
Cancer Drug Sensitivity Prediction Based on Deep Transfer Learning


Abstract
:1. Introduction
2. Results
2.1. Performance Comparison with Other Algorithms
2.2. Blind Test
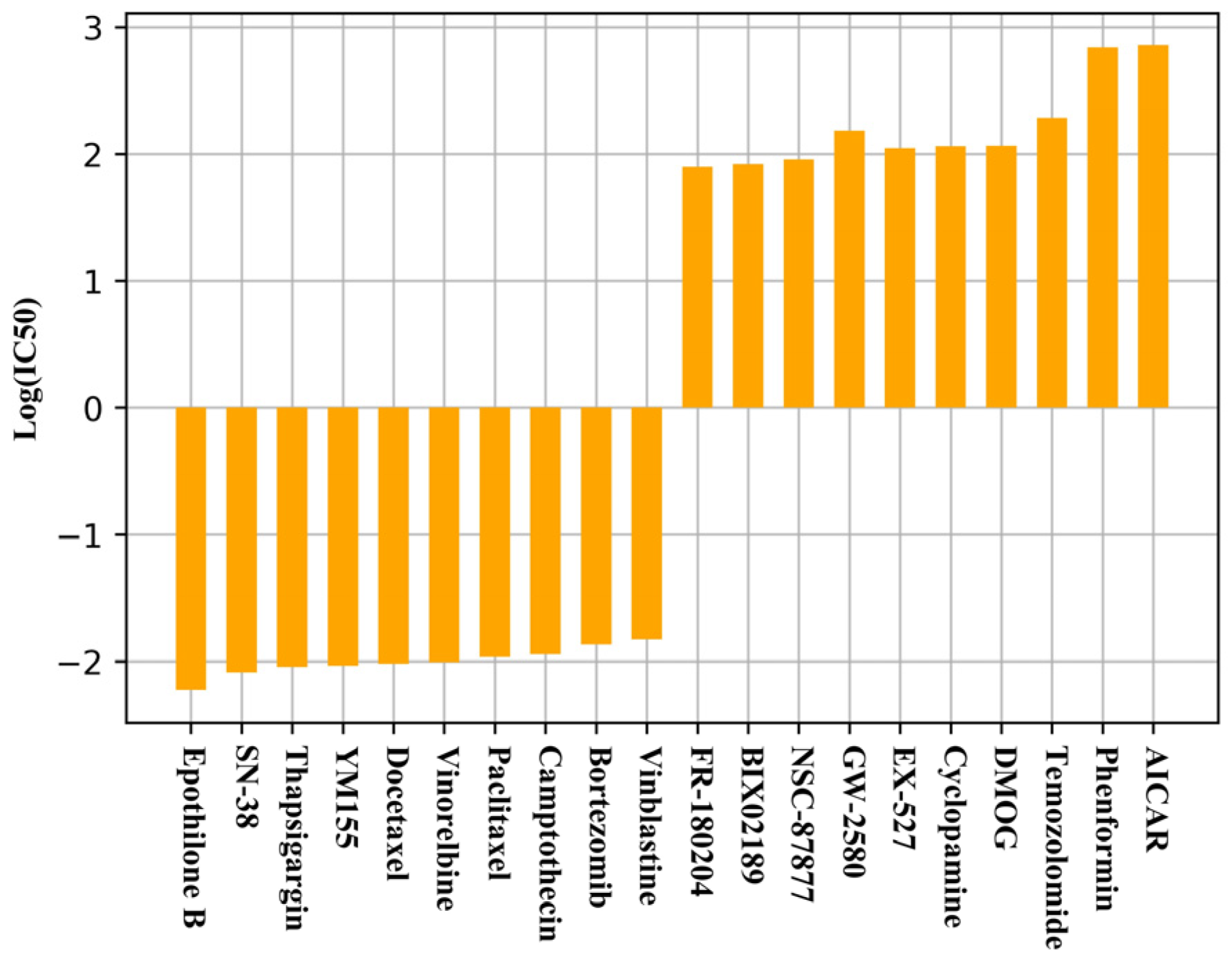
2.3. Comparison of the Characteristics of Different Drugs
2.4. Uknown Drug Response Prediction
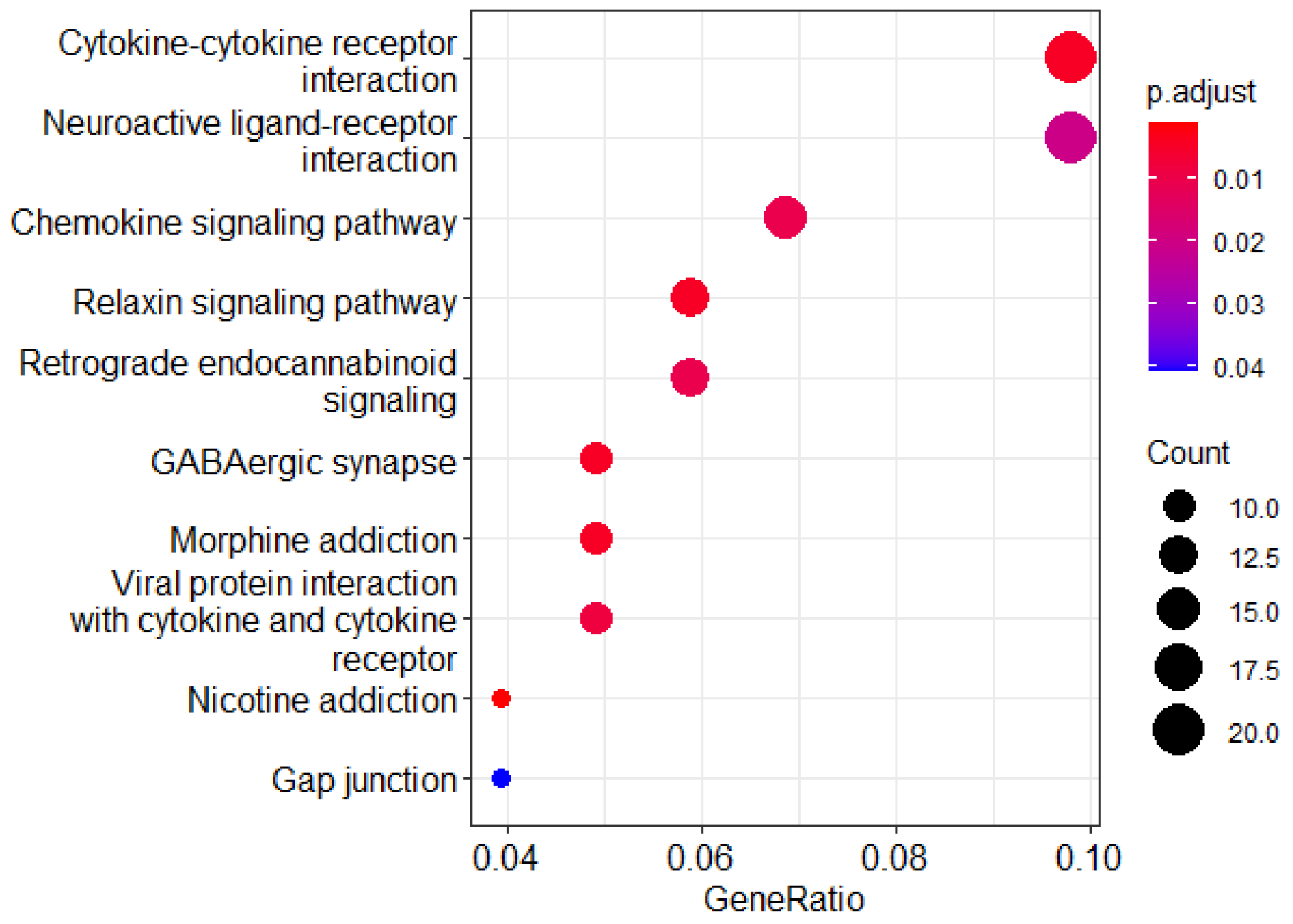
2.5. Predicting Critical Genes for Drug Responsiveness
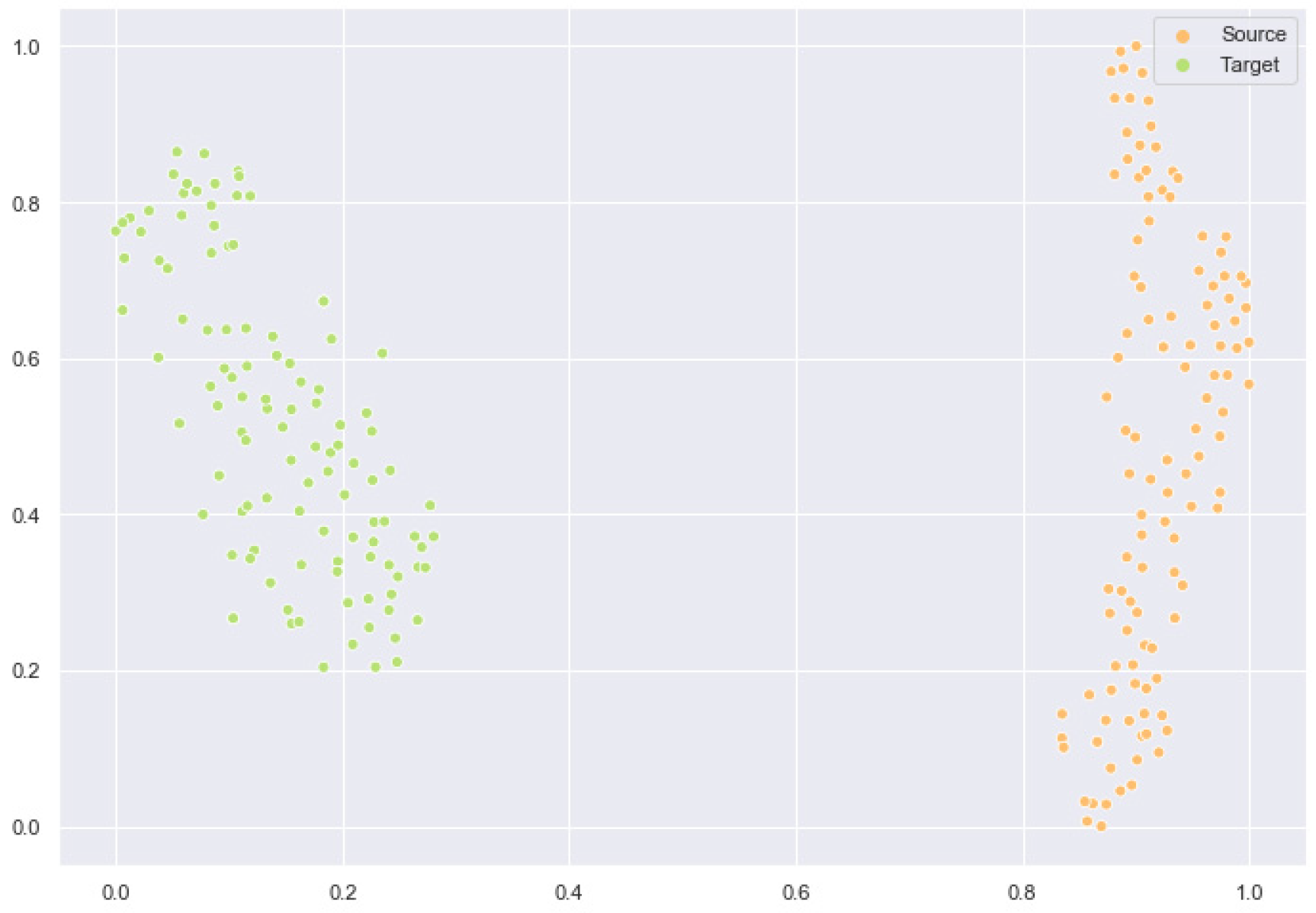
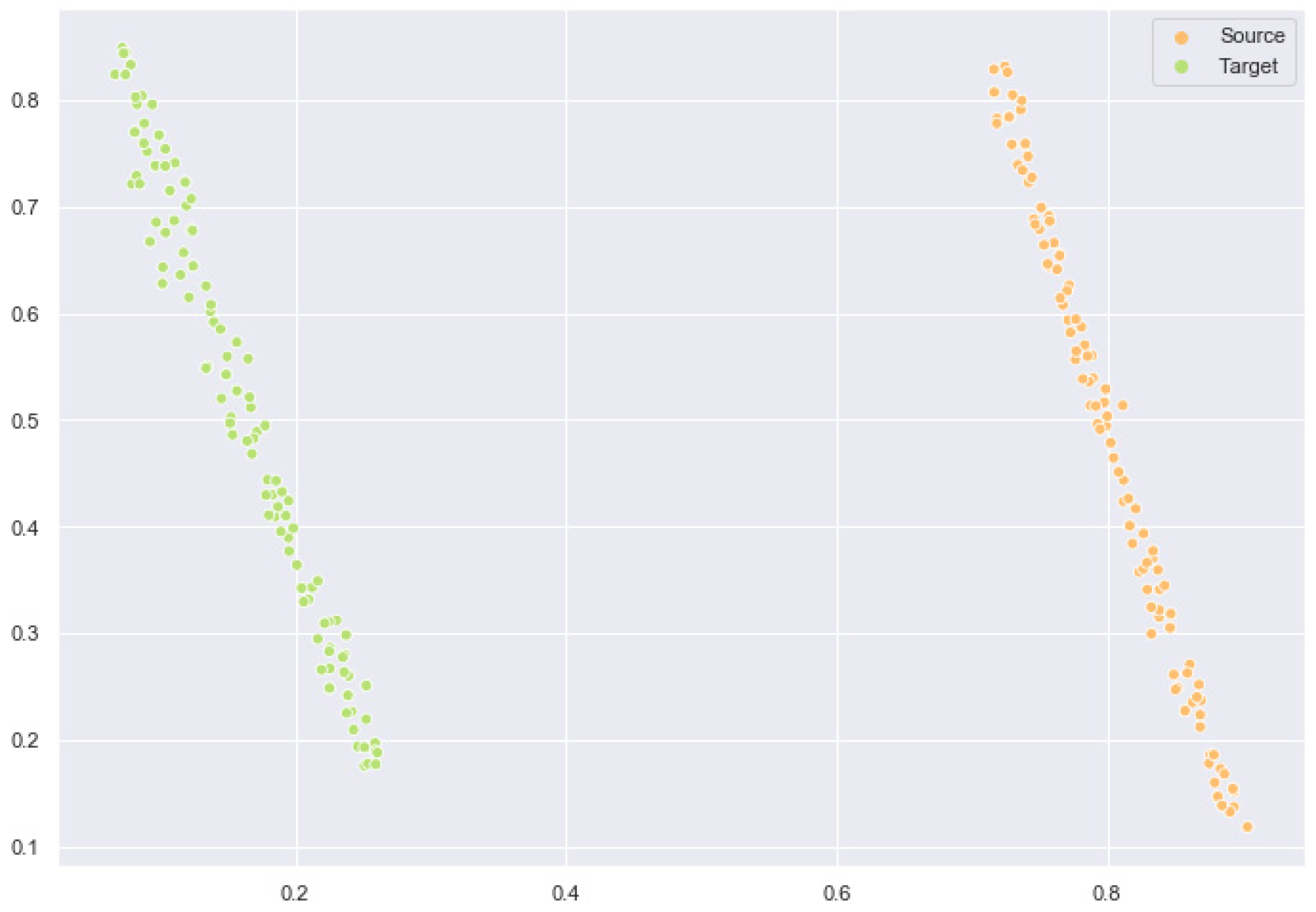
2.6. Feature Space Comparison After Domain Adaptation
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Model Input Data
- (1)
- The gene expression profiles of cancer cell lines, represented as d, where 16,016 is the number of shared genes and N is the number of training samples in a batch.
- (2)
- Drug features, represented as , where 256 is the length of the hashed Morgan fingerprint. Subsequent chapters will conduct experimental analysis on different ways of representing drug features.
- (3)
- Cancer cell line–drug sensitivity data, represented in the format [Cell Line ID, Drug ID, IC50 value].
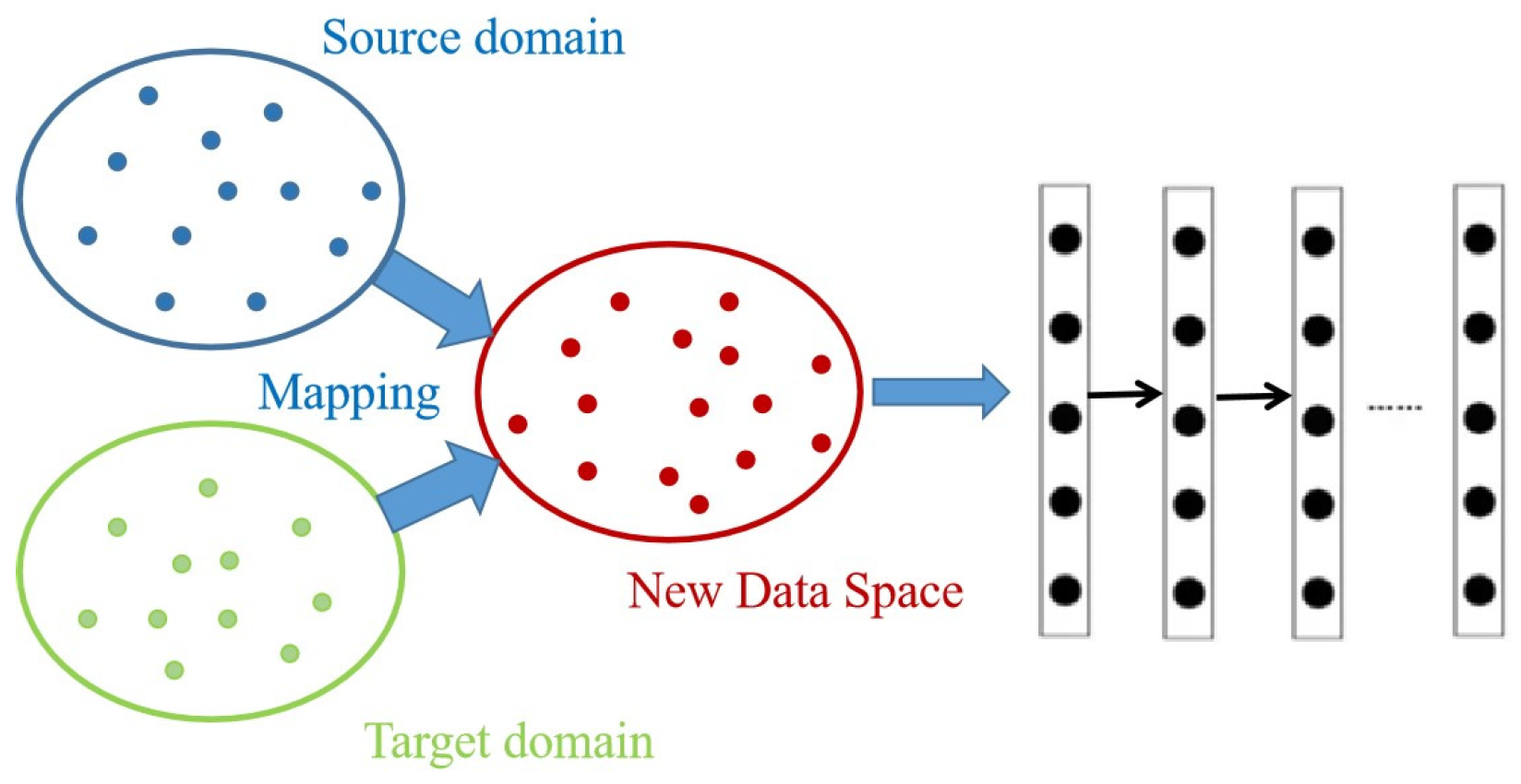
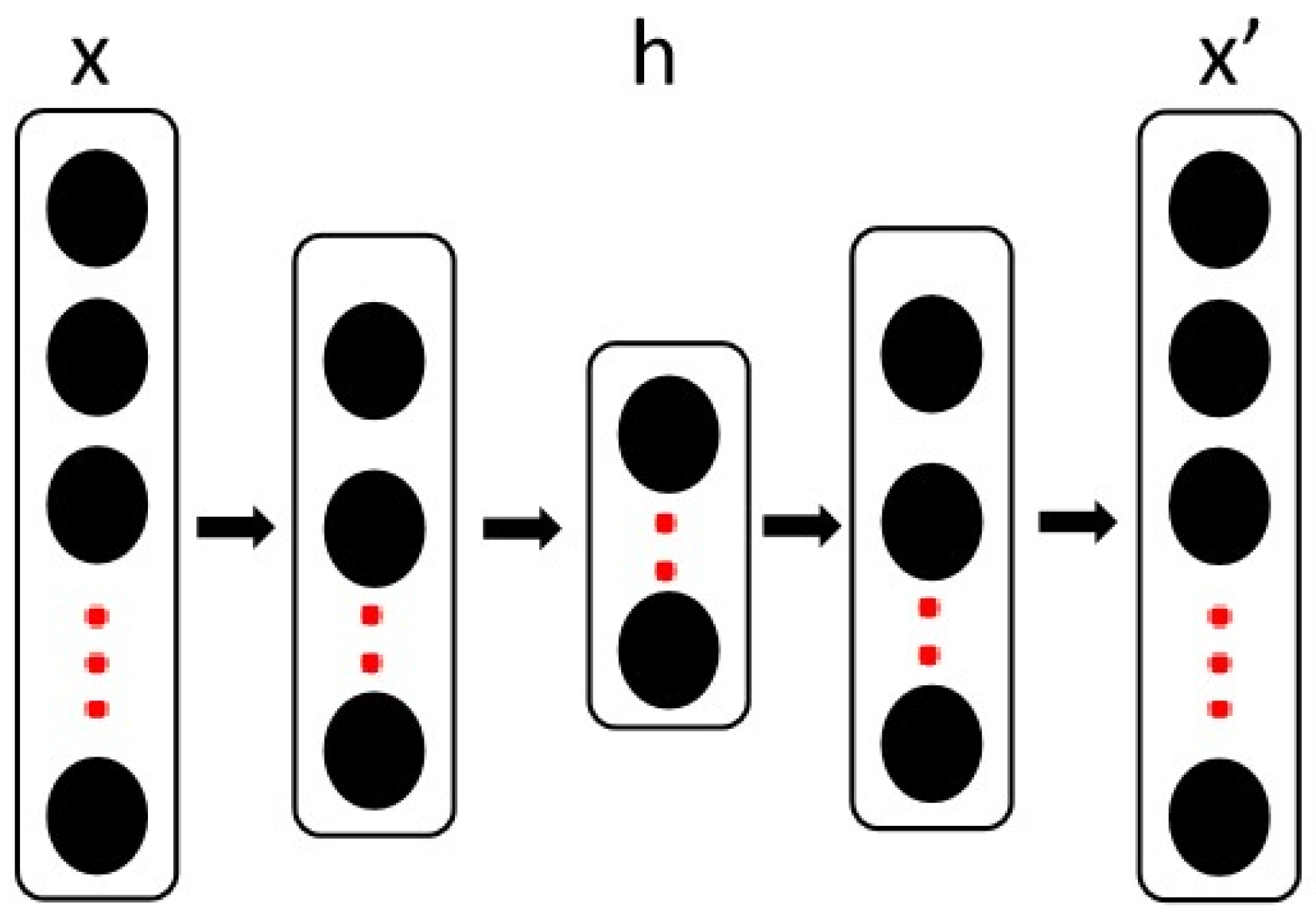
4.3. Deep Transfer Learning and Autoencoder
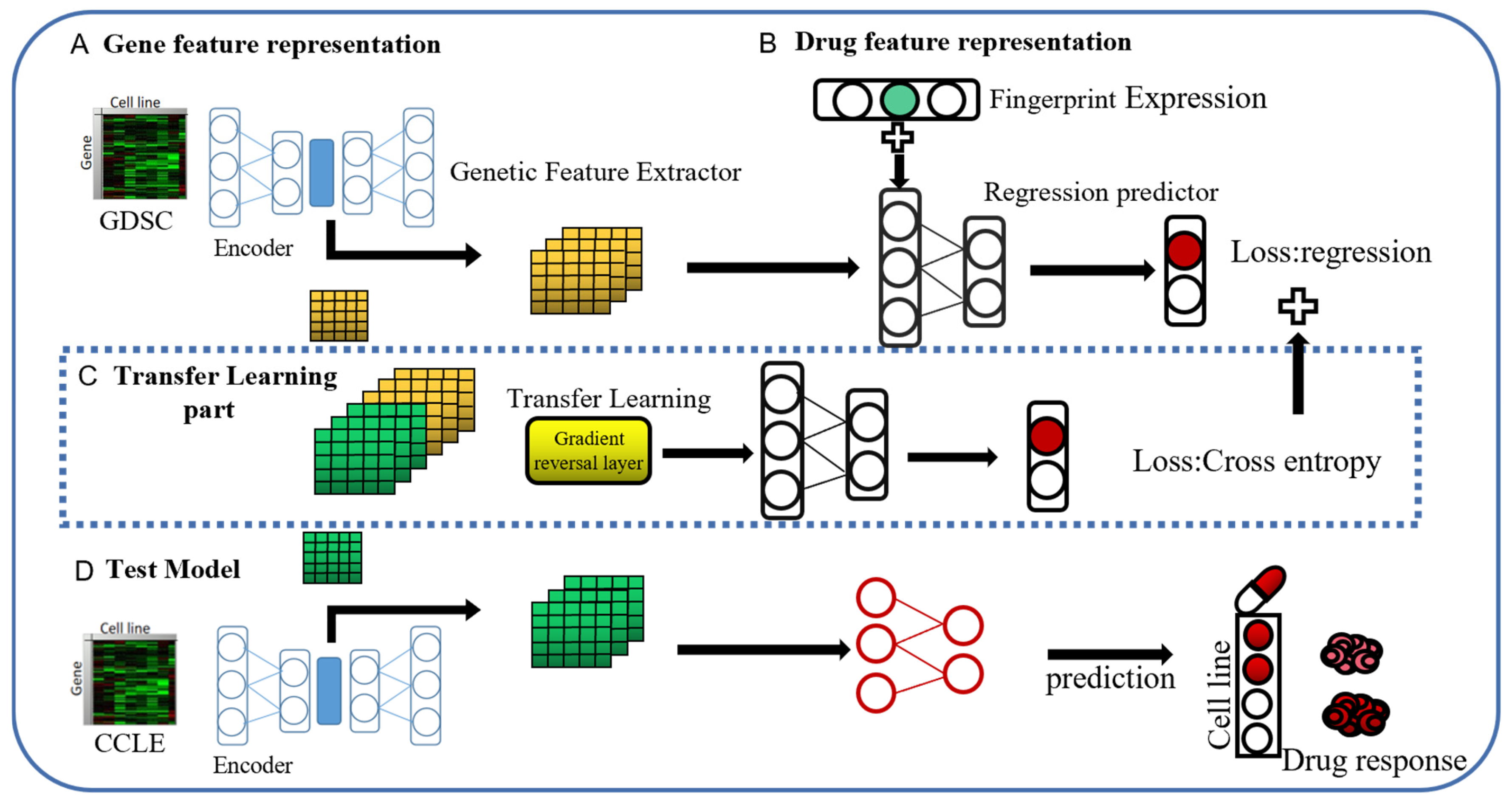
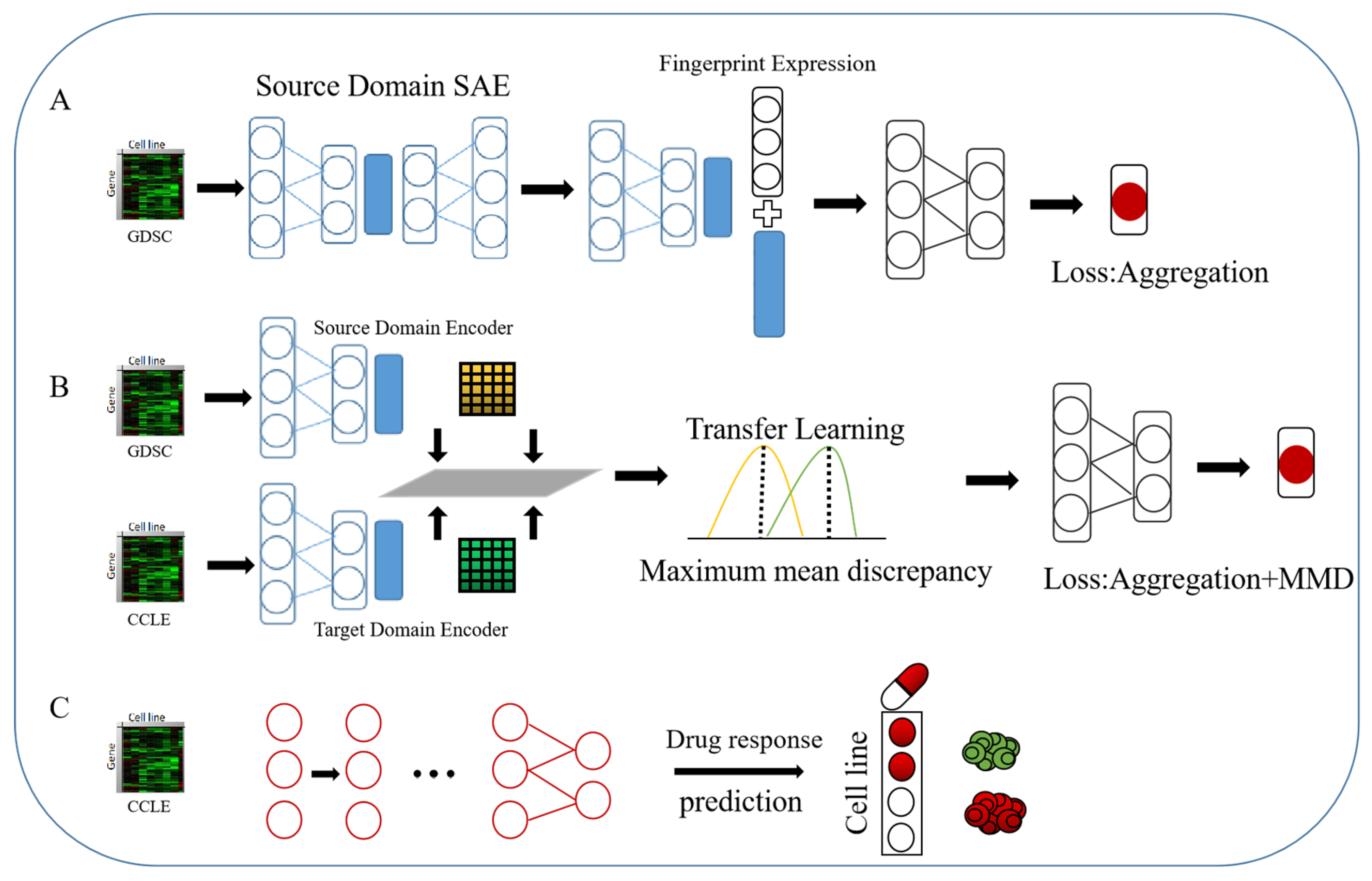
4.4. Our Method
4.4.1. Adversarial-Based Domain Adaptation Models
4.4.2. Deep Transfer Models Based on Autoencoders and Difference Metrics
- The feature extractor and regressor are trained using the source domain data to achieve the best possible performance for the source domain data on the regression task;
- The target domain data are input into another stacked autoencoder [90]. We share the encoder parameters of the source domain data under the condition of freezing the regressor parameters, the reconstruction loss of the training target domain data layer by layer and the source domain data of the MMD loss.
- Overall fine-tuning, freezing of the feedforward parameters of all feature extraction layers, and training of the regressor are performed. The loss at this time is only the MSE loss.
4.5. Performance Metrics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Gao, D.; Xie, X.; Qin, J.; Li, J.; Lin, H.; Yan, D.; Deng, K. DeepIDC: A Prediction Framework of Injectable Drug Combination Based on Heterogeneous Information and Deep Learning. Clin. Pharmacokinet. 2022, 61, 1749–1759. [Google Scholar] [CrossRef]
- Shaker, B.; Tran, K.M.; Jung, C.; Na, D. Introduction of Advanced Methods for Structure-based Drug Discovery. Curr. Bioinform. 2021, 16, 351–363. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]
- Zeng, X.; Wang, F.; Luo, Y.; Kang, S.-G.; Tang, J.; Lightstone, F.C.; Fang, E.F.; Cornell, W.; Nussinov, R.; Cheng, F. Deep generative molecular design reshapes drug discovery. Cell Rep. Med. 2022, 3, 100794. [Google Scholar] [CrossRef]
- Long, J.; Yang, H.; Yang, Z.; Jia, Q.; Liu, L.; Kong, L.; Cui, H.; Ding, S.; Qin, Q.; Zhang, N.; et al. Integrated biomarker profiling of the metabolome associated with impaired fasting glucose and type 2 diabetes mellitus in large-scale Chinese patients. Clin. Transl. Med. 2021, 11, e432. (In English) [Google Scholar] [CrossRef]
- Cao, C.; Wang, J.; Kwok, D.; Cui, F.; Zhang, Z.; Zhao, D.; Li, M.J.; Zou, Q. webTWAS: A resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res. 2021, 50, D1123–D1130. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F.; Zou, Q. Identification of drug–target interactions via multiple kernel-based triple collaborative matrix factorization. Brief. Bioinform. 2022, 23, bbab582. [Google Scholar] [CrossRef]
- Wang, Y.; Pang, C.; Wang, Y.; Jin, J.; Zhang, J.; Zeng, X.; Su, R.; Zou, Q.; Wei, L. Retrosynthesis prediction with an interpretable deep-learning framework based on molecular assembly tasks. Nat. Commun. 2023, 14, 6155. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef]
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef]
- Zeng, X.; Xiang, H.; Yu, L.; Wang, J.; Li, K.; Nussinov, R.; Cheng, F. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat. Mach. Intell. 2022, 4, 1004–1016. [Google Scholar] [CrossRef]
- Ru, X.Q.; Ye, X.C.; Sakurai, T.; Zou, Q. NerLTR-DTA: Drug-target binding affinity prediction based on neighbor relationship and learning to rank. Bioinformatics 2022, 38, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.C.; Boroni, M.; Amazonas, M.K.; Vargas, F.R. New drug candidates for osteosarcoma: Drug repurposing based on gene expression signature. Comput. Biol. Med. 2021, 134, 104470. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Al, E. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Mervin, L.; Bender, A. Current Trends in Drug Sensitivity Prediction. Curr. Pharm. Des. 2016, 22, 6918–6927. [Google Scholar] [CrossRef]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine Learning Prediction of Cancer Cell Sensitivity to Drugs Based on Genomic and Chemical Properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X.; Liu, X.S. Predicting Anticancer Drug Responses Using a Dual-Layer Integrated Cell Line-Drug Network Model. PLoS Comput. Biol. 2015, 11, e1004498. [Google Scholar] [CrossRef]
- Mehmet, G.; Margolin, A.A. Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning. Bioinformatics 2014, 30, 556–563. [Google Scholar]
- Su, R.; Liu, X.; Wei, L.; Zou, Q. Deep-Resp-Forest: A deep forest model to predict anti-cancer drug response. Methods 2019, 166, 91–102. (In English) [Google Scholar] [CrossRef]
- Lecun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436. [Google Scholar] [CrossRef]
- Madugula, S.S.; John, L.; Nagamani, S.; Gaur, A.S.; Poroikov, V.V.; Sastry, G.N. Molecular descriptor analysis of approved drugs using unsupervised learning for drug repurposing. Comput. Biol. Med. 2021, 138, 104856. [Google Scholar] [CrossRef]
- Jin, J.; Yu, Y.; Wang, R.; Zeng, X.; Pang, C.; Jiang, Y.; Li, Z.; Dai, Y.; Su, R.; Zou, Q.; et al. iDNA-ABF: Multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 2022, 23, 219. [Google Scholar] [CrossRef]
- Li, H.-L.; Pang, Y.-H.; Liu, B. BioSeq-BLM: A platform for analyzing DNA, RNA and protein sequences based on biological language models. Nucleic Acids Res. 2021, 49, e129. [Google Scholar] [CrossRef]
- Li, H.; Liu, B. BioSeq-Diabolo: Biological sequence similarity analysis using Diabolo. PLoS Comput. Biol. 2023, 19, e1011214. [Google Scholar] [CrossRef]
- Chiu, Y.C.; Chen, H.I.H.; Zhang, T.; Zhang, S.; Gorthi, A.; Wang, L.J.; Huang, Y.; Chen, Y. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med. Genom. 2019, 12, 143–155. [Google Scholar]
- Li, M.; Wang, Y.; Zheng, R.; Shi, X.; Li, Y.; Wu, F.-X.; Wang, J. DeepDSC: A Deep Learning Method to Predict Drug Sensitivity of Cancer Cell Lines. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 575–582. [Google Scholar] [CrossRef]
- Nguyen, T.; Nguyen, G.; Nguyen, T.V.; Le, D.H. Graph Convolutional Networks for Drug Response Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 146–154. [Google Scholar] [CrossRef]
- Haibe-Kains, B.; El-Hachem, N.; Birkbak, N.J.; Jin, A.C.; Beck, A.H.; Aerts, H.J.; Quackenbush, J. Inconsistency in large pharmacogenomic studies. Nature 2019, 504, 389–393. [Google Scholar] [CrossRef]
- Stransky, N.; Ghandi, M.; Kryukov, G.V.; Garraway, L.A.; Saez-Rodriguez, J. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar]
- Dhruba, S.R.; Rahman, R.; Matlock, K.; Ghosh, S.; Pal, R. Application of transfer learning for cancer drug sensitivity prediction. BMC Bioinform. 2018, 19, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Song, Y.; Zhong, E.; Qiang, Y. Transitive Transfer Learning. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Sydney, Australia, 10–13 August 2015. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kleinbaum, D.G.; Dietz, K.; Gail, M.; Klein, M.; Klein, M. Logistic Regression; Springer: New York, NY, USA, 2002. [Google Scholar]
- Smola, A.J.; Schölkopf, B. A tutorial on support vector regression. Stat. Comput. 2004, 14, 199–222. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Eckert, H.; Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov. Today 2007, 12, 225–233. [Google Scholar] [CrossRef]
- Liu, P.; Li, H.; Li, S.; Leung, K.-S. Improving prediction of phenotypic drug response on cancer cell lines using deep convolutional network. BMC Bioinform. 2019, 20, 408. [Google Scholar] [CrossRef]
- Ramsundar, B.; Eastman, P.; Walters, P.; Pande, V. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More; O’Reilly Media: Sebastopol, CA, USA, 2019. [Google Scholar]
- Vita, V.T.D.; Hellman, S.; Rosenberg, S.A. Cancer: Principles & practice of oncology. Eur. J. Cancer Care 2005. [Google Scholar]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? FEBS J. 2010, 229, 558–565. [Google Scholar] [CrossRef]
- Sundararajan, M.; Taly, A.; Yan, Q. Axiomatic attribution for deep networks. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 3319–3328. [Google Scholar]
- Xu, G.; Fan, L.; Zhao, S.; OuYang, C. MT1G inhibits the growth and epithelial-mesenchymal transition of gastric cancer cells by regulating the PI3K/AKT signaling pathway. Genet. Mol. Biol. 2022, 45, e20210067. [Google Scholar] [CrossRef]
- Jadhav, R.R.; Ye, Z.; Huang, R.-L.; Liu, J.; Hsu, P.-Y.; Huang, Y.-W.; Rangel, L.B.; Lai, H.-C.; Roa, J.C.; Kirma, N.B.; et al. Genome-wide DNA methylation analysis reveals estrogen-mediated epigenetic repression of metallothionein-1 gene cluster in breast cancer. Clin. Epigenetics 2015, 7, 13. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ru, X.; Wang, L.; Li, L.; Ding, H.; Ye, X.; Zou, Q. Exploration of the correlation between GPCRs and drugs based on a learning to rank algorithm. Comput. Biol. Med. 2020, 119, 103660. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Liu, E.; Kinnebrew, G.; Zhang, X.; Stover, D.; Huo, Y.; Zeng, Z.; Jiang, W.; Cheng, L.; et al. Identification of Alternatively-Activated Pathways between Primary Breast Cancer and Liver Metastatic Cancer Using Microarray Data. Genes 2019, 10, 753. [Google Scholar] [CrossRef]
- Dang, Y.-W.; Lin, P.; Liu, L.-M.; He, R.-Q.; Zhang, L.-J.; Peng, Z.-G.; Li, X.-J.; Chen, G. In silico analysis of the potential mechanism of telocinobufagin on breast cancer MCF-7 cells. Pathol.-Res. Pract. 2018, 214, 631–643. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A.; Wakefield, L.; Hunter, K. Chemokine networks and breast cancer metastasis. Breast Dis. 2007, 26, 75. [Google Scholar] [CrossRef]
- Khabele, D.; Lopez-Jones, M.; Yang, W.; Arango, D.; Gross, S.J.; Augenlicht, L.H.; Goldberg, G.L. Tumor necrosis factor-α related gene response to Epothilone B in ovarian cancer. Gynecol. Oncol. 2004, 93, 19–26. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Zhuang, F.; Qi, Z.; Duan, K.; Xi, D.; Zhu, Y.; Zhu, H.; Xiong, H.; He, Q. A Comprehensive Survey on Transfer Learning. Proc. IEEE 2021, 109, 43–76. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- O’boyle, N.M. Towards a Universal SMILES representation—A standard method to generate canonical SMILES based on the InChI. J. Cheminforma. 2012, 4, 22. [Google Scholar] [CrossRef]
- Bento, A.P.; Hersey, A.; Felix, E.; Landrum, G.; Leach, A.R. An Open Source Chemical Structure Curation Pipeline using RDKit. J. Cheminform. 2020, 12, 51. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2016, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, C.; Ding, L.; Li, Z.; Pan, Y.; Gao, X. Deep learning in bioinformatics: Introduction, application, and perspective in the big data era. Methods 2019, 166, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Zulfiqar, H.; Huang, Q.-L.; Lv, H.; Sun, Z.-J.; Dao, F.-Y.; Lin, H. Deep-4mCGP: A Deep Learning Approach to Predict 4mC Sites in Geobacter pickeringii by Using Correlation-Based Feature Selection Technique. Int. J. Mol. Sci. 2022, 23, 1251. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Dao, F.; Lin, H. DeepKla: An attention mechanism-based deep neural network for protein lysine lactylation site prediction. iMeta 2022, 1, e11. [Google Scholar] [CrossRef]
- Liu, M.; Li, C.; Chen, R.; Cao, D.; Zeng, X. Geometric Deep Learning for Drug Discovery. Expert Syst. Appl. 2023, 240, 122498. [Google Scholar] [CrossRef]
- Xu, J.; Xu, J.; Meng, Y.; Lu, C.; Cai, L.; Zeng, X.; Nussinov, R.; Cheng, F. Graph embedding and Gaussian mixture variational autoencoder network for end-to-end analysis of single-cell RNA sequencing data. Cell Rep. Methods 2023, 3, 100382. [Google Scholar] [CrossRef]
- Wang, R.; Jiang, Y.; Jin, J.; Yin, C.; Yu, H.; Wang, F.; Feng, J.; Su, R.; Nakai, K.; Zou, Q.; et al. DeepBIO: An automated and interpretable deep-learning platform for high-throughput biological sequence prediction, functional annotation and visualization analysis. Nucleic Acids Res. 2023, 51, 3017–3029. [Google Scholar] [CrossRef]
- Tang, Y.-J.; Pang, Y.-H.; Liu, B. IDP-Seq2Seq: Identification of intrinsically disordered regions based on sequence to sequence learning. Bioinformatics 2020, 36, 5177–5186. [Google Scholar] [CrossRef]
- Yan, K.; Lv, H.; Guo, Y.; Peng, W.; Liu, B. sAMPpred-GAT: Prediction of Antimicrobial Peptide by Graph Attention Network and Predicted Peptide Structure. Bioinformatics 2023, 39, btac715. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Hornik, K.; Stinchcombe, M.; White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 1989, 2, 359–366. [Google Scholar] [CrossRef]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to Sequence Learning with Neural Networks. Adv. Neural Inf. Process. Syst. 2014. [Google Scholar]
- Long, M.; Cao, Y.; Cao, Z.; Wang, J.; Jordan, M.I. Transferable Representation Learning with Deep Adaptation Networks. IEEE Trans. Pattern Anal. Mach. Intell. 2018, 41, 3071–3085. [Google Scholar] [CrossRef]
- Ganin, Y.; Ustinova, E.; Ajakan, H.; Germain, P.; Larochelle, H.; Laviolette, F.; Marchand, M.; Lempitsky, V. Domain-Adversarial Training of Neural Networks. J. Mach. Learn. Res. 2016, 17, 1–35. [Google Scholar]
- Joshi, P.; Vedhanayagam, M.; Ramesh, R. An Ensembled SVM Based Approach for Predicting Adverse Drug Reactions. Curr. Bioinform. 2021, 16, 422–432. [Google Scholar] [CrossRef]
- You, K.; Long, M.; Cao, Z.; Wang, J.; Jordan, M.I. Universal domain adaptation. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–17 June 2019; pp. 2720–2729. [Google Scholar]
- Wang, M.; Deng, W. Deep visual domain adaptation: A survey. Neurocomputing 2018, 312, 135–153. [Google Scholar] [CrossRef]
- Baldi, P.; Guyon, G.; Dror, V.; Lemaire, G.; Taylor, G.; Silver, D. Autoencoders, unsupervised learning and deep architectures. In Proceedings of the UTLW’11 2011 International Conference on Unsupervised and Transfer Learning Workshop, Washington, DC, USA, 2 July 2011; Volume 27. [Google Scholar]
- Gehring, J.; Miao, Y.; Metze, F.; Waibel, A. Extracting deep bottleneck features using stacked auto-encoders. In Proceedings of the ICASSP, IEEE International Conference on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, 26–31 May 2013; pp. 3377–3381. [Google Scholar]
- An, N.; Zhao, W.; Wang, J.; Shang, D.; Zhao, E. Using multi-output feedforward neural network with empirical mode decomposition based signal filtering for electricity demand forecasting. Energy 2013, 49, 279–288. [Google Scholar] [CrossRef]
- Bhaskar, K.; Singh, S.N. AWNN-Assisted Wind Power Forecasting Using Feed-Forward Neural Network. IEEE Trans. Sustain. Energy 2012, 3, 306–315. [Google Scholar] [CrossRef]
- Tran, D.; Tan, Y.K. Sensorless Illumination Control of a Networked LED-Lighting System Using Feedforward Neural Network. IEEE Trans. Ind. Electron. 2013, 61, 2113–2121. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Ni, P.; Zhao, K.; Wu, F.-X.; Pan, Y. Computational Drug Repositioning with Random Walk on a Heterogeneous Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, H.; Ai, C.; Ding, Y.; Guo, F.; Tang, J. MVML-MPI: Multi-View Multi-Label Learning for Metabolic Pathway Inference. Brief. Bioinform. 2023, 24, bbad393. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Ding, J.; Chen, G.; Duan, J.; Guo, F.; Tang, J. IK-DDI: A novel framework based on instance position embedding and key external text for DDI extraction. Brief. Bioinform. 2023, 24, bbad099. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, S.; Ahn, J. RefDNN: A reference drug based neural network for more accurate prediction of anticancer drug resistance. Sci. Rep. 2020, 10, 1861. [Google Scholar] [CrossRef]
- Yang, H.; Luo, Y.-M.; Ma, C.-Y.; Zhang, T.-Y.; Zhou, T.; Ren, X.-L.; He, X.-L.; Deng, K.-J.; Yan, D.; Tang, H.; et al. A gender specific risk assessment of coronary heart disease based on physical examination data. Npj Digit. Med. 2023, 6, 136. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Ning, L.; Ye, X.; Yang, Y.H.; Futamura, Y.; Sakurai, T.; Lin, H. iLoc-miRNA: Extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief. Bioinform. 2022, 23, bbac395. [Google Scholar] [CrossRef]
- Wang, Y.; Zhai, Y.; Ding, Y.; Zou, Q. SBSM-Pro: Support Bio-sequence Machine for Proteins. arXiv 2023, arXiv:2308.10275. [Google Scholar] [CrossRef]
- Jang, I.S.; Neto, E.C.; Guinney, J.; Friend, S.H.; Margolin, A.A. Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. Biocomputing 2013, 19, 63–74. [Google Scholar]
- Borgwardt, K.M.; Gretton, A.; Rasch, M.J.; Kriegel, H.-P.; Schölkopf, B.; Smola, A.J. Integrating structured biological data by Kernel Maximum Mean Discrepancy. Bioinformatics 2006, 22, e49–e57. [Google Scholar] [CrossRef]
- Tzeng, E.; Hoffman, J.; Zhang, N.; Saenko, K.; Darrell, T. Deep Domain Confusion: Maximizing for Domain Invariance. Computer Science. arXiv 2014, arXiv:1412.3474. [Google Scholar]
- Yosinski, J.; Clune, J.; Bengio, Y.; Lipson, H. How Transferable Are Features in Deep Neural Networks? MIT Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Zhuang, F.; Cheng, X.; Luo, P.; Pan, S.J.; He, Q. Supervised Representation Learning with Double Encoding-Layer Autoencoder for Transfer Learning. ACM Trans. Intell. Syst. Technol. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Caruana, R. Multitask Learning. Mach. Learn. 1997, 28, 41–75. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | RMSE | R2 |
|---|---|---|
| DADSP-A | 0.64 | 0.43 |
| DADSPA- | 0.71 | 0.31 |
| DADSP-B | 0.69 | 0.35 |
| DeepDSC-1 | 0.82 | 0.11 |
| DeepDSC-2 | 0.72 | 0.29 |
| SLA | 0.82 | 0.10 |
| RF | 0.75 | 0.27 |
| LR | 0.75 | 0.26 |
| SVR | 0.73 | 0.29 |
| Method | RMSE | R2 |
|---|---|---|
| DADSP-A | 0.69 | 0.32 |
| DADSP-B | 0.92 | 0.01 |
| DeepDSC-1 | 0.70 | 0.29 |
| SLA | 0.72 | 0.30 |
| Method | RMSE | R2 |
|---|---|---|
| DADSP-A | 0.64 | 0.43 |
| DADSP-A + SSP | 0.67 | 0.35 |
| DADSP-A + CNN | 0.76 | 0.29 |
| DADSP-A + GCN | 0.74 | 0.23 |
| MKN7 | ZR-75-30 | MEL-HO | |||
|---|---|---|---|---|---|
| Critical Gene | Score | Critical Gene | Score | Critical Gene | Score |
| ENSG00000205364 | 0.002803 | ENSG00000111700 | 0.003027 | ENSG00000205364 | 0.003027 |
| ENSG00000187908 | 0.002338 | ENSG00000183032 | 0.002608 | ENSG00000164821 | 0.002608 |
| ENSG00000164821 | 0.002276 | ENSG00000158023 | 0.002451 | ENSG00000158023 | 0.002451 |
| ENSG00000174469 | 0.002231 | ENSG00000103316 | 0.002122 | ENSG00000187908 | 0.002122 |
| ENSG00000158023 | 0.002206 | ENSG00000183668 | 0.002003 | ENSG00000183032 | 0.002003 |
| ENSG00000111404 | 0.002066 | ENSG00000111249 | 0.001953 | ENSG00000110077 | 0.001953 |
| ENSG00000183032 | 0.002022 | ENSG00000187908 | 0.001919 | ENSG00000111404 | 0.001919 |
| ENSG00000183668 | 0.001981 | ENSG00000165168 | 0.001918 | ENSG00000183668 | 0.001918 |
| ENSG00000167083 | 0.001887 | ENSG00000164821 | 0.001834 | ENSG00000166049 | 0.001834 |
| ENSG00000120162 | 0.001884 | ENSG00000166049 | 0.001831 | ENSG00000111249 | 0.001831 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, W.; Xu, X.; Xiao, Z.; Gao, L.; Yu, L. Cancer Drug Sensitivity Prediction Based on Deep Transfer Learning. Int. J. Mol. Sci. 2025, 26, 2468. https://doi.org/10.3390/ijms26062468
Meng W, Xu X, Xiao Z, Gao L, Yu L. Cancer Drug Sensitivity Prediction Based on Deep Transfer Learning. International Journal of Molecular Sciences. 2025; 26(6):2468. https://doi.org/10.3390/ijms26062468
Chicago/Turabian StyleMeng, Weijun, Xinyu Xu, Zhichao Xiao, Lin Gao, and Liang Yu. 2025. "Cancer Drug Sensitivity Prediction Based on Deep Transfer Learning" International Journal of Molecular Sciences 26, no. 6: 2468. https://doi.org/10.3390/ijms26062468
APA StyleMeng, W., Xu, X., Xiao, Z., Gao, L., & Yu, L. (2025). Cancer Drug Sensitivity Prediction Based on Deep Transfer Learning. International Journal of Molecular Sciences, 26(6), 2468. https://doi.org/10.3390/ijms26062468