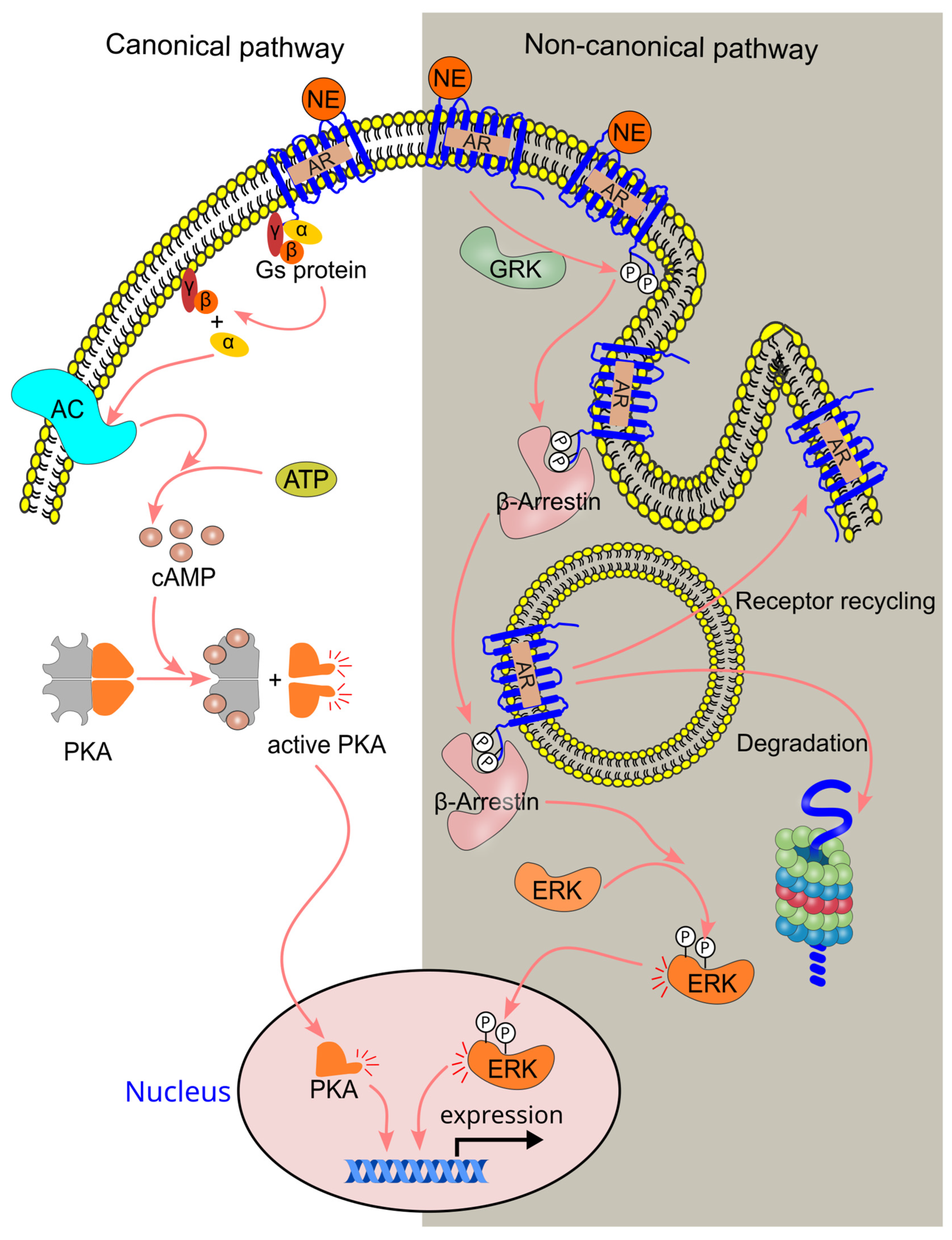
4.3. β-Adrenergic Signaling Pathway
5. Normal Range of Circulating Norepinephrine
6. Effect of Sympathetic Activation on Circulating Leukocyte Numbers
6.1. Effect of Norepinephrine on Numbers of Circulating Innate Immune Cells
6.2. Effect of Norepinephrine on Circulating Lymphocyte Numbers
7. Norepinephrine Is a Chemoattractant for Monocytes, Macrophages, Stem, and Progenitor Cells
8. Effect of Sympathetic Activity on Immune Cell Trafficking
8.1. Effect of Sympathetic Activity on Monocyte and Macrophage Trafficking
8.2. Effect of Sympathetic Activity on Neutrophil Trafficking
8.3. Effect of Sympathetic Activity on NK Cell Trafficking
8.4. Effect of Sympathetic Activity on Lymphocyte Trafficking
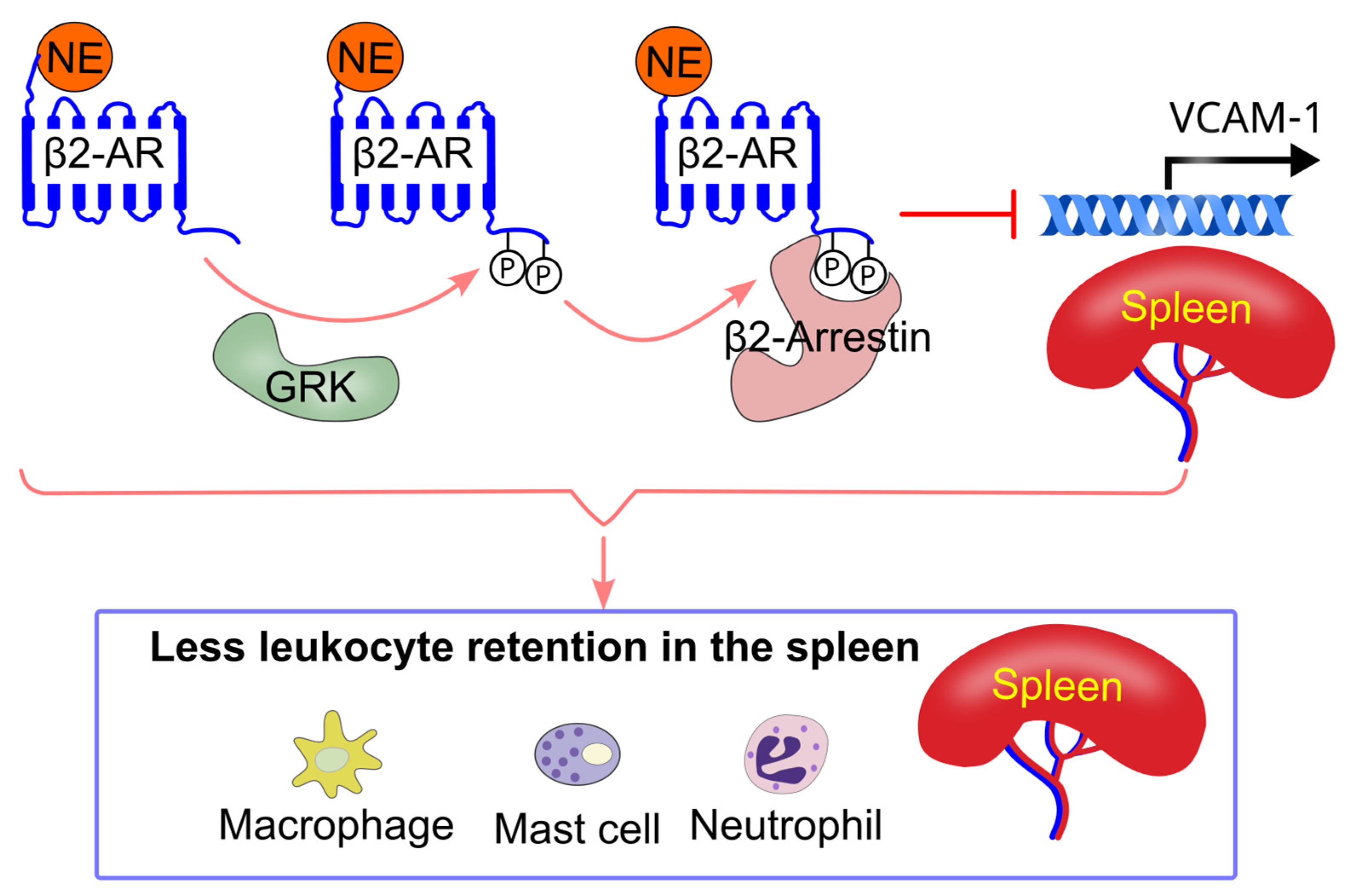
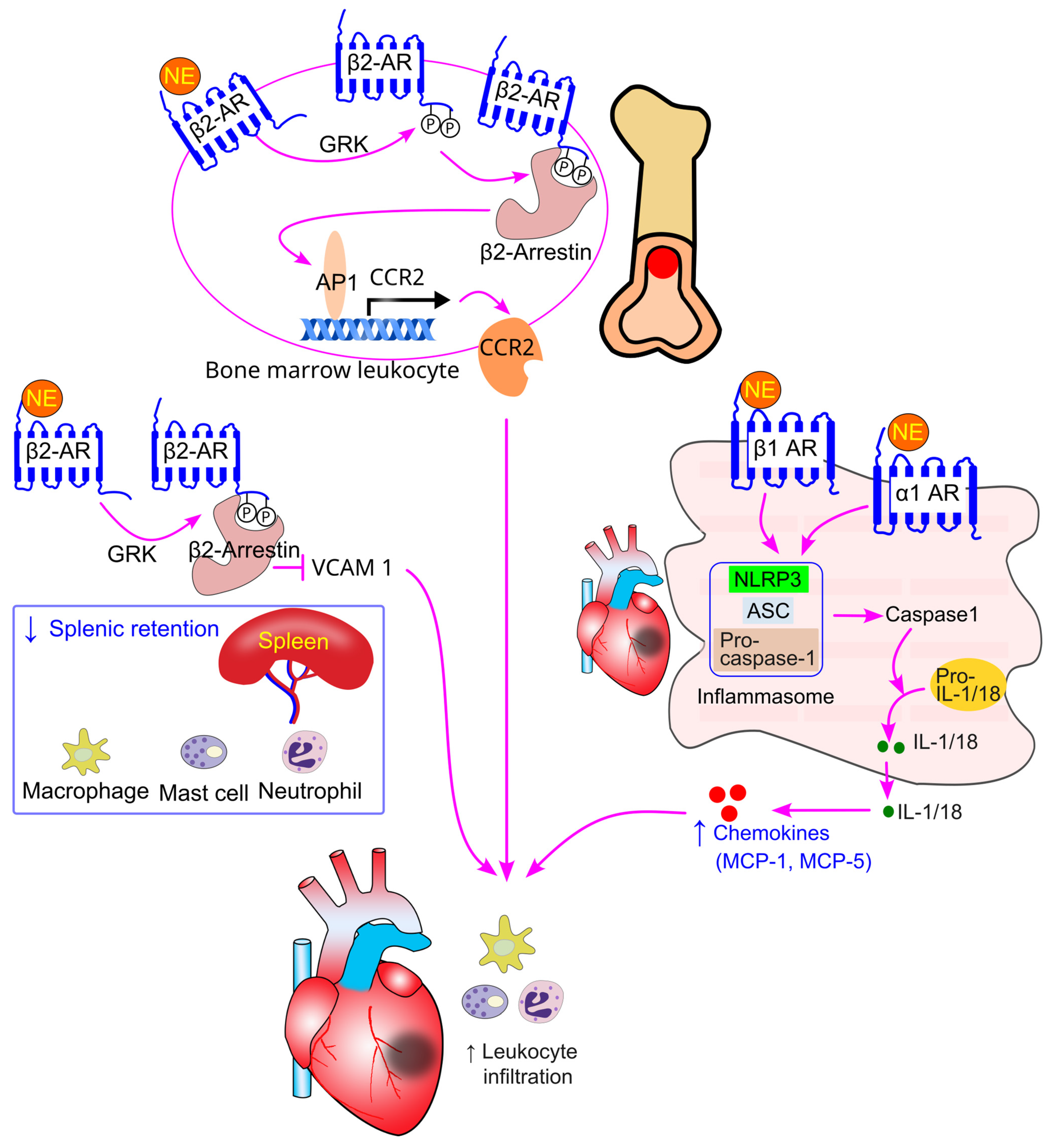
8.5. Effect of Sympathetic Activity on Immune Cell Splenic Retention
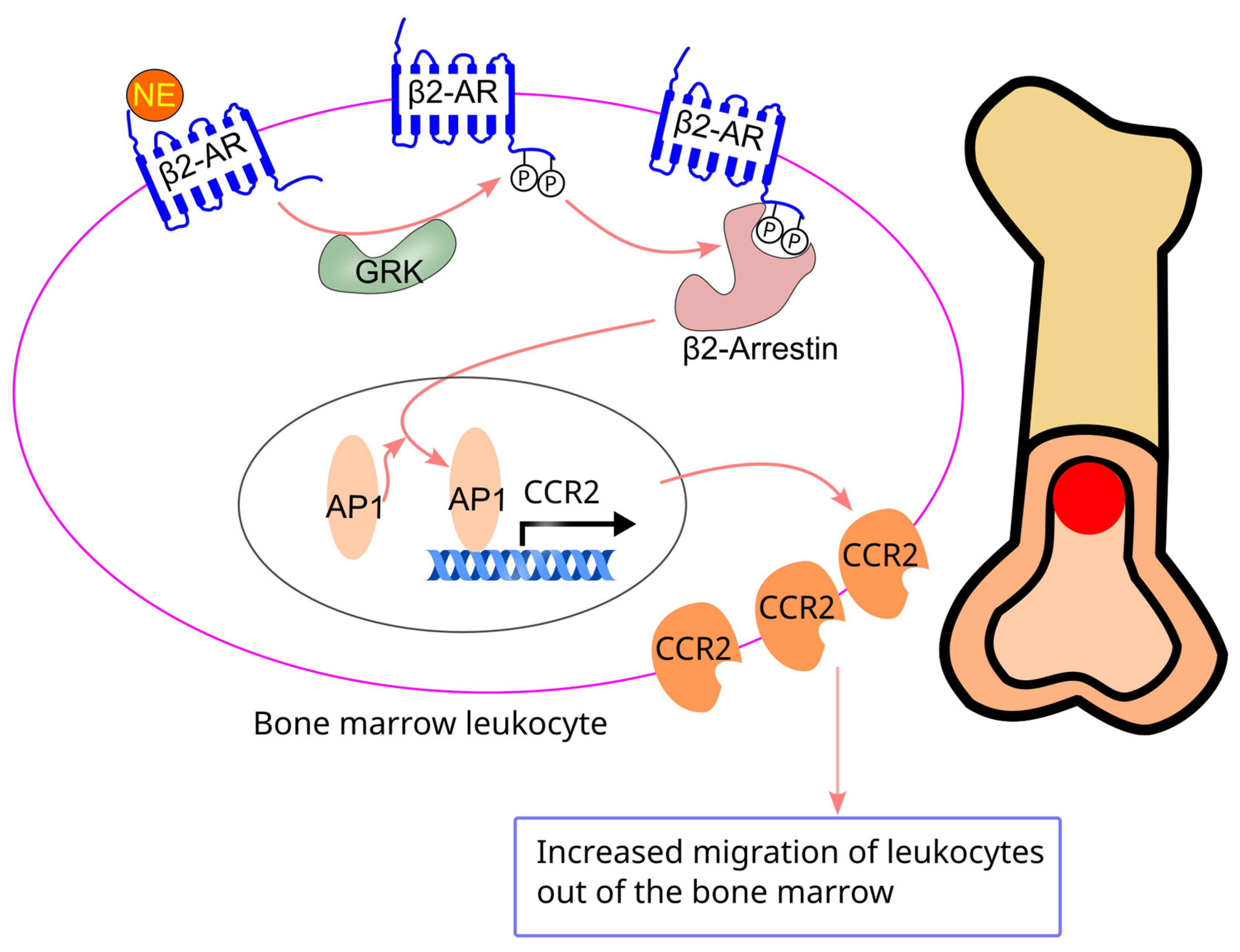
8.6. Effect of Sympathetic Activity on Bone Marrow Cell Migration
8.7. Effect of Sympathetic Activity on Peripheral Blood Leukocyte Migration
8.8. Effect of Sympathetic Activity on Motility of Lymph Node Lymphocytes
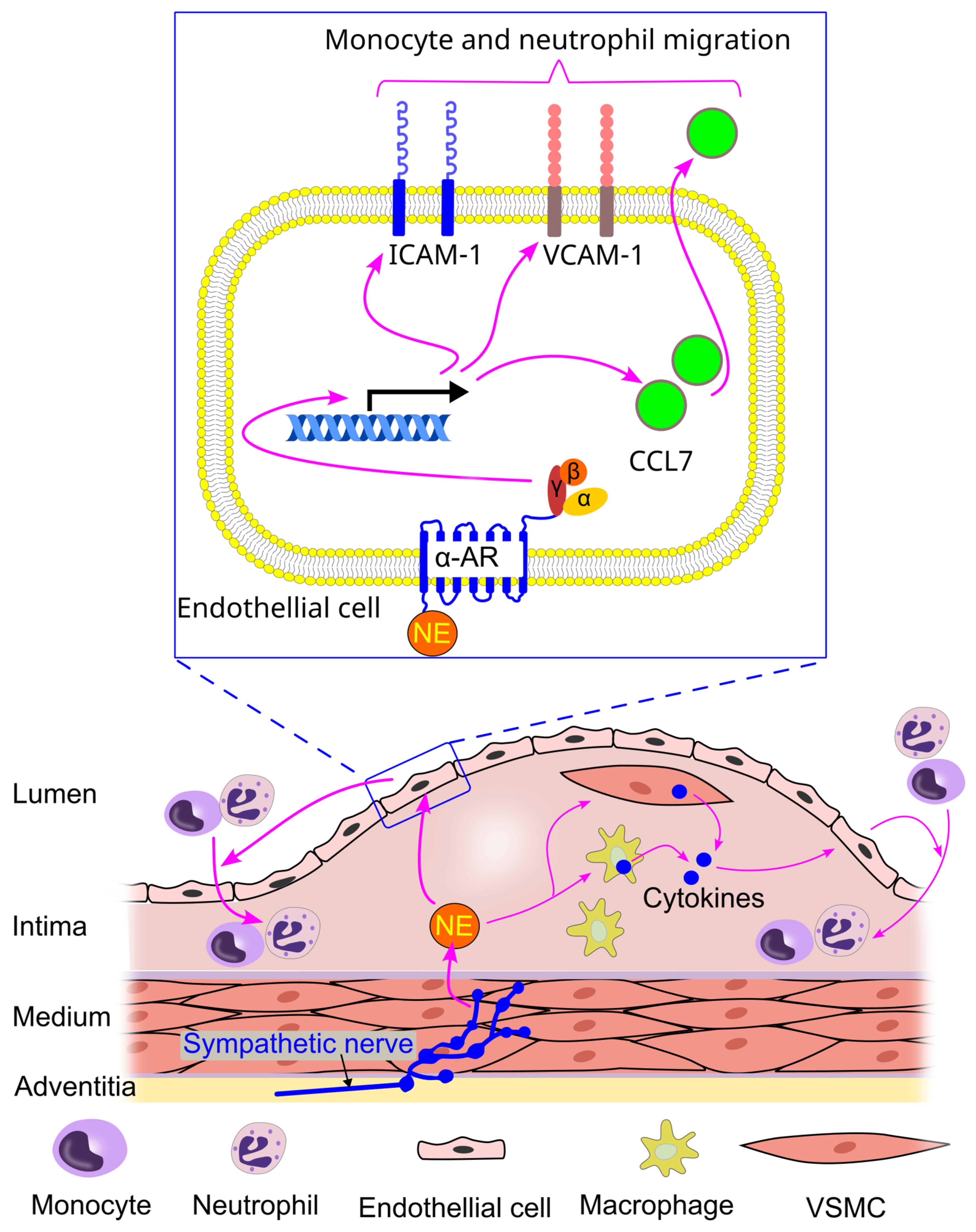
8.9. Effect of Sympathetic Activity on Immune Cell Interaction with Endothelial Cells
9. Sympathetic Activation and Immune Cell Trafficking in Cardiovascular Diseases (CVDs)
9.1. Sympathetic Activation and Immune Cell Trafficking in Atherosclerosis
9.2. Sympathetic Activation and Immune Cell Trafficking in Hypertension
9.3. Sympathetic Activation and Immune Cell Trafficking in Cardiac Fibrosis, Hypertrophy, and Arrhythmia
9.4. Sympathetic Activation and Immune Cell Trafficking in Myocardial Infarction
9.5. Sympathetic Activation and Immune Cell Trafficking in Heart Failure
9.6. Sympathetic Activation and Immune Cell Trafficking in Stroke
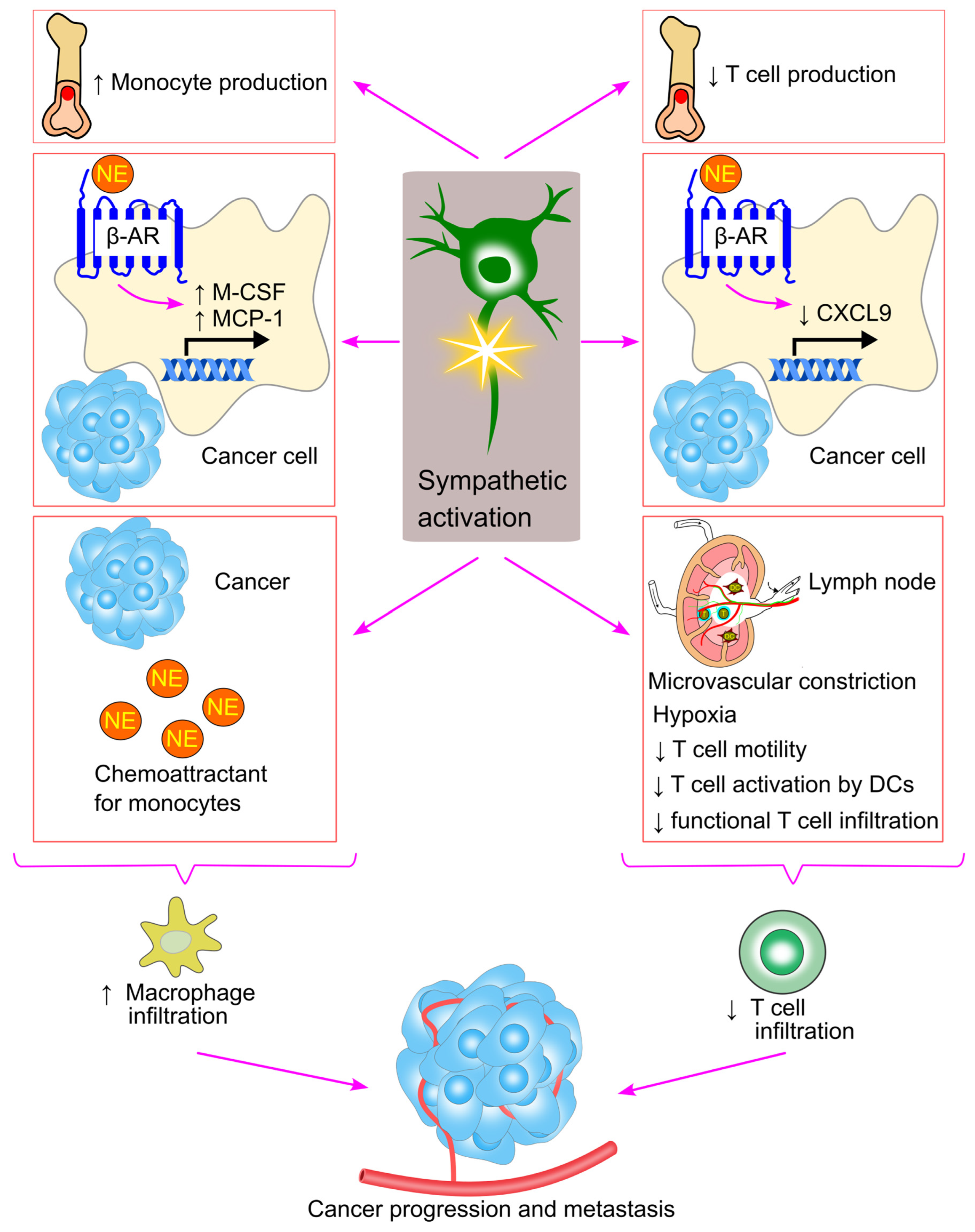
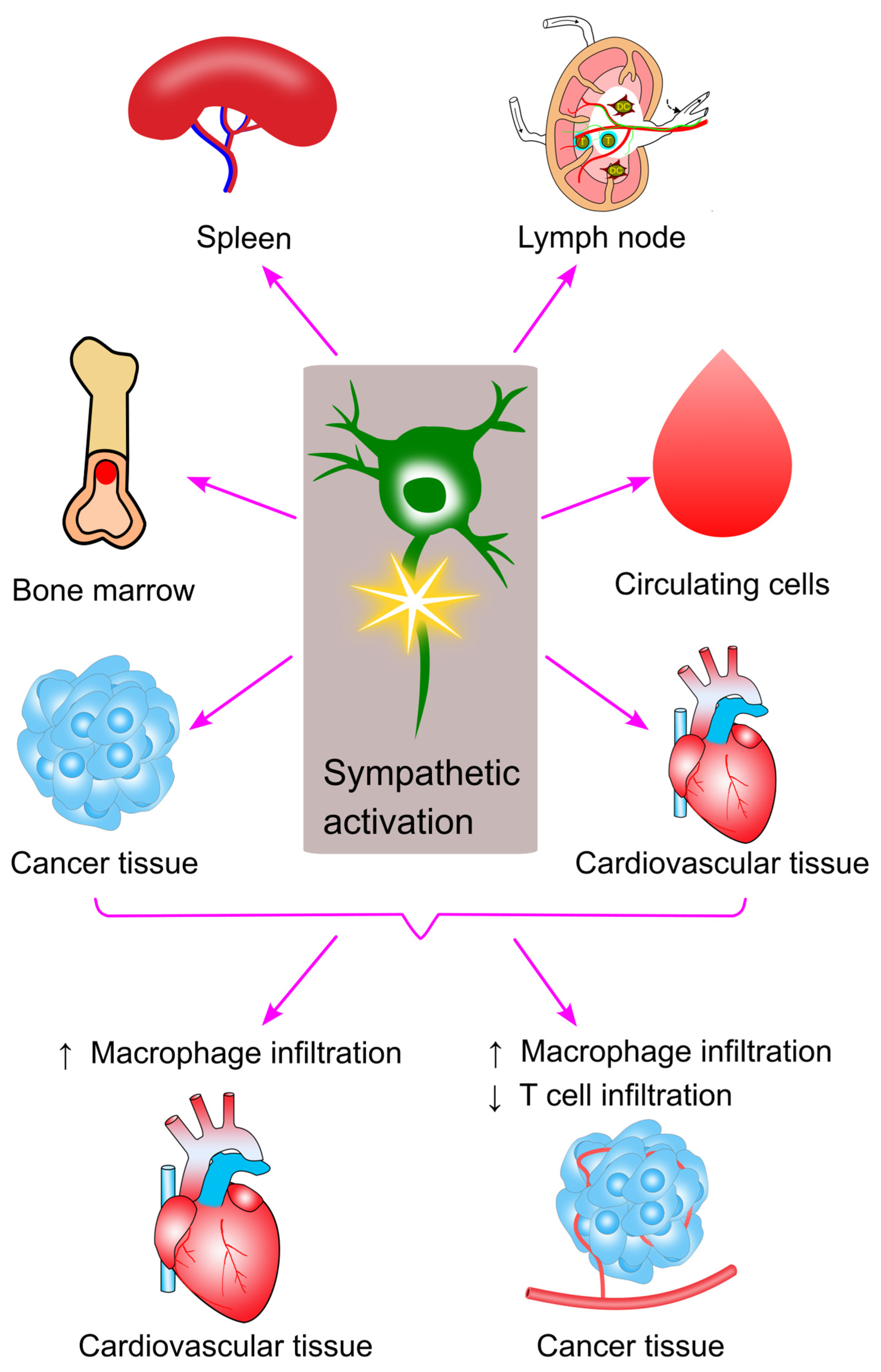
10. Sympathetic Activation and Immune Cell Trafficking in Cancer
11. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Nicholls, A.J.; Wen, S.W.; Hall, P.; Hickey, M.J.; Wong, C.H.Y. Activation of the sympathetic nervous system modulates neutrophil function. J. Leukoc. Biol. 2018, 103, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Golledge, J. Neuronal nitric oxide synthase and sympathetic nerve activity in neurovascular and metabolic systems. Curr. Neurovasc. Res. 2013, 10, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Chhatar, S.; Lal, G. Role of adrenergic receptor signalling in neuroimmune communication. Curr. Res. Immunol. 2021, 2, 202–217. [Google Scholar] [CrossRef]
- Wang, Y.; Denton, K.M. Special Issue “Sympathetic Nerves and Cardiovascular Diseases”. Int. J. Mol. Sci. 2024, 25, 2633. [Google Scholar] [CrossRef] [PubMed]
- WHO. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)#:~:text=What%20are%20the%20risk%20factors,and%20harmful%20use%20of%20alcohol (accessed on 14 November 2024).
- Wang, Y.; Magliano, D.J. Special Issue: “New Trends in Diabetes, Hypertension, and Cardiovascular Diseases—2nd Edition”. Int. J. Mol. Sci. 2025, 26, 449. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; Sobey, C.G.; Drummond, G.R. Prior cancer diagnosis and mortality profile in US adults. Am. J. Med. Sci. 2023, 365, 176–183. [Google Scholar] [CrossRef]
- Bellinger, D.L.; Millar, B.A.; Perez, S.; Carter, J.; Wood, C.; ThyagaRajan, S.; Molinaro, C.; Lubahn, C.; Lorton, D. Innervation of lymphoid organs: Clinical implications. Clin. Neurosci. Res. 2006, 6, 3–33. [Google Scholar] [CrossRef]
- Prisby, R.D. Bone Marrow Microvasculature. Compr. Physiol. 2020, 10, 1009–1046. [Google Scholar]
- Weih, F.; Gräbner, R.; Hu, D.; Beer, M.; Habenicht, A.J. Control of Dichotomic Innate and Adaptive Immune Responses by Artery Tertiary Lymphoid Organs in Atherosclerosis. Front. Physiol. 2012, 3, 226. [Google Scholar] [CrossRef]
- Hu, D.; Mohanta, S.K.; Yin, C.; Peng, L.; Ma, Z.; Srikakulapu, P.; Grassia, G.; MacRitchie, N.; Dever, G.; Gordon, P.; et al. Artery Tertiary Lymphoid Organs Control Aorta Immunity and Protect against Atherosclerosis via Vascular Smooth Muscle Cell Lymphotoxin β Receptors. Immunity 2015, 42, 1100–1115. [Google Scholar] [CrossRef]
- Riffard, C.; Letaïef, L.; Azar, S.; Casrouge, A.; Brunet, I.; Teillaud, J.-L.; Dieu-Nosjean, M.-C. Absence of sympathetic innervation hampers the generation of tertiary lymphoid structures upon acute lung inflammation. Sci. Rep. 2024, 14, 11749. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J.; Horowitz, M.C. Nutrient regulation of bone marrow adipose tissue: Skeletal implications of weight loss. Nat. Rev. Endocrinol. 2023, 19, 626–638. [Google Scholar] [CrossRef]
- Jung, W.-C.; Levesque, J.-P.; Ruitenberg, M.J. It takes nerve to fight back: The significance of neural innervation of the bone marrow and spleen for immune function. Semin. Cell Dev. Biol. 2017, 61, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Calvo, W. The innervation of the bone marrow in laboratory animals. Am. J. Anat. 1968, 123, 315–328. [Google Scholar] [CrossRef]
- Chartier, S.R.; Mitchell, S.A.T.; Majuta, L.A.; Mantyh, P.W. The Changing Sensory and Sympathetic Innervation of the Young, Adult and Aging Mouse Femur. Neuroscience 2018, 387, 178–190. [Google Scholar] [CrossRef]
- Courties, G.; Herisson, F.; Sager, H.B.; Heidt, T.; Ye, Y.; Wei, Y.; Sun, Y.; Severe, N.; Dutta, P.; Scharff, J.; et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res. 2015, 116, 407–417. [Google Scholar] [CrossRef]
- Mignini, F.; Sabbatini, M.; Mattioli, L.; Cosenza, M.; Artico, M.; Cavallotti, C. Neuro-immune modulation of the thymus microenvironment (Review). Int. J. Mol. Med. 2014, 33, 1392–1400. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, H.; Yang, J. Neuroanatomy of lymphoid organs: Lessons learned from whole-tissue imaging studies. Eur. J. Immunol. 2023, 53, 2250136. [Google Scholar] [CrossRef]
- Bulloch, K.; Pomerantz, W. Autonomic nervous system innervation of thymic-related lymphoid tissue in wildtype and nude mice. J. Comp. Neurol. 1984, 228, 57–68. [Google Scholar] [CrossRef]
- Huang, S.; Ziegler, C.G.K.; Austin, J.; Mannoun, N.; Vukovic, M.; Ordovas-Montanes, J.; Shalek, A.K.; von Andrian, U.H. Lymph nodes are innervated by a unique population of sensory neurons with immunomodulatory potential. Cell 2021, 184, 441–459.e25. [Google Scholar] [CrossRef]
- Steiniger, B.S.; Pfeffer, H.; Guthe, M.; Lobachev, O. Exploring human splenic red pulp vasculature in virtual reality: Details of sheathed capillaries and the open capillary network. Histochem. Cell Biol. 2021, 155, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D. Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)? Int. J. Mol. Sci. 2018, 19, 1188. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, H.; Qian, X.; Han, X.; Yang, L.; Cao, Y.; Wang, Q.; Yang, J. Panicle-Shaped Sympathetic Architecture in the Spleen Parenchyma Modulates Antibacterial Innate Immunity. Cell Rep. 2019, 27, 3799–3807.e3. [Google Scholar] [CrossRef]
- Ackerman, K.D.; Felten, S.Y.; Dijkstra, C.D.; Livnat, S.; Felten, D.L. Parallel development of noradrenergic innervation and cellular compartmentation in the rat spleen. Exp. Neurol. 1989, 103, 239–255. [Google Scholar] [CrossRef]
- Felten, D.L.; Ackerman, K.D.; Wiegand, S.J.; Felten, S.Y. Noradrenergic sympathetic innervation of the spleen: I. Nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J. Neurosci. Res. 1987, 18, 28–36. [Google Scholar] [CrossRef]
- Bujoreanu, I.; Gupta, V. Anatomy, Lymph Nodes. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557717/ (accessed on 17 January 2024).
- Weihe, E.; Nohr, D.; Michel, S.; Müller, S.; Zentel, H.J.; Fink, T.; Krekel, J. Molecular anatomy of the neuro-immune connection. Int. J. Neurosci. 1991, 59, 1–23. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Mohanta, S.K.; Yin, C.; Peng, L.; Srikakulapu, P.; Bontha, V.; Hu, D.; Weih, F.; Weber, C.; Gerdes, N.; Habenicht, A.J.R. Artery Tertiary Lymphoid Organs Contribute to Innate and Adaptive Immune Responses in Advanced Mouse Atherosclerosis. Circ. Res. 2014, 114, 1772–1787. [Google Scholar] [CrossRef]
- Mohanta, S.K.; Yin, C.; Weber, C.; Godinho-Silva, C.; Veiga-Fernandes, H.; Xu, Q.J.; Chang, R.B.; Habenicht, A.J.R. Cardiovascular Brain Circuits. Circ. Res. 2023, 132, 1546–1565. [Google Scholar] [CrossRef]
- Mohanta, S.K.; Peng, L.; Li, Y.; Lu, S.; Sun, T.; Carnevale, L.; Perrotta, M.; Ma, Z.; Förstera, B.; Stanic, K.; et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature 2022, 605, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Anesi, J.; Maier, M.C.; Myers, M.A.; Oqueli, E.; Sobey, C.G.; Drummond, G.R.; Denton, K.M. Sympathetic Nervous System and Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 13132. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, L.; Zhao, S. Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules 2021, 11, 936. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.R. Subtypes of functional alpha1-adrenoceptor. Cell. Mol. Life Sci. 2010, 67, 405–417. [Google Scholar] [CrossRef]
- Sheng, Y.; Zhu, L. The crosstalk between autonomic nervous system and blood vessels. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 17–28. [Google Scholar]
- Krief, S.; Lönnqvist, F.; Raimbault, S.; Baude, B.; Van Spronsen, A.; Arner, P.; Strosberg, A.D.; Ricquier, D.; Emorine, L.J. Tissue distribution of beta 3-adrenergic receptor mRNA in man. J. Clin. Investig. 1993, 91, 344–349. [Google Scholar] [CrossRef]
- Enocksson, S.; Shimizu, M.; Lönnqvist, F.; Nordenström, J.; Arner, P. Demonstration of an in vivo functional beta 3-adrenoceptor in man. J. Clin. Investig. 1995, 95, 2239–2245. [Google Scholar] [CrossRef]
- Matzkin, M.E.; Riviere, E.; Rossi, S.P.; Ponzio, R.; Puigdomenech, E.; Levalle, O.; Terradas, C.; Calandra, R.S.; Mayerhofer, A.; Frungieri, M.B. β-adrenergic receptors in the up-regulation of COX2 expression and prostaglandin production in testicular macrophages: Possible relevance to male idiopathic infertility. Mol. Cell. Endocrinol. 2019, 498, 110545. [Google Scholar] [CrossRef]
- Zieziulewicz, T.J.; Mondal, T.K.; Gao, D.; Lawrence, D.A. Stress-induced effects, which inhibit host defenses, alter leukocyte trafficking. Cell Stress Chaperones 2013, 18, 279–291. [Google Scholar] [CrossRef]
- Laukova, M.; Vargovic, P.; Csaderova, L.; Chovanova, L.; Vlcek, M.; Imrich, R.; Krizanova, O.; Kvetnansky, R. Acute stress differently modulates β1, β2 and β3 adrenoceptors in T cells, but not in B cells, from the rat spleen. Neuroimmunomodulation 2012, 19, 69–78. [Google Scholar] [CrossRef]
- Motulsky, H.J.; Insel, P.A. Adrenergic receptors in man: Direct identification, physiologic regulation, and clinical alterations. N. Engl. J. Med. 1982, 307, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Benschop, R.J.; Rodriguez-Feuerhahn, M.; Schedlowski, M. Catecholamine-induced leukocytosis: Early observations, current research, and future directions. Brain Behav. Immun. 1996, 10, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, M.; Fietta, A.M.; Ferrari, M.; Rasini, E.; Bombelli, R.; Carcano, E.; Saporiti, F.; Meloni, F.; Marino, F.; Lecchini, S. Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 2007, 109, 632–642. [Google Scholar] [CrossRef]
- Guereschi, M.G.; Araujo, L.P.; Maricato, J.T.; Takenaka, M.C.; Nascimento, V.M.; Vivanco, B.C.; Reis, V.O.; Keller, A.C.; Brum, P.C.; Basso, A.S. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur. J. Immunol. 2013, 43, 1001–1012. [Google Scholar] [CrossRef]
- Lubahn, C.L.; Lorton, D.; Schaller, J.A.; Sweeney, S.J.; Bellinger, D.L. Targeting α-and β-adrenergic receptors differentially shifts Th1, Th2, and inflammatory cytokine profiles in immune organs to attenuate adjuvant arthritis. Front. Immunol. 2014, 5, 346. [Google Scholar] [CrossRef]
- Pilipović, I.; Vujnović, I.; Stojić-Vukanić, Z.; Petrović, R.; Kosec, D.; Nacka-Aleksić, M.; Jasnić, N.; Leposavić, G. Noradrenaline modulates CD4+ T cell priming in rat experimental autoimmune encephalomyelitis: A role for the α1-adrenoceptor. Immunol. Res. 2019, 67, 223–240. [Google Scholar] [CrossRef]
- Yuki, K. The immunomodulatory mechanism of dexmedetomidine. Int. Immunopharmacol. 2021, 97, 107709. [Google Scholar] [CrossRef]
- Sanders, V.M. The beta2-adrenergic receptor on T and B lymphocytes: Do we understand it yet? Brain Behav. Immun. 2012, 26, 195–200. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Perez, D.M.; Porter, J.E. Modulation of immune cell function by α1-adrenergic receptor activation. Curr. Top. Membr. 2011, 67, 113–138. [Google Scholar]
- Jetschmann, J.-U.; Benschop, R.J.; Jacobs, R.; Kemper, A.; Oberbeck, R.; Schmidt, R.E.; Schedlowski, M. Expression and in-vivo modulation of α-and β-adrenoceptors on human natural killer (CD16+) cells. J. Neuroimmunol. 1997, 74, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Surface molecules regulating rolling and adhesion to endothelium of neutrophil granulocytes and MDA-MB-468 breast carcinoma cells and their interaction. Cell. Mol. Life Sci. 2007, 64, 3306–3316. [Google Scholar] [CrossRef]
- Strell, C.; Sievers, A.; Bastian, P.; Lang, K.; Niggemann, B.; Zänker, K.S.; Entschladen, F. Divergent effects of norepinephrine, dopamine and substance P on the activation, differentiation and effector functions of human cytotoxic T lymphocytes. BMC Immunol. 2009, 10, 62. [Google Scholar] [CrossRef]
- Silberman, D.M.; Wald, M.R.; Genaro, A.M. Acute and chronic stress exert opposing effects on antibody responses associated with changes in stress hormone regulation of T-lymphocyte reactivity. J. Neuroimmunol. 2003, 144, 53–60. [Google Scholar] [CrossRef]
- Madden, K.S.; Felten, D.L. Experimental basis for neural-immune interactions. Physiol. Rev. 1995, 75, 77–106. [Google Scholar] [CrossRef]
- Ben-Shalom, N.; Sandbank, E.; Abramovitz, L.; Hezroni, H.; Levine, T.; Trachtenberg, E.; Fogel, N.; Mor, M.; Yefet, R.; Stoler-Barak, L.; et al. β2-adrenergic signaling promotes higher-affinity B cells and antibodies. Brain Behav. Immun. 2023, 113, 66–82. [Google Scholar] [CrossRef]
- Honke, N.; Wiest, C.J.; Pongratz, G. β2-Adrenergic Receptor Expression and Intracellular Signaling in B Cells Are Highly Dynamic during Collagen-Induced Arthritis. Biomedicines 2022, 10, 1950. [Google Scholar] [CrossRef]
- Marino, F.; Cosentino, M. Adrenergic modulation of immune cells: An update. Amino Acids 2013, 45, 55–71. [Google Scholar] [CrossRef]
- Miksa, M.; Das, P.; Zhou, M.; Wu, R.; Dong, W.; Ji, Y.; Goyert, S.M.; Ravikumar, T.S.; Wang, P. Pivotal role of the α2A-adrenoceptor in producing inflammation and organ injury in a rat model of sepsis. PLoS ONE 2009, 4, e5504. [Google Scholar] [CrossRef]
- Li, M.; Yang, X.; Zhuang, C.; Cao, Z.; Ren, L.; Xiu, C.; Li, Y.; Zhu, Y. NE strengthens the immunosuppression induced by AlCl3 through β2-AR/cAMP pathway in cultured rat peritoneal macrophages. Biol. Trace Elem. Res. 2015, 164, 234–241. [Google Scholar] [CrossRef]
- Kavelaars, A. Regulated expression of α-1 adrenergic receptors in the immune system. Brain Behav. Immun. 2002, 16, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, G.J. Dendritic cell migration controlled by α1b-adrenergic receptors. J. Immunol. 2000, 165, 6743–6747. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, G.J.; Mazzola, P. Langerhans cells β2-adrenoceptors: Role in migration, cytokine production, Th priming and contact hypersensitivity. J. Neuroimmunol. 2003, 144, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Kirabo, A.; Wu, J.; Saleh, M.A.; Zhu, L.; Wang, F.; Takahashi, T.; Loperena, R.; Foss, J.D.; Mernaugh, R.L.; et al. Renal Denervation Prevents Immune Cell Activation and Renal Inflammation in Angiotensin II-Induced Hypertension. Circ. Res. 2015, 117, 547–557. [Google Scholar] [CrossRef]
- García-Prieto, J.; Villena-Gutiérrez, R.; Gómez, M.; Bernardo, E.; Pun-García, A.; García-Lunar, I.; Crainiciuc, G.; Fernández-Jiménez, R.; Sreeramkumar, V.; Bourio-Martínez, R. Neutrophil stunning by metoprolol reduces infarct size. Nat. Commun. 2017, 8, 14780. [Google Scholar] [CrossRef]
- Kim, M.-H.; Gorouhi, F.; Ramirez, S.; Granick, J.L.; Byrne, B.A.; Soulika, A.M.; Simon, S.I.; Isseroff, R.R. Catecholamine stress alters neutrophil trafficking and impairs wound healing by β2-adrenergic receptor–mediated upregulation of IL-6. J. Investig. Dermatol. 2014, 134, 809–817. [Google Scholar] [CrossRef]
- Marino, F.; Scanzano, A.; Pulze, L.; Pinoli, M.; Rasini, E.; Luini, A.; Bombelli, R.; Legnaro, M.; de Eguileor, M.; Cosentino, M. β2-Adrenoceptors inhibit neutrophil extracellular traps in human polymorphonuclear leukocytes. J. Leukoc. Biol. 2018, 104, 603–614. [Google Scholar] [CrossRef]
- Gan, X.; Zhang, L.; Solomon, G.F.; Bonavida, B. Mechanism of norepinephrine-mediated inhibition of human NK cytotoxic functions: Inhibition of cytokine secretion, target binding, and programming for cytotoxicity. Brain Behav. Immun. 2002, 16, 227–246. [Google Scholar] [CrossRef]
- Shakhar, G.; Ben-Eliyahu, S. In vivo β-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats. J. Immunol. 1998, 160, 3251–3258. [Google Scholar] [CrossRef]
- Lang, K.; Drell, T.L.; Niggemann, B.; Zänker, K.S.; Entschladen, F. Neurotransmitters regulate the migration and cytotoxicity in natural killer cells. Immunol. Lett. 2003, 90, 165–172. [Google Scholar] [CrossRef]
- Takamoto, T.; Hori, Y.; Koga, Y.; Toshima, H.; Hara, A.; Yokoyama, M.M. Norepinephrine inhibits human natural killer cell activity in vitro. Int. J. Neurosci. 1991, 58, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Alexandre, Y.O.; Loi, J.K.; Gillis, R.; Ghazanfari, N.; Creed, S.J.; Holz, L.E.; Shackleford, D.; Mackay, L.K.; Heath, W.R.; et al. Adrenergic regulation of the vasculature impairs leukocyte interstitial migration and suppresses immune responses. Immunity 2021, 54, 1219–1230.e7. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Traynham, C.J.; Repas, A.A.; Gao, E.; Koch, W.J.; Tilley, D.G. β2-Adrenergic receptor-dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc. Natl. Acad. Sci. USA 2016, 113, 15126–15131. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef]
- Geng, Q.; Li, L.; Shen, Z.; Zheng, Y.; Wang, L.; Xue, R.; Xue, W.; Peng, M.; Zhao, J. Norepinephrine inhibits CD8+ T-cell infiltration and function, inducing anti-PD-1 mAb resistance in lung adenocarcinoma. Br. J. Cancer 2023, 128, 1223–1235. [Google Scholar] [CrossRef]
- Xiao, H.; Li, H.; Wang, J.J.; Zhang, J.S.; Shen, J.; An, X.B.; Zhang, C.C.; Wu, J.M.; Song, Y.; Wang, X.Y.; et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart J. 2018, 39, 60–69. [Google Scholar] [CrossRef]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.G.; Morizono, K.; Karanikolas, B.D.W.; Wu, L.; et al. The Sympathetic Nervous System Induces a Metastatic Switch in Primary Breast Cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef]
- Xin, J.Z.; Wu, J.M.; Hu, G.M.; Gu, H.J.; Feng, Y.N.; Wang, S.X.; Cong, W.W.; Li, M.Z.; Xu, W.L.; Song, Y.; et al. α1-AR overactivation induces cardiac inflammation through NLRP3 inflammasome activation. Acta Pharmacol. Sin. 2020, 41, 311–318. [Google Scholar] [CrossRef]
- García, J.J.; del Carmen Sáez, M.; De la Fuente, M.; Ortega, E. Noradrenaline and its end metabolite 3-methoxy-4-hydroxyphenylglycol inhibit lymphocyte chemotaxis: Role of alpha- and beta-adrenoreceptors. Mol. Cell. Biochem. 2003, 254, 305–309. [Google Scholar] [CrossRef]
- Pagano, F.; Angelini, F.; Siciliano, C.; Tasciotti, J.; Mangino, G.; De Falco, E.; Carnevale, R.; Sciarretta, S.; Frati, G.; Chimenti, I. Beta2-adrenergic signaling affects the phenotype of human cardiac progenitor cells through EMT modulation. Pharmacol. Res. 2018, 127, 41–48. [Google Scholar] [CrossRef]
- Van Tits, L.J.; Michel, M.C.; Grosse-Wilde, H.; Happel, M.; Eigler, F.W.; Soliman, A.; Brodde, O.E. Catecholamines increase lymphocyte beta 2-adrenergic receptors via a beta 2-adrenergic, spleen-dependent process. Am. J. Physiol. 1990, 258 Pt 1, E191–E202. [Google Scholar] [CrossRef]
- Murray, D.R.; Irwin, M.; Rearden, C.A.; Ziegler, M.; Motulsky, H.; Maisel, A.S. Sympathetic and immune interactions during dynamic exercise. Mediation via a beta 2-adrenergic-dependent mechanism. Circulation 1992, 86, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Higashikuni, Y.; Liu, W.; Numata, G.; Tanaka, K.; Fukuda, D.; Tanaka, Y.; Hirata, Y.; Imamura, T.; Takimoto, E.; Komuro, I.; et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation 2023, 147, 338–355. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; de Lucia, C.; Thomas, T.P.; Stark, A.; Strony, J.T.; Myers, V.D.; Beretta, R.; Yu, D.; Sardu, C.; Marfella, R.; et al. Prior beta blocker treatment decreases leukocyte responsiveness to injury. JCI Insight 2019, 5, e99485. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Gumpert, A.M.; Traynham, C.J.; Gorsky, J.E.; Repas, A.A.; Gao, E.; Carter, R.L.; Yu, D.; Calvert, J.W.; García, A.P.; et al. Leukocyte-Expressed β2-Adrenergic Receptors Are Essential for Survival After Acute Myocardial Injury. Circulation 2016, 134, 153–167. [Google Scholar] [CrossRef]
- Stanley, D.; Mason, L.J.; Mackin, K.E.; Srikhanta, Y.N.; Lyras, D.; Prakash, M.D.; Nurgali, K.; Venegas, A.; Hill, M.D.; Moore, R.J.; et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat. Med. 2016, 22, 1277–1284. [Google Scholar] [CrossRef]
- Chimenti, I.; Pagano, F.; Cavarretta, E.; Angelini, F.; Peruzzi, M.; Barretta, A.; Greco, E.; De Falco, E.; Marullo, A.G.; Sciarretta, S.; et al. Β-blockers treatment of cardiac surgery patients enhances isolation and improves phenotype of cardiosphere-derived cells. Sci. Rep. 2016, 6, 36774. [Google Scholar] [CrossRef]
- Wong, C.H.Y.; Jenne, C.N.; Lee, W.-Y.; Léger, C.; Kubes, P. Functional Innervation of Hepatic iNKT Cells Is Immunosuppressive Following Stroke. Science 2011, 334, 101–105. [Google Scholar] [CrossRef]
- Ozdemir, E. Adrenergic receptor system as a pharmacological target in the treatment of epilepsy (Review). Med. Int. 2024, 4, 20. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ADRA1A Adrenoceptor Alpha 1A [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/148#:~:text=Alpha%2D1%2Dadrenergic%20receptors%20(,provided%20by%20RefSeq%2C%20Jul%202008%5D (accessed on 1 December 2024).
- Fan, N.W.; Yu, M.; Wang, S.; Blanco, T.; Luznik, Z.; Chauhan, S.K.; Viswanath, V.; Gil, D.; Held, K.; Chen, Y.; et al. Activation of α2B/2C adrenergic receptor ameliorates ocular surface inflammation through enhancing regulatory T cell function. Mucosal Immunol. 2024, 18, 176–187. [Google Scholar] [CrossRef]
- Durand, M.; Hagimont, E.; Louis, H.; Asfar, P.; Frippiat, J.P.; Singer, M.; Gauchotte, G.; Labat, C.; Lacolley, P.; Levy, B.; et al. The β1-Adrenergic Receptor Contributes to Sepsis-Induced Immunosuppression Through Modulation of Regulatory T-Cell Inhibitory Function. Crit. Care Med. 2022, 50, e707–e718. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, G.G.; Takagi, J.; Fujiwara, Y.; Sano, T.; Note, H. Catecholamines Attenuate LPS-Induced Inflammation through β2 Adrenergic Receptor Activation- and PKA Phosphorylation-Mediated TLR4 Downregulation in Macrophages. Curr. Issues Mol. Biol. 2024, 46, 11336–11348. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.M. α1-Adrenergic Receptors in Neurotransmission, Synaptic Plasticity, and Cognition. Front. Pharmacol. 2020, 11, 581098. [Google Scholar] [CrossRef]
- Umemura, S.; Smyth, D.D.; Pettinger, W.A. Alpha 2-adrenoceptor stimulation and cellular cAMP levels in microdissected rat glomeruli. Am. J. Physiol. 1986, 250 Pt 2, F103–F108. [Google Scholar] [CrossRef]
- Szabo, B. Presynaptic Adrenoceptors. Handb. Exp. Pharmacol. 2024, 285, 185–245. [Google Scholar]
- Avet, C.; Mancini, A.; Breton, B.; Le Gouill, C.; Hauser, A.S.; Normand, C.; Kobayashi, H.; Gross, F.; Hogue, M.; Lukasheva, V.; et al. Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs. eLife 2022, 11, e74101. [Google Scholar] [CrossRef]
- Liao, J.K.; Homey, C.J. The release of endothelium-derived relaxing factor via alpha 2-adrenergic receptor activation is specifically mediated by Gi alpha 2. J. Biol. Chem. 1993, 268, 19528–19533. [Google Scholar] [CrossRef]
- Chao, M.L.; Luo, S.; Zhang, C.; Zhou, X.; Zhou, M.; Wang, J.; Kong, C.; Chen, J.; Lin, Z.; Tang, X.; et al. S-nitrosylation-mediated coupling of G-protein alpha-2 with CXCR5 induces Hippo/YAP-dependent diabetes-accelerated atherosclerosis. Nat. Commun. 2021, 12, 4452. [Google Scholar] [CrossRef]
- Guo, R.; Liu, T.; Shasaltaneh, M.D.; Wang, X.; Imani, S.; Wen, Q. Targeting Adenylate Cyclase Family: New Concept of Targeted Cancer Therapy. Front. Oncol. 2022, 12, 829212. [Google Scholar] [CrossRef]
- Ostrom, K.F.; LaVigne, J.E.; Brust, T.F.; Seifert, R.; Dessauer, C.W.; Watts, V.J.; Ostrom, R.S. Physiological roles of mammalian transmembrane adenylyl cyclase isoforms. Physiol. Rev. 2022, 102, 815–857. [Google Scholar] [CrossRef]
- Taylor, S.S.; Stafford, P.H. Characterization of adenosine 3′:5′-monophosphate-dependent protein kinase and its dissociated subunits from porcine skeletal muscle. J. Biol. Chem. 1978, 253, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.; Mahavadi, S.; Huang, J.; Zhou, H.; Sriwai, W. Phosphorylation of GRK2 by PKA augments GRK2-mediated phosphorylation, internalization, and desensitization of VPAC2 receptors in smooth muscle. Am. J. Physiol. Cell Physiol. 2008, 294, C477–C487. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G Protein–Coupled Receptor Kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef]
- Zhai, R.; Snyder, J.; Montgomery, S.; Sato, P.Y. Double life: How GRK2 and β-arrestin signaling participate in diseases. Cell. Signal. 2022, 94, 110333. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Z.; Yang, M.; Ding, Y.; Zhao, M.; Wu, H.; Zhang, Y.; Lu, Q. The Roles of Orphan G Protein-Coupled Receptors in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2021, 60, 220–243. [Google Scholar] [CrossRef]
- Luttrell, L.M. Composition and function of G protein-coupled receptor signalsomes controlling mitogen-activated protein kinase activity. J. Mol. Neurosci. 2005, 26, 253–264. [Google Scholar] [CrossRef]
- Bjørgo, E.; Solheim, S.A.; Abrahamsen, H.; Baillie, G.S.; Brown, K.M.; Berge, T.; Okkenhaug, K.; Houslay, M.D.; Taskén, K. Cross talk between phosphatidylinositol 3-kinase and cyclic AMP (cAMP)-protein kinase A signaling pathways at the level of a protein kinase B/β-arrestin/cAMP phosphodiesterase 4 complex. Mol. Cell. Biol. 2010, 30, 1660–1672. [Google Scholar] [CrossRef]
- Rich, T.C.; Leavesley, S.J.; Brandon, A.P.; Evans, C.A.; Raju, S.V.; Wagener, B.M. Phosphodiesterase 4 mediates interleukin-8-induced heterologous desensitization of the β2 -adrenergic receptor. FASEB J. 2021, 35, e21946. [Google Scholar] [CrossRef]
- Icahn School of Medicine at Mount Sinai. Catecholamine Blood Test. Available online: https://www.mountsinai.org/health-library/tests/catecholamine-blood-test (accessed on 1 December 2024).
- Izzo, J., Jr. Cardiovascular hormonal effects of circulating norepinephrine. Hypertension 1983, 5, 787–789. [Google Scholar] [CrossRef]
- Bote, M.E.; Garcia, J.J.; Hinchado, M.D.; Ortega, E. Fibromyalgia: Anti-inflammatory and stress responses after acute moderate exercise. PLoS ONE 2013, 8, e74524. [Google Scholar] [CrossRef] [PubMed]
- Djurhuus, S.S.; Schauer, T.; Simonsen, C.; Toft, B.G.; Jensen, A.R.D.; Erler, J.T.; Røder, M.A.; Hojman, P.; Brasso, K.; Christensen, J.F. Effects of acute exercise training on tumor outcomes in men with localized prostate cancer: A randomized controlled trial. Physiol. Rep. 2022, 10, e15408. [Google Scholar] [CrossRef] [PubMed]
- Felten, D.L.; Felten, S.Y.; Bellinger, D.L.; Carlson, S.L.; Ackerman, K.D.; Madden, K.S.; Olschowki, J.A.; Livnat, S. Noradrenergic sympathetic neural interactions with the immune system: Structure and function. Immunol. Rev. 1987, 100, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Qureshi, M.A.; McCorkle, F.M. Profile of chicken macrophage functions after exposure to catecholamines in vitro. Immunopharmacol. Immunotoxicol. 1994, 16, 611–625. [Google Scholar] [CrossRef]
- Samuels, A.J.; Hecht, H.H.; Tyler, F.; Carlisle, R. Leukocyte changes following intra-muscular injection of epinephrine and epinephrine congeners, with observations on the alterations induced by adrenergic blocking agents. Am. J. Med. 1950, 8, 533–534. [Google Scholar] [CrossRef]
- Schedlowski, M.; Falk, A.; Rohne, A.; Wagner, T.O.; Jacobs, R.; Tewes, U.; Schmidt, R.E. Catecholamines induce alterations of distribution and activity of human natural killer (NK) cells. J. Clin. Immunol. 1993, 13, 344–351. [Google Scholar] [CrossRef]
- Gader, A.M.; Cash, J.D. The effect of adrenaline, noradrenaline, isoprenaline and salbutamol on the resting levels of white blood cells in man. Scand. J. Haematol. 1975, 14, 5–10. [Google Scholar] [CrossRef]
- Samuels, A.J. Primary and secondary leucocyte changes following the intramuscular injection of epinephrine hydrochloride. J. Clin. Investig. 1951, 30, 941–947. [Google Scholar] [CrossRef]
- Schedlowski, M.; Hosch, W.; Oberbeck, R.; Benschop, R.J.; Jacobs, R.; Raab, H.R.; Schmidt, R.E. Catecholamines modulate human NK cell circulation and function via spleen-independent beta 2-adrenergic mechanisms. J. Immunol. 1996, 156, 93–99. [Google Scholar] [CrossRef]
- Benschop, R.J.; Jacobs, R.; Sommer, B.; Schürmeyer, T.H.; Raab, J.R.; Schmidt, R.E.; Schedlowski, M. Modulation of the immunologic response to acute stress in humans by beta-blockade or benzodiazepines. FASEB J. 1996, 10, 517–524. [Google Scholar] [CrossRef]
- Ernström, U.; Sandberg, G. Effects of adrenergic alpha- and beta-receptor stimulation on the release of lymphocytes and granulocytes from the spleen. Scand. J. Haematol. 1973, 11, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Mayer, M.; Kreutz, M.; Leeb, S.; Schölmerich, J.; Falk, W. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J. Leukoc. Biol. 2000, 67, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhou, J.Y.; Zhong, H.J.; Wang, H.Y.; Yan, J.; Liu, Q.; Huang, S.N.; Jiang, J.X. Exogenous norepinephrine correlates with macrophage endoplasmic reticulum stress response in association with XBP-1. J. Surg. Res. 2011, 168, 262–271. [Google Scholar] [CrossRef]
- Spiegel, A.; Shivtiel, S.; Kalinkovich, A.; Ludin, A.; Netzer, N.; Goichberg, P.; Azaria, Y.; Resnick, I.; Hardan, I.; Ben-Hur, H.; et al. Catecholaminergic neurotransmitters regulate migration and repopulation of immature human CD34+ cells through Wnt signaling. Nat. Immunol. 2007, 8, 1123–1131. [Google Scholar] [CrossRef]
- Volpe, S.; Thelen, S.; Pertel, T.; Lohse, M.J.; Thelen, M. Polarization of migrating monocytic cells is independent of PI 3-kinase activity. PLoS ONE 2010, 5, e10159. [Google Scholar] [CrossRef]
- García, J.J.; del Carmen Sáez, M.; De la Fuente, M.; Ortega, E. Regulation of phagocytic process of macrophages by noradrenaline and its end metabolite 4-hydroxy-3-metoxyphenyl-glycol. Role of alpha- and beta-adrenoreceptors. Mol. Cell. Biochem. 2003, 254, 299–304. [Google Scholar] [CrossRef]
- Ortega, E.; García, J.J.; De la Fuente, M. Modulation of adherence and chemotaxis of macrophages by norepinephrine. Influence of ageing. Mol. Cell. Biochem. 2000, 203, 113–117. [Google Scholar] [CrossRef]
- Giraldo, E.; Hinchado, M.D.; Ortega, E. Combined activity of post-exercise concentrations of NA and eHsp72 on human neutrophil function: Role of cAMP. J. Cell. Physiol. 2013, 228, 1902–1906. [Google Scholar] [CrossRef]
- Hill, H.R.; Estensen, R.D.; Quie, P.G.; Hogan, N.A.; Goldberg, N.D. Modulation of human neutrophil chemotactic responses by cyclic 3′,5′-guanosine monophosphate and cyclic 3′,5′-adenosine monophosphate. Metabolism 1975, 24, 447–456. [Google Scholar] [CrossRef]
- Hinchado, M.D.; Giraldo, E.; Ortega, E. Adrenoreceptors are involved in the stimulation of neutrophils by exercise-induced circulating concentrations of Hsp72: cAMP as a potential “intracellular danger signal”. J. Cell. Physiol. 2012, 227, 604–608. [Google Scholar] [CrossRef]
- Deitch, E.A.; Bridges, R.M. Stress hormones modulate neutrophil and lymphocyte activity in vitro. J. Trauma 1987, 27, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Salazar, C.; Bou-Puerto, R.; Mujal, A.M.; Lau, C.M.; von Hoesslin, M.; Zehn, D.; Sun, J.C. Cell-intrinsic adrenergic signaling controls the adaptive NK cell response to viral infection. J. Exp. Med. 2020, 217, e20190549. [Google Scholar] [CrossRef] [PubMed]
- Basílio-Queirós, D.; Mischak-Weissinger, E. Natural killer cells- from innate cells to the discovery of adaptability. Front. Immunol. 2023, 14, 1172437. [Google Scholar] [CrossRef]
- Bosch, J.A.; Berntson, G.G.; Cacioppo, J.T.; Marucha, P.T. Differential mobilization of functionally distinct natural killer subsets during acute psychologic stress. Psychosom. Med. 2005, 67, 366–375. [Google Scholar] [CrossRef]
- Tang, Y.; Shankar, R.; Gamboa, M.; Desai, S.; Gamelli, R.L.; Jones, S.B. Norepinephrine modulates myelopoiesis after experimental thermal injury with sepsis. Ann. Surg. 2001, 233, 266–275. [Google Scholar] [CrossRef]
- Cohen, M.J.; Shankar, R.; Stevenson, J.; Fernandez, R.; Gamelli, R.L.; Jones, S.B. Bone marrow norepinephrine mediates development of functionally different macrophages after thermal injury and sepsis. Ann. Surg. 2004, 240, 132–141. [Google Scholar] [CrossRef]
- Powell, N.D.; Sloan, E.K.; Bailey, M.T.; Arevalo, J.M.; Miller, G.E.; Chen, E.; Kobor, M.S.; Reader, B.F.; Sheridan, J.F.; Cole, S.W. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16574–16579. [Google Scholar] [CrossRef]
- Shirvaikar, N.; Marquez-Curtis, L.A.; Shaw, A.R.; Turner, A.R.; Janowska-Wieczorek, A. MT1-MMP association with membrane lipid rafts facilitates G-CSF−induced hematopoietic stem/progenitor cell mobilization. Exp. Hematol. 2010, 38, 823–835. [Google Scholar] [CrossRef]
- Suzuki, K.; Hayano, Y.; Nakai, A.; Furuta, F.; Noda, M. Adrenergic control of the adaptive immune response by diurnal lymphocyte recirculation through lymph nodes. J. Exp. Med. 2016, 213, 2567–2574. [Google Scholar] [CrossRef]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.B.; Springer, T.A. Leukocytes roll on a selectin at physiologic flow rates: Distinction from and prerequisite for adhesion through integrins. Cell 1991, 65, 859–873. [Google Scholar] [CrossRef] [PubMed]
- von Andrian, U.H.; Chambers, J.D.; McEvoy, L.M.; Bargatze, R.F.; Arfors, K.E.; Butcher, E.C. Two-step model of leukocyte-endothelial cell interaction in inflammation: Distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 7538–7542. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Montresor, A.; Toffali, L.; Constantin, G.; Laudanna, C. Chemokines and the signaling modules regulating integrin affinity. Front. Immunol. 2012, 3, 127. [Google Scholar] [CrossRef]
- Kim, S.H.J.; Hammer, D.A. Integrin cross-talk modulates stiffness-independent motility of CD4+ T lymphocytes. Mol. Biol. Cell 2021, 32, 1749–1757. [Google Scholar] [CrossRef]
- Hinterdobler, J.; Schott, S.; Jin, H.; Meesmann, A.; Steinsiek, A.L.; Zimmermann, A.S.; Wobst, J.; Müller, P.; Mauersberger, C.; Vilne, B.; et al. Acute mental stress drives vascular inflammation and promotes plaque destabilization in mouse atherosclerosis. Eur. Heart J. 2021, 42, 4077–4088. [Google Scholar] [CrossRef]
- Wang, Y.; Magliano, D.J. Special Issue: “New Trends in Diabetes, Hypertension, and Cardiovascular Diseases”. Int. J. Mol. Sci. 2024, 25, 2711. [Google Scholar] [CrossRef]
- Wang, Y.; Panicker, I.S.; Anesi, J.; Sargisson, O.; Atchison, B.; Habenicht, A.J.R. Animal Models, Pathogenesis, and Potential Treatment of Thoracic Aortic Aneurysm. Int. J. Mol. Sci. 2024, 25, 901. [Google Scholar] [CrossRef]
- Wang, Y.; Emeto, T.I.; Lee, J.; Marshman, L.; Moran, C.; Seto, S.W.; Golledge, J. Mouse models of intracranial aneurysm. Brain Pathol. 2015, 25, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Omega-3 Fatty Acids Effect on Major Cardiovascular Events in Patients at High Cardiovascular Risk. JAMA 2021, 325, 1333. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; Witting, P.K.; Charchar, F.J.; Sobey, C.G.; Drummond, G.R.; Golledge, J. Dietary fatty acids and mortality risk from heart disease in US adults: An analysis based on NHANES. Sci. Rep. 2023, 13, 1614. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.S.; Dooley, E.E.; Master, H.; Spartano, N.L.; Brittain, E.L.; Pettee Gabriel, K. Physical Activity over the Lifecourse and Cardiovascular Disease. Circ. Res. 2023, 132, 1725–1740. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Ning, H.; Sinha, A.; Wilkins, J.; Allen, N.B.; Vu, T.H.T.; Berry, J.D.; Lloyd-Jones, D.M.; Sweis, R. Cigarette Smoking and Competing Risks for Fatal and Nonfatal Cardiovascular Disease Subtypes Across the Life Course. J. Am. Heart. Assoc. 2021, 10, e021751. [Google Scholar] [CrossRef]
- Biddinger, K.J.; Emdin, C.A.; Haas, M.E.; Wang, M.; Hindy, G.; Ellinor, P.T.; Kathiresan, S.; Khera, A.V.; Aragam, K.G. Association of Habitual Alcohol Intake with Risk of Cardiovascular Disease. JAMA Netw. Open 2022, 5, e223849. [Google Scholar] [CrossRef]
- Yang, G.; Qian, T.; Sun, H.; Xu, Q.; Hou, X.; Hu, W.; Zhang, G.; Drummond, G.R.; Sobey, C.G.; Witting, P.K.; et al. Adjustment for body mass index changes inverse associations of HDL-cholesterol with blood pressure and hypertension to positive associations. J. Hum. Hypertens. 2022, 36, 570–579. [Google Scholar] [CrossRef]
- Qian, T.; Sun, H.; Xu, Q.; Hou, X.; Hu, W.; Zhang, G.; Drummond, G.R.; Sobey, C.G.; Charchar, F.J.; Golledge, J.; et al. Hyperuricemia is independently associated with hypertension in men under 60 years in a general Chinese population. J. Hum. Hypertens. 2021, 35, 1020–1028. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; Magliano, D.J.; Charchar, F.J.; Sobey, C.G.; Drummond, G.R.; Golledge, J. Fasting triglycerides are positively associated with cardiovascular mortality risk in people with diabetes. Cardiovasc. Res. 2023, 119, 826–834. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; Aberson, C.L.; Charchar, F.J.; Ceriello, A. Postprandial Plasma Glucose between 4 and 7.9 h May Be a Potential Diagnostic Marker for Diabetes. Biomedicines 2024, 12, 1313. [Google Scholar] [CrossRef]
- Yang, G.; Qian, T.; Sun, H.; Xu, Q.; Hou, X.; Hu, W.; Zhang, G.; Fang, Y.; Song, D.; Chai, Z.; et al. Both low and high levels of low-density lipoprotein cholesterol are risk factors for diabetes diagnosis in Chinese adults. Diabetes Epidemiol. Manag. 2022, 6, 100050. [Google Scholar] [CrossRef]
- Wang, Y. Higher fasting triglyceride predicts higher risks of diabetes mortality in US adults. Lipids Health Dis. 2021, 20, 181. [Google Scholar] [CrossRef] [PubMed]
- Judkins, C.P.; Wang, Y.; Jelinic, M.; Bobik, A.; Vinh, A.; Sobey, C.G.; Drummond, G.R. Association of constipation with increased risk of hypertension and cardiovascular events in elderly Australian patients. Sci. Rep. 2023, 13, 10943. [Google Scholar] [CrossRef] [PubMed]
- Willerson, J.T.; Ridker, P.M. Inflammation as a Cardiovascular Risk Factor. Circulation 2004, 109 (Suppl. 1), II-2–II-10. [Google Scholar] [CrossRef]
- Wang, Y.; Dinh, T.N.; Nield, A.; Krishna, S.M.; Denton, K.; Golledge, J. Renal Denervation Promotes Atherosclerosis in Hypertensive Apolipoprotein E-Deficient Mice Infused with Angiotensin II. Front. Physiol. 2017, 8, 215. [Google Scholar] [CrossRef]
- Arnold, N.; Lechner, K.; Waldeyer, C.; Shapiro, M.D.; Koenig, W. Inflammation and Cardiovascular Disease: The Future. Eur. Cardiol. 2021, 16, e20. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, D.T.; Anesi, J.; Alramahi, A.; Witting, P.K.; Chai, Z.; Khan, A.W.; Kelly, J.; Denton, K.M.; Golledge, J. Moxonidine Increases Uptake of Oxidised Low-Density Lipoprotein in Cultured Vascular Smooth Muscle Cells and Inhibits Atherosclerosis in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2023, 24, 3857. [Google Scholar] [CrossRef]
- Wang, Y.; Tikellis, C.; Thomas, M.C.; Golledge, J. Angiotensin converting enzyme 2 and atherosclerosis. Atherosclerosis 2013, 226, 3–8. [Google Scholar] [CrossRef]
- Gräbner, R.; Lötzer, K.; Döpping, S.; Hildner, M.; Radke, D.; Beer, M.; Spanbroek, R.; Lippert, B.; Reardon, C.A.; Getz, G.S.; et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J. Exp. Med. 2009, 206, 233–248. [Google Scholar] [CrossRef]
- Moos, M.P.; John, N.; Gräbner, R.; Nossmann, S.; Günther, B.; Vollandt, R.; Funk, C.D.; Kaiser, B.; Habenicht, A.J. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2386–2391. [Google Scholar] [CrossRef]
- Wang, Y.; Lim, K.; Denton, K.M. Editorial: Function of Renal Sympathetic Nerves. Front. Physiol. 2017, 8, 642. [Google Scholar] [CrossRef] [PubMed]
- Akhavanpoor, M.; Gleissner, C.A.; Akhavanpoor, H.; Lasitschka, F.; Doesch, A.O.; Katus, H.A.; Erbel, C. Adventitial tertiary lymphoid organ classification in human atherosclerosis. Cardiovasc. Pathol. 2018, 32, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sargisson, O.; Nguyen, D.T.; Parker, K.; Pyke, S.J.R.; Alramahi, A.; Thihlum, L.; Fang, Y.; Wallace, M.E.; Berzins, S.P.; et al. Effect of Hydralazine on Angiotensin II-Induced Abdominal Aortic Aneurysm in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2023, 24, 15955. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.P.; Hahn, M.; Changsirivathanathamrong, D.; Wu, B.J.; Ball, H.J.; Thomas, S.R.; et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285. [Google Scholar] [CrossRef]
- Wang, Y. Stage 1 hypertension and risk of cardiovascular disease mortality in United States adults with or without diabetes. J. Hypertens. 2022, 40, 794–803. [Google Scholar] [CrossRef]
- Zhang, R.M.; McNerney, K.P.; Riek, A.E.; Bernal-Mizrachi, C. Immunity and Hypertension. Acta Physiol. 2021, 231, e13487. [Google Scholar] [CrossRef]
- Zhang, J.; Crowley, S.D. Role of T lymphocytes in hypertension. Curr. Opin. Pharmacol. 2015, 21, 14–19. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Ahmari, N.; Hayward, L.F.; Zubcevic, J. The importance of bone marrow and the immune system in driving increases in blood pressure and sympathetic nerve activity in hypertension. Exp. Physiol. 2020, 105, 1815–1826. [Google Scholar] [CrossRef]
- Michell, D.L.; Shihata, W.A.; Andrews, K.L.; Abidin, N.A.Z.; Jefferis, A.M.; Sampson, A.K.; Lumsden, N.G.; Huet, O.; Parat, M.O.; Jennings, G.L.; et al. High intraluminal pressure promotes vascular inflammation via caveolin-1. Sci. Rep. 2021, 11, 5894. [Google Scholar] [CrossRef]
- Banek, C.T.; Knuepfer, M.M.; Foss, J.D.; Fiege, J.K.; Asirvatham-Jeyaraj, N.; Van Helden, D.; Shimizu, Y.; Osborn, J.W. Resting Afferent Renal Nerve Discharge and Renal Inflammation: Elucidating the Role of Afferent and Efferent Renal Nerves in Deoxycorticosterone Acetate Salt Hypertension. Hypertension 2016, 68, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Ahmari, N.; Santisteban, M.M.; Miller, D.R.; Geis, N.M.; Larkin, R.; Redler, T.; Denson, H.; Khoshbouei, H.; Baekey, D.M.; Raizada, M.K.; et al. Elevated bone marrow sympathetic drive precedes systemic inflammation in angiotensin II hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H279–H289. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Qian, T.; Sun, H.; Xu, Q.; Hou, X.; Hu, W.; Zhang, G.; Drummond, G.R.; Sobey, C.G.; et al. Reduced renal function may explain the higher prevalence of hyperuricemia in older people. Sci. Rep. 2021, 11, 1302. [Google Scholar] [CrossRef]
- Wang, Y.; Seto, S.W.; Golledge, J. Therapeutic effects of renal denervation on renal failure. Curr. Neurovasc. Res. 2013, 10, 172–184. [Google Scholar] [CrossRef]
- Raikwar, N.; Braverman, C.; Snyder, P.M.; Fenton, R.A.; Meyerholz, D.K.; Abboud, F.M.; Harwani, S.C. Renal denervation and CD161a immune ablation prevent cholinergic hypertension and renal sodium retention. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H517–H530. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Ahmari, N.; Carvajal, J.M.; Zingler, M.B.; Qi, Y.; Kim, S.; Joseph, J.; Garcia-Pereira, F.; Johnson, R.D.; Shenoy, V.; et al. Involvement of bone marrow cells and neuroinflammation in hypertension. Circ. Res. 2015, 117, 178–191. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, W.; Lu, C.; Ma, Z.; Jiang, S.; Gu, C.; Acuña-Castroviejo, D.; Yang, Y. Targeting NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome in Cardiovascular Disorders. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2765–2779. [Google Scholar] [CrossRef]
- Pasqua, T.; Pagliaro, P.; Rocca, C.; Angelone, T.; Penna, C. Role of NLRP-3 Inflammasome in Hypertension: A Potential Therapeutic Target. Curr. Pharm. Biotechnol. 2018, 19, 708–714. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP 3 Inflammasome in Atherosclerosis. J. Am. Heart. Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef]
- Hohensinner, P.J.; Kaun, C.; Rychli, K.; Ben-Tal Cohen, E.; Kastl, S.P.; Demyanets, S.; Pfaffenberger, S.; Speidl, W.S.; Rega, G.; Ullrich, R.; et al. Monocyte chemoattractant protein (MCP-1) is expressed in human cardiac cells and is differentially regulated by inflammatory mediators and hypoxia. FEBS Lett. 2006, 580, 3532–3538. [Google Scholar] [CrossRef]
- Zhang, M.; Alemasi, A.; Zhao, M.; Xu, W.; Zhang, Y.; Gao, W.; Yu, H.; Xiao, H. Exercise Training Attenuates Acute β-Adrenergic Receptor Activation-Induced Cardiac Inflammation via the Activation of AMP-Activated Protein Kinase. Int. J. Mol. Sci. 2023, 24, 9263. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Zhong, B.; Luo, Q.; Liu, Q.; Chen, X.; Cao, D.; Li, X.; Yang, S. Phillyrin attenuates norepinephrine-induced cardiac hypertrophy and inflammatory response by suppressing p38/ERK1/2 MAPK and AKT/NF-kappaB pathways. Eur. J. Pharmacol. 2022, 927, 175022. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.K.; Xiao, L.; Pimental, D.R.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Reactive Oxygen Species Mediate Alpha-adrenergic Receptor-stimulated Hypertrophy in Adult Rat Ventricular Myocytes. J. Mol. Cell. Cardiol. 2001, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Inflammation in cardiac injury, repair and regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2020, 117, 1450–1488. [Google Scholar] [CrossRef]
- Xiao, Z.; Kong, B.; Yang, H.; Dai, C.; Fang, J.; Qin, T.; Huang, H. Key Player in Cardiac Hypertrophy, Emphasizing the Role of Toll-like Receptor 4. Front. Cardiovasc. Med. 2020, 7, 579036. [Google Scholar] [CrossRef]
- Castoldi, G.; Carletti, R.; Ippolito, S.; Colzani, M.; Pelucchi, S.; Zerbini, G.; Perseghin, G.; Zatti, G.; di Gioia, C.R.T. Cardioprotective Effects of Sodium Glucose Cotransporter 2 Inhibition in Angiotensin II-Dependent Hypertension Are Mediated by the Local Reduction of Sympathetic Activity and Inflammation. Int. J. Mol. Sci. 2023, 24, 10710. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Chen, M.; Li, X.; Wang, S.; Yu, L.; Tang, J.; Zhou, S. The Role of Cardiac Macrophage and Cytokines on Ventricular Arrhythmias. Front. Physiol. 2020, 11, 1113. [Google Scholar] [CrossRef]
- Lombardi, F.; Sandrone, G.; Spinnler, M.T.; Torzillo, D.; Lavezzaro, G.C.; Brusca, A.; Malliani, A. Heart rate variability in the early hours of an acute myocardial infarction. Am. J. Cardiol. 1996, 77, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G. Sympathetic activation and prognosis in cardiovascular disease. Eur. Soc. Cardiol. 2006, 5, 11–28. [Google Scholar]
- Wei, Y.Z.; Yang, S.; Li, W.; Tang, Y.H. Gefapixant, a Novel P2X3 Antagonist, Protects against Post Myocardial Infarction Cardiac Dysfunction and Remodeling Via Suppressing NLRP3 Inflammasome. Curr. Med. Sci. 2023, 43, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xiong, J.; Zou, Q.; Wang, X.; Hu, K.; Zhao, Q. Sinapic Acid Attenuated Cardiac Remodeling After Myocardial Infarction by Promoting Macrophage M2 Polarization Through the PPARγ Pathway. Front. Cardiovasc. Med. 2022, 9, 915903. [Google Scholar] [CrossRef]
- Lyu, J.; Huang, J.; Wu, J.; Yu, T.; Wei, X.; Lei, Q. Lack of Macrophage Migration Inhibitory Factor Reduces Susceptibility to Ventricular Arrhythmias During the Acute Phase of Myocardial Infarction. J. Inflamm. Res. 2021, 14, 1297–1311. [Google Scholar] [CrossRef]
- Chen, H.; Wang, R.; Li, Q.; Yin, J.; Ge, Z.; Xu, F.; Zang, T.; Pei, Z.; Li, C.; Shen, L.; et al. Immediate Renal Denervation After Acute Myocardial Infarction Mitigates the Progression of Heart Failure via the Modulation of IL-33/ST2 Signaling. Front. Cardiovasc. Med. 2021, 8, 746934. [Google Scholar] [CrossRef]
- García-Ruiz, J.M.; Fernández-Jiménez, R.; García-Alvarez, A.; Pizarro, G.; Galán-Arriola, C.; Fernández-Friera, L.; Mateos, A.; Nuno-Ayala, M.; Aguero, J.; Sánchez-González, J.; et al. Impact of the Timing of Metoprolol Administration During STEMI on Infarct Size and Ventricular Function. J. Am. Coll. Cardiol. 2016, 67, 2093–2104. [Google Scholar] [CrossRef]
- Joo, S.J. Beta-blocker therapy in patients with acute myocardial infarction: Not all patients need it. Acute Crit. Care 2023, 38, 251–260. [Google Scholar] [CrossRef]
- Bangalore, S.; Makani, H.; Radford, M.; Thakur, K.; Toklu, B.; Katz, S.D.; DiNicolantonio, J.J.; Devereaux, P.J.; Alexander, K.P.; Wetterslev, J.; et al. Clinical outcomes with β-blockers for myocardial infarction: A meta-analysis of randomized trials. Am. J. Med. 2014, 127, 939–953. [Google Scholar] [CrossRef]
- American Heart Association. Heart Failure. Available online: https://www.heart.org/en/health-topics/heart-failure (accessed on 15 January 2025).
- Malik, A.; Chhabra, L. Congestive Heart Failure. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430873/ (accessed on 15 January 2025).
- Gronda, E.; Dusi, V.; D’Elia, E.; Iacoviello, M.; Benvenuto, E.; Vanoli, E. Sympathetic activation in heart failure. Eur. Heart J. Suppl. 2022, 24 (Suppl. E), E4–E11. [Google Scholar] [CrossRef]
- Swedberg, K.; Hjalmarson, A.; Waagstein, F.; Wallentin, I. Prolongation of survival in congestive cardiomyopathy by beta-receptor blockade. Lancet 1979, 1, 1374–1376. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Schefold, J.C.; Jankowska, E.; Doehner, W.; Springer, J.; Strohschein, K.; Genth-Zotz, S.; Volk, H.D.; Poole-Wilson, P.; Anker, S.D. Leukocyte redistribution: Effects of beta blockers in patients with chronic heart failure. PLoS ONE 2009, 4, e6411. [Google Scholar] [CrossRef]
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 1 December 2024).
- Dorrance, A.M.; Fink, G. Effects of Stroke on the Autonomic Nervous System. Compr. Physiol. 2015, 5, 1241–1263. [Google Scholar]
- Wang, H.; Li, J.; Wu, G.; Lin, X.; Chen, J.; Liang, J.; Zhang, J.; Luo, X.; Mao, H.; Xie, J.; et al. Activated sympathetic nerve post stroke downregulates Toll-like receptor 5 and disrupts the gut mucosal barrier. Cell Rep. Med. 2024, 5, 101754. [Google Scholar] [CrossRef]
- Xu, Y.; Ge, Y.; Zhou, M.; Zhang, Z. Clenbuterol, a Selective β2-Adrenergic Receptor Agonist, Inhibits or Limits Post-Stroke Pneumonia, but Increases Infarct Volume in MCAO Mice. J. Inflamm. Res. 2022, 15, 295–309. [Google Scholar] [CrossRef]
- Myers, M.G.; Norris, J.W.; Hachniski, V.C.; Sole, M.J. Plasma norepinephrine in stroke. Stroke 1981, 12, 200–204. [Google Scholar] [CrossRef]
- Jackman, K.A.; Brait, V.H.; Wang, Y.; Maghzal, G.J.; Ball, H.J.; McKenzie, G.; De Silva, T.M.; Stocker, R.; Sobey, C.G. Vascular expression, activity and function of indoleamine 2,3-dioxygenase-1 following cerebral ischaemia-reperfusion in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 471–481. [Google Scholar] [CrossRef]
- Lindsberg, P.J.; Grau, A.J. Inflammation and Infections as Risk Factors for Ischemic Stroke. Stroke 2003, 34, 2518–2532. [Google Scholar] [CrossRef]
- Kimura, K.; Minematsu, K.; Kazui, S.; Yamaguchi, T. Mortality and cause of death after hospital discharge in 10,981 patients with ischemic stroke and transient ischemic attack. Cerebrovasc. Dis. 2005, 19, 171–178. [Google Scholar] [CrossRef]
- Choi, Y.H.; Laaker, C.; Hsu, M.; Cismaru, P.; Sandor, M.; Fabry, Z. Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke. Int. J. Mol. Sci. 2021, 22, 9486. [Google Scholar] [CrossRef] [PubMed]
- Maier, I.L.; Becker, J.C.; Leyhe, J.R.; Schnieder, M.; Behme, D.; Psychogios, M.-N.; Liman, J. Influence of beta-blocker therapy on the risk of infections and death in patients at high risk for stroke induced immunodepression. PLoS ONE 2018, 13, e0196174. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, T.; Slowik, A.; Pera, J.; Szczudlik, A. Beta-blockers reduce the risk of early death in ischemic stroke. J. Neurol. Sci. 2007, 252, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef]
- Wang, J.; Yu, L.; Jiang, C.; Fu, X.; Liu, X.; Wang, M.; Ou, C.; Cui, X.; Zhou, C.; Wang, J. Cerebral ischemia increases bone marrow CD4+CD25+FoxP3+ regulatory T cells in mice via signals from sympathetic nervous system. Brain Behav. Immun. 2015, 43, 172–183. [Google Scholar] [CrossRef]
- Borlongan, C.V. Concise Review: Stem Cell Therapy for Stroke Patients: Are We There Yet? Stem Cells Transl. Med. 2019, 8, 983–988. [Google Scholar] [CrossRef]
- Borlongan, C.V. Bone marrow stem cell mobilization in stroke: A ‘bonehead’ may be good after all! Leukemia 2011, 25, 1674–1686. [Google Scholar] [CrossRef]
- WHO. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 3 February 2025).
- Wang, Y.; Fang, Y.; Habenicht, A.J.R.; Golledge, J.; Giovannucci, E.L.; Ceriello, A. Postprandial Plasma Glucose with a Fasting Time of 4–7.9 h Is Positively Associated with Cancer Mortality in US Adults. Diabetes Metab. Res. Rev. 2024, 40, e70008. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef]
- Armaiz-Pena, G.N.; Gonzalez-Villasana, V.; Nagaraja, A.S.; Rodriguez-Aguayo, C.; Sadaoui, N.C.; Stone, R.L.; Matsuo, K.; Dalton, H.J.; Previs, R.A.; Jennings, N.B.; et al. Adrenergic regulation of monocyte chemotactic protein 1 leads to enhanced macrophage recruitment and ovarian carcinoma growth. Oncotarget 2015, 6, 4266–4273. [Google Scholar] [CrossRef]
- Torisu, H.; Ono, M.; Kiryu, H.; Furue, M.; Ohmoto, Y.; Nakayama, J.; Nishioka, Y.; Sone, S.; Kuwano, M. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: Possible involvement of TNFalpha and IL-1alpha. Int. J. Cancer 2000, 85, 182–188. [Google Scholar] [CrossRef]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef] [PubMed]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; Liu, H.; Kokolus, K.M.; Eng, J.W.; Messmer, M.N.; et al. β-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8+ T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, I.; Sarantopoulos, A.; Liverezas, A. Does sympathetic nervous system modulate tumor progression? A narrative review of the literature. J. Drug Assess. 2020, 9, 106–116. [Google Scholar] [CrossRef]
- Wrobel, L.J.; Bod, L.; Lengagne, R.; Kato, M.; Prévost-Blondel, A.; Le Gal, F.A. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget 2016, 7, 77825–77837. [Google Scholar] [CrossRef]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.B.; et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Ann. Oncol. 2019, 30, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10. [Google Scholar] [CrossRef]
- Steinberger, K.J.; Bailey, M.T.; Gross, A.C.; Sumner, L.A.; Voorhees, J.L.; Crouser, N.; Curry, J.M.; Wang, Y.; DeVries, A.C.; Marsh, C.B.; et al. Stress-induced Norepinephrine Downregulates CCL2 in Macrophages to Suppress Tumor Growth in a Model of Malignant Melanoma. Cancer Prev. Res. 2020, 13, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, L.J.; Le Gal, F.A. Inhibition of Human Melanoma Growth by a Non-Cardioselective β-Blocker. J. Investig. Dermatol. 2015, 135, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-Y.; Sun, B.-W. Recent advances in neutrophil chemotaxis abnormalities during sepsis. Chin. J. Traumatol. 2022, 25, 317–324. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | α1 AR | α2 AR | β1 AR | β2 AR | β3 AR | Reference |
|---|---|---|---|---|---|---|
| T cells | √ | √ | √ | √ | [42,46,47,48,49,50,51] | |
| CD4+ T cells | √ | √ | √ | [46,47,48,49,50,51] | ||
| CD8+ T cells | √ | √ | √ | √ | [52,53,54,55] | |
| B cells | √ | √ | √ | [42,51,56,57,58,59] | ||
| Macrophages | √ | √ | √ | √ | √ | [25,40,50,52,60,61,62] |
| Monocytes | √ | √ | √ | √ | [41,50,57,60,63] | |
| Dendritic cells | √ | √ | √ | √ | [52,64,65,66] | |
| Neutrophils | √ | √ | √ | √ | √ | [1,41,50,67,68,69] |
| NK cells | √ | √ | √ | [52,53,70,71,72,73] |
| Drugs | α1 AR | α2 AR | β1 AR | β2 AR | Reference |
|---|---|---|---|---|---|
| Agonists | |||||
| Phenylephrine | √ | [74] | |||
| Clonidine | √ | [74] | |||
| Dobutamine | √ | [74] | |||
| Salbutamol | √ | [75] | |||
| Salmeterol | √ | [74] | |||
| Formoterol | √ | [74] | |||
| Clenbuterol | √ | [76] | |||
| Isoproterenol | √ | √ | [74,77,78,79] | ||
| Antagonists | |||||
| Prazosin | √ | [80] | |||
| Phentolamine | √ | [74,81] | |||
| Butoxamine | √ | [82] | |||
| Atenolol | √ | [77] | |||
| Bisoprolol | √ | [83,84,85] | |||
| Metoprolol | √ | [77,86,87,88] | |||
| Nebivolol | √ | [34] | |||
| ICI 118,551 | √ | [83,86,87] | |||
| Nipradilol | √ | √ | [34] | ||
| Propranolol | √ | √ | [74,76,78,81,84,89,90] | ||
| Carvedilol | √ | √ | √ | [34,86] | |
| Dose | Route | Time | Effect | Reference |
|---|---|---|---|---|
| 7 μg/min, for 30 min | iv | During infusion Immediately after infusion | ↑ Lymphocytes | [121] |
| N/R | im | 15 min after injection | ↑ Lymphocytes | [119] |
| 0.15 μg/kg/min, for 20 min | iv | During infusion Immediately after infusion | ↑ CD3+ T cells ↑ CD4+ T cells ↑ CD8+ T cells ↔ CD20+ B cells | [123] |
| 0.15 μg/kg/min, for 20 min | iv | 30 min after infusion | ↔ CD3+ T cells ↔ CD4+ T cells ↔ CD8+ T cells ↔ CD20+ B cells | [123] |
| 7 μg/min, for 30 min | iv | 30 min after infusion | ↔ Lymphocytes | [121] |
| 50 ng/kg/min, for 90 min | iv | 30, 60, and 90 min after injection | ↔ Helper T cells ↔ Cytolytic T cells ↔ B cells | [83] |
| 10 μg/kg | sc | 5, 15, 30, 60, and 120 min after injection | ↔ CD3+ T cells ↔ CD4+ T cells ↔ CD8+ T cells | [120] |
| Cell Source | NE Concentration, Molar | Chemotactic? | Reference |
|---|---|---|---|
| Monocytes, human | 10−9–10−11 | Yes | [126] |
| Macrophages, human | 10−9–10−11 | Yes | [126] |
| Macrophages, mice | 1 × 10−8 and 6 × 10−8 | Yes | [127] |
| Macrophages, mice | 3 × 10−7 and 6 × 10−7 | No | [127] |
| CD34+ stem and progenitor cells, human | 10−8 or 10−6 | Yes | [128] |
| Cell Source | [NE], Molar | Migration Inducer | Effect on Migration | Mechanism | Ref |
|---|---|---|---|---|---|
| Macrophages, mice | 10−12 | fMLP | ↑ | α-ARs | [130] |
| Macrophages, mice aged 12 and 22 w | 10−12 | fMLP | ↑ | N/R | [131] |
| Macrophages, mice | 10−5 | fMLP | ↔ | N/R | [130] |
| Macrophages, mice aged 12 and 22 w | 10−5 | fMLP | ↔ | N/R | [131] |
| Macrophages, mice aged 48 and 72 w | 10−12 | fMLP | ↔ | N/R | [131] |
| Macrophages, mice aged 72 w | 10−5 | fMLP | ↓ | N/R | [131] |
| Macrophages, rats, treated with AlCl3 | 10−9 and 10−8 | fMLP | ↓ | β2-AR/cAMP | [62] |
| Cell Source | NE Concentration | Migration Inducer | Migration Effect | Mechanism | Reference |
|---|---|---|---|---|---|
| Human | 4 × 10−7 M | fMLP | ↑ | N/R | [132] |
| Mice | 10−7–10−5 M | fMLP | ↓ | N/R | [1] |
| Human | 10−5–10−3 M | BCF | ↓ | β AR and cAMP | [133] |
| Human | 10−7 M | Serum | ↓ | N/R | [135] |
| Mice | 10−5 M | fMLP | ↓ in vivo | N/R | [1] |
| Cell Source | NE Concentration | Migration | Mechanism | Ref |
|---|---|---|---|---|
| Activated CD8+ T cells, human | 10−5 M | ↔ | N/R | [54] |
| CD8+ T cells, human | 10−6 M | ↔ | N/R | [72] |
| Naïve CD8+ T cells, human | 10−5 M | ↔ | N/R | [77] |
| Lymphocytes, mice | 10−5 M | ↓ | α AR and β AR | [81] |
| Activated CD8+ T cells, human | 10−5 M | ↓ | N/R | [55] |
| CD8+ T cells, human | Medium from 10−5 M NE-treated TCs | ↓ | β2 AR, ↓ CXCL9 secretion by TCs | [77] |
| CD8+ T cells, tumor-bearing mice | 2 mg/mg/2 days, i.p., 7 times | ↓ | ↓ CXCL9 secretion by tumor cells, ↓ CD8+ T cell infiltration | [77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Anesi, J.C.; Panicker, I.S.; Cook, D.; Bista, P.; Fang, Y.; Oqueli, E. Neuroimmune Interactions and Their Role in Immune Cell Trafficking in Cardiovascular Diseases and Cancer. Int. J. Mol. Sci. 2025, 26, 2553. https://doi.org/10.3390/ijms26062553
Wang Y, Anesi JC, Panicker IS, Cook D, Bista P, Fang Y, Oqueli E. Neuroimmune Interactions and Their Role in Immune Cell Trafficking in Cardiovascular Diseases and Cancer. International Journal of Molecular Sciences. 2025; 26(6):2553. https://doi.org/10.3390/ijms26062553
Chicago/Turabian StyleWang, Yutang, Jack C. Anesi, Indu S. Panicker, Darcy Cook, Prapti Bista, Yan Fang, and Ernesto Oqueli. 2025. "Neuroimmune Interactions and Their Role in Immune Cell Trafficking in Cardiovascular Diseases and Cancer" International Journal of Molecular Sciences 26, no. 6: 2553. https://doi.org/10.3390/ijms26062553
APA StyleWang, Y., Anesi, J. C., Panicker, I. S., Cook, D., Bista, P., Fang, Y., & Oqueli, E. (2025). Neuroimmune Interactions and Their Role in Immune Cell Trafficking in Cardiovascular Diseases and Cancer. International Journal of Molecular Sciences, 26(6), 2553. https://doi.org/10.3390/ijms26062553