
A Comprehensive Review of Metabolic Dysfunction-Associated Steatotic Liver Disease: Its Mechanistic Development Focusing on Methylglyoxal and Counterbalancing Treatment Strategies


Abstract

1. Introduction
2. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD)
2.1. Current Definition of MASLD
2.2. The Pathogenesis of MASLD
2.2.1. Multiple-Hit Hypothesis
2.2.2. Adipose Tissue-Liver Axis
2.2.3. Hypoxia
2.2.4. Gut–Liver Axis
2.2.5. Dietary Fructose
2.2.6. Autophagy Versus Cell Death
2.2.7. AMP-Activated Protein Kinase
2.2.8. Advanced Glycation End Products
2.2.9. Hereditary Predisposition for MASLD
3. Methylglyoxal (MGO)
4. MGO in MASLD
4.1. MGO in the Early MASLD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model | Detailed Observations | Major Findings in the Liver (Tissue/Cells) | Ref. Year |
|---|---|---|---|
| A. Early MASLD | |||
| The model of early MASLD: seven-week-old male Wistar rats (WRs) divided into 2 groups: (1) WR injected with (0.3 mL/kg/week of 40%) CCl4 (in soy bean oil) for 4 weeks. (2) WR injected with the same volume of soybean oil (control group) | Serum (as compared with control group): D-lactate ↑; AST, ALT and MGO =. Liver: MGO level and D-lactate ↑. Urine: D-lactate ↑; MGO =. | MGO ↑ D-lactate ↑ | [97] 2018 |
| The model of hypertriglyceridemia/prediabetes: (1) Six-month-old male hereditary hypertriglyceridemic rats (HHTg) as the non-obese prediabetic model treated or not-treated with salsalate (2) WR as the control group | In HHTg rats (in comparison with WR and attenuated by salsalate); Liver: TAGs, Chol and MGO ↑; oxidative stress ↑ (TBARS ↑, GSH/GSSG ↓, SOD ↓). Upon salsalate treatment in HHTg: Glo1 gene ↑ associated with MGO ↓. | MGO ↑ Lipids ↑ oxidative stress ↑ | [103] 2023 |
| The model of postmenopausal MetS: Female Wistar rats (WRs) divided into 2 groups: (1) Ovariectomized WR used as a model of postmenopausal MetS (W-OVX); (2) Sham-operated WR as a control (W-sham) | In W-OVX rats (in comparison with W-sham rats); Serum: leptin, FAs, HDL-Chol, MCP-1 ↑; TAGs and Chol =. Liver: MGO and TAGs ↑; Glo1 (mRNA and activity) and Chol =; oxidative stress ↑ (TBARS ↑, GSH/GSSG ↓, GPx ↓); Muscle: TAGs ↑. | MGO ↑ TAGs ↑ oxidative stress ↑ | [104] 2021 |
| The model of MGO-enriched high-fat diet: Male WR divided into four groups: (1) control (Ct) with standard diet A03 (5% triglycerides and 45% carbohydrates) (2) methylglyoxal group (MG) with a standard diet and MGO administration (rats fed 75 mg MGO kg-1 daily for 18 weeks) (3) high-fat diet-fed group (HFD) (40% triglycerides and 10% carbohydrates) (4) high-fat diet group with MGO supplementation (rats fed 75 mg MGO kg-1 daily for 18 weeks) (HFDMG) | Effect of MGO supplementation (HFDMG group compared to control and/or MG or HFD rats); Blood plasma: NEFAs ↑; albumin ↓; adiponectin ↓ (as compared to adiponectin ↑ in HFD). Liver: Inflammatory cells ↑ (F4/80 ↑—a marker of macrophages/Kupffer cells); MAGEs ↑ (MG-H1 ↑, CEL ↑, but ArgP =); Insulin receptor phosphorylation at Tyr1163 ↓; Phosphorylation of ACC ↓ (ACC activity ↑); Phosphorylation of AMPK ↓ (AMPK activity ↓) Cardiolipin 70:2 ↓; Expression of FAS ↑ and AceCS ↑; membrane RAGE =; Glo1 expression = (but Glo1 activity ↑ in MG; Glo1 activity ↓ in HFDMG) | Inflammation ↑ MAGE ↑ IR ↑ ACC ↑ AMPK ↓ | [101] 2019 |
| The model of genistein effect evaluation in high-fat diet: Male C57BL/6J mice divided into 8 groups: Study 1 (mice fed for 16 weeks with): (1) low-fat diet (10% fat energy) (LF) (2) very-high-fat diet (60% fat energy) (VHF) (3) very-high-fat diet with 0.25% genistein (VHF-G). Study 2 (mice fed for 18 weeks with): (4) low-fat diet (10% fat energy) (LF) (5) moderately high-fat diet (HF) (6) moderately high-fat diet with MGO (110–145 mg/kg/day) (HFM) (7) moderately high-fat diet with MGO and 0.067% genistein (HFM-GL) (8) moderately high-fat diet with MGO and 0.2% genistein (HFM-GH) | Genistein effect (VHF-G vs. VHF and HFM-GH vs. HFM); Blood plasma: MGO ↓, AGEs ↓, Glc ↓, Chol ↓, ALT ↓, AST ↓. Liver and kidney: AGEs ↓; Glo1/2 expression ↑, aldose reductase expression ↑; RAGE expression ↓. Liver: TAGs level ↓. | Glo1/2 ↑ TAGs ↓ RAGE↓ AGEs ↓ | [105] 2019 |
| Fru/MGO effect on rat hepatocytes: (1) Primary rat hepatocytes (isolated from WR) (PRH) incubated with Glc (8 mM) and inulin (0.12%) with or without inulinase in the absence or presence of insulin for up to 4 h. (2) PRH incubated with Glc (8 mM) and inulin (0.12%) and MGO (20 µM) in the absence or presence of insulin for 4 h. | Effect of Fru on PRH (in comparison with Glc-exposed PRH): MGO ↑ (~ 2-fold). Effects of Fru or MGO on PRH: phosphorylation of MKK7 ↑; phosphorylation of JNK ↑; phosphorylation of serine307 on IRS-1 ↑ (in the absence and presence of insulin); insulin-stimulated tyrosine phosphorylation of IRS-1 and IRS-2 ↓. | Fru effect: MGO ↑ Fru/MGO effect: IR ↑ | [98] 2013 |
| B. Liver cirrhosis | |||
| The model of liver cirrhosis: (1) Male WR treated with CCl4 and phenobarbital for 8 weeks (early cirrhosis without ascites) or 12–14 weeks (advanced cirrhosis with ascites) (2) Male WR treated with CCl4 for 12–14 weeks, and Glo1 inhibitor (ethyl pyruvate—EP) starting from week 8. Primary rat hepatocytes (pHEP), primary hepatic stellate cells (pHSC) and primary liver sinusoidal endothelial cells (pLSEC) isolated from control and cirrhotic WR. Normal hepatic stellate cells (HSZ-B-S1). | In comparison with pHEP: Glo1 expression in pHSC and pLSEC derived from control WR ↓. In the whole liver, and pHEP, pHSC, and pLSEC in cirrhosis (in comparison with healthy WR): Glo1 expression ↓ (and lower in advanced cirrhosis as compared to early cirrhosis). In pHSC and pLSEC in cirrhosis (in comparison with healthy WR): Glo1 activity ↓. In the whole liver and pHEP in cirrhosis (in comparison with healthy WR): Glo1 activity ↑ In the whole liver in cirrhosis (in comparison with healthy WR): MGO level ↑ (and higher increase in advanced cirrhosis as compared to early cirrhosis) Upon LPS induction of HSZ-B-S1: Glo1 activity ↑. Upon EP or MGO treatment of LPS-induced HSZ-B-S1: TNF-α ↓, collagen-I ↓, α-SMA ↓. Upon EP treatment of LPS-induced HSZ-B-S1: LPS-induced NF-κB stimulation ↓, LPS-induced reduction in Nrf2 ↓, LPS-induced pERK ↓, ERK expression =. Effect of EP treatment on cirrhotic WR (compared to cirrhotic livers without EP treatment): fibrotic tissue ↓, α-SMA ↓, TGF-β ↓, NF-κB expression ↓, Nrf2 expression ↑. | MGO ↑ Glo1 expression ↓ Glo1 activity ↑ (liver and hepatocytes) Glo1 activity ↓ (pHSC and pLSEC) | [108] 2017 |
| C. Hepatocellular carcinoma | |||
| Human HCC cell lines: Huh-7, HepG2 and Hep3B. | Effect of 1 µM MGO on Huh-7 and HepG2 cells (but not Hep3B): cells adhesion to collagen ↓, cells invasion through Matrigel ↓ (via promoting p53 localization in the nucleus). | MGO effect: invasiveness ↓ | [109] 2013 |
| Human HCC cell lines: Hep3B, SK-HEP-1 and SMMC-7721 | Effect of Glo1 knock-down in all 3 cell lines: cells proliferation ↓. Effect of Glo1 over-expression in all 3 cell lines: cells growth =. | Glo1 silencing effect: proliferation ↓ | [110] 2014 |
| Human HCC cell lines: Huh-7 and HepG2 Murine hepatocyte cell line AML12 | In comparison with normal AML12 cells: Glo1 in Huh-7 ↑ (mRNA, protein and activity); Glo1 in HepG2 ↑ (only mRNA); Effects of Glo1 inhibition in Huh-7 cells (by 1–20 mM ethyl pyruvate or 1–10 µM BrBzGSHCp2): proliferation ↓, migration ↓, colony formation ↓; PDGFR-β ↓, VEGFR2 ↓, VEGF ↓, pERK/ERK ↓, NF-κB ↓; Nrf2 ↑. Effects of 2.5–10 µM sorafenib (a multi-tyrosine kinase inhibitor approved for the therapy of advanced HCC): Glo1 ↑, MGO ↑. | Glo1 silencing effect: proliferation ↓ migration ↓ invasiveness ↓ | [111] 2019 |
| Human HCC cell line HepG2 incubated with palmitic or oleic acids for 24 h. | Glo1 ↓ in oleic acid treated HepG2. MGO ↑ in both palmitic and oleic acids treated HepG2 and their culture media. | FAs effect: MGO ↑ Glo1 ↓ | [106] 2018 |
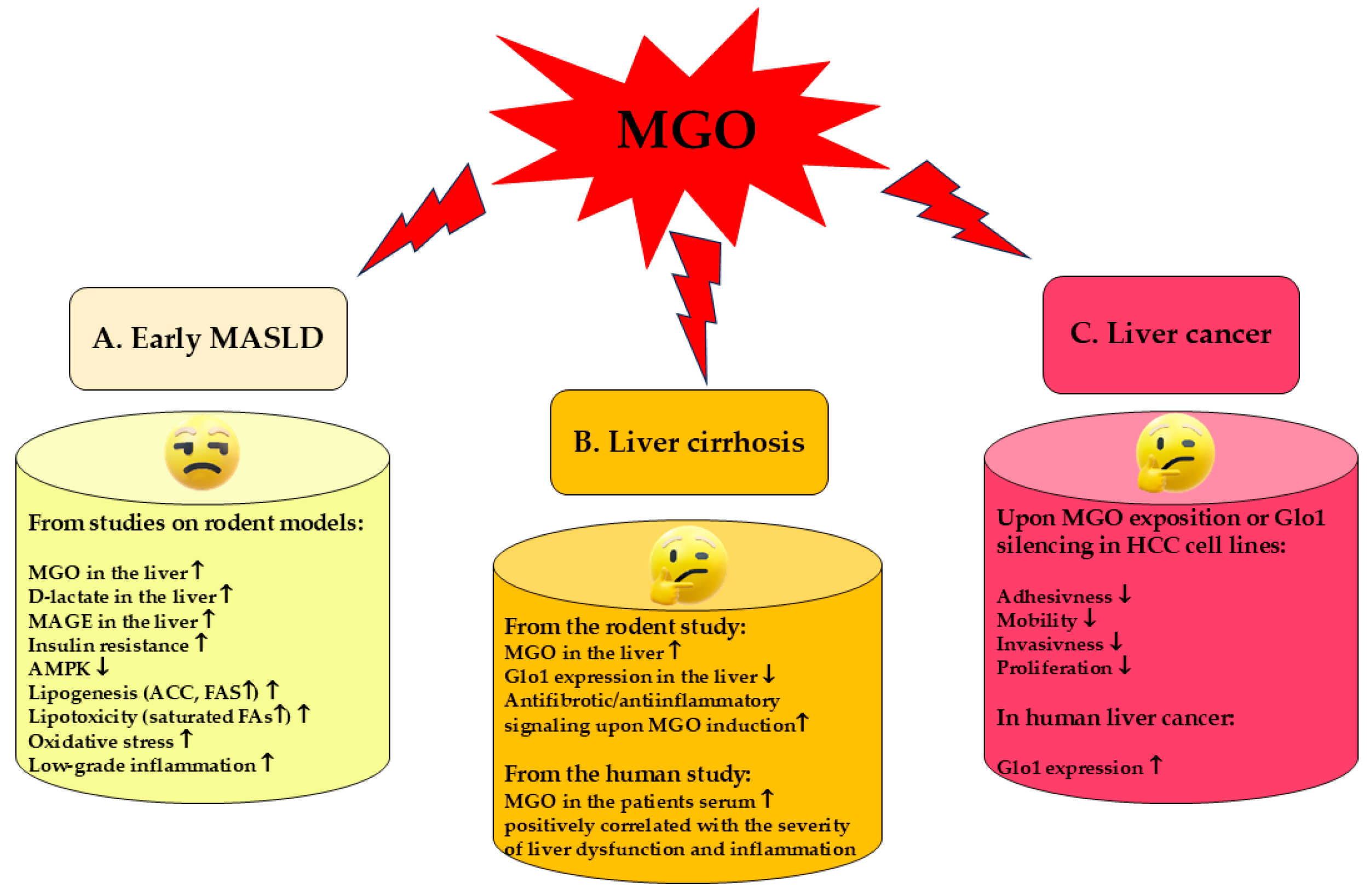
4.2. MGO in Liver Cirrhosis
4.3. MGO in Liver Cancer
5. Contribution of Fructose-Derived MGO to MASLD Development
6. Approved and Potential Therapies in MASLD
6.1. Recommended Therapies and Medications
6.2. MGO, AGEs, and Gut Microbiota as Therapeutic Targets
6.3. MASLD Therapy with MGO Scavengers and Antiglycation Agents
6.4. MGO Scavengers Protect the Activity of AMPK and Promote Autophagy in the Liver
7. Methodology
8. Conclusions and Remarks for Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| AceCS | acetyl-CoA synthetase |
| AGEs | advanced glycation end products |
| AIFM2 | factor mitochondria associated 2A |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| ArgP | Argpyrimidine |
| ApoE−/− | apolipoprotein E knockout |
| AST | aspartate aminotransferase |
| ATAUC | adipose tissuearea under the curve |
| α-SMA | alpha-smooth muscle actin |
| BCAA | Branched-chain amino acids |
| CaMKKβ | Ca21/calmodulin-dependent protein kinase kinase β |
| CCl4 | carbon tetrachloride |
| CD43 | leukosialin (mucin-like protein expressed on the surface of most hematopoietic cells) |
| CEdG | N2-carboxyethyl-20–deoxyguanosine |
| CEL | Nε-(1-carboxyethyl)lysine = N6-(1-carboxyethyl)lysine |
| Chol | Cholesterol |
| ChREBP | carbohydrate-responsive element-binding protein |
| CRP | C-reactive protein |
| CTGF | connective tissue growth factor |
| CVD | cardiovascular disease |
| DAGs | Diacylglycerols |
| DAMPs | damage-associated molecular patterns |
| DMT1 | divalent metal transporter 1 |
| DNL | de novo lipogenesis |
| ECM | extracellular matrix |
| EVsFAs | extracellular vesiclesfatty acids |
| FAS | fatty acid synthase |
| Fru | Fructose |
| FXR | farnesoid X receptor |
| GA | Glyceraldehyde |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GGT | γ-glutamyl transpeptidase |
| GIT | gastrointestinal tract |
| Glc | Glucose |
| Glo1 | glyoxalase 1 |
| Glo2 | glyoxalase 2 |
| GLUT-4 | insulin-dependent glucose transporters in skeletal muscle and adipose tissue |
| GPx | glutathione peroxidase |
| GPX4 | glutathione peroxidase 4 |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| HCC | hepatocellular carcinoma |
| HDL-Chol | high-density lipoproteins cholesterol |
| Hep G2 | epithelial hepatoblastoma cell line |
| HFCS | high-fructose corn syrup |
| HFD | high-fat diet |
| HHTg | hereditary hypertriglyceridemic rats |
| HIF | hypoxia-inducible factor |
| HO | heme oxygenase |
| HOMA | homeostatic model assessment |
| HSCs | hepatic stellate cells |
| IR | insulin resistance |
| IRS-1,2 | insulin receptor substrate 1,2 |
| JNK | c-jun NH2-terminal kinase |
| KCs | Kupffer cells |
| LKB1 | liver kinase B1 |
| LPO | lipid peroxidation |
| LPS | Lipopolysaccharide |
| LSECs | liver sinusoidal endothelial cells |
| MAGEs | MGO-derived advanced glycation end products |
| MAPKs | mitogen-activated protein kinases |
| MASLD | metabolic dysfunction-associated fatty liver disease |
| MCP-1 | monocyte chemoattractant protein 1 |
| MDA | Malondialdehyde |
| MetS | metabolic syndrome |
| MG-dG | 3-(20–deoxyribosyl)-6,7-dihydro-6,7-dihydroxy-6/7-methylimidazo-[2,3-b]purin-9(8)one |
| MG-H1 | Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine |
| MG-H2 | 2-amino-5-(2-amino-5-hydro-5-methyl-4- imidazolon-1-yl)-pentanoic acid |
| MG-H3 | 2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)-pentanoic acid |
| MGO | Methylglyoxal |
| MKK7 | mitogen-activated protein kinase kinase 7 |
| NEFAs | non-esterified fatty acids |
| NF-κB | nuclear factor-kB |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PARP | poly(ADP-ribose) polymerase |
| PRRs | pattern recognition receptors |
| PUFAs | polyunsaturated fatty acids |
| p38 MAPK | p38 mitogen-activated protein kinase |
| RAGE | advanced glycation end products receptor |
| RCS | reactive carbonyl species |
| RCT | randomized controlled trial |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SCFAs | short chain fatty acids |
| SMAD3 | a protein involved in TGF-β signal transduction |
| SOD | superoxide dismutase |
| SREBP | sterol regulatory element-binding protein |
| TAGs | Triacylglycerols |
| TAK1 | TGF-β-activated kinase 1 |
| TBARS | thiobarbituric acid reactive substances |
| TC | total cholesterol |
| TCA | tricarboxylic acid cycle (Krebs cycle) |
| T2DM | type 2 diabetes mellitus |
| TfR1 | transferrin receptor 1 |
| TGF-β | transforming growth factor β |
| THP | Tetrahydropyrimidine |
| TNFα | tumor necrosis factor alfa |
| THR-β | thyroid hormone receptor β |
| Trx | Thioredoxin |
| WR | Wistar rats |
| Glossary | |
| Term | Definition |
| AMPK | AMP-activated protein kinase is an energy sensor which regulates metabolism, switching the processes between catabolic and anabolic depending on the energetic status of the cell. AMPK requires threonine (Thr172) phosphorylation for its activation, which is achieved by three different kinases (liver kinase B1—LKB1, Ca21/calmodulin-dependent protein kinase kinase β—CaMKKβ, and TGF-β-activated kinase 1—TAK1) induced by various signals [48]. AMPK is sustained in this active phosphorylated formed at low energy level by AMP (and ADP) binding, whereas higher ATP concentration inactivates the enzyme. Therefore, at low energy levels reflected by high AMP/ATP ratio, AMPK is active and regulates specific target enzymes, increasing lipid oxidation and mitochondrial biogenesis, whereas the synthesis of lipids and glycogen is inhibited. In such a way, energy-consuming anabolic pathways are attenuated in favor of induced catabolic pathways aimed at the replenishment of energy. One of the important targets of AMPK is acetyl-CoA carboxylase (ACC) which produces malonyl-CoA. Malonyl-CoA is a substrate for palmitic acid synthesis, but also it is an inhibitor of carnitine palmitoyl-transferase 1 (CPT1)—an enzyme involved in the transport of long-chain fatty acids to the mitochondrium for β-oxidation. AMPK phosphorylates and inhibits ACC which leads to a decrease in malonyl-CoA, thus attenuating palmitic acid synthesis. Simultaneously, a drop in malonyl-CoA level releases the inhibition of CPT1 which enables entry of FAs to the mitochondrium for β-oxidation. Consequently, the synthesis of fatty acids (DNL) in the liver decreases, whereas mitochondrial β-oxidation of FAs increases [48,49]. Additionally, AMPK phosphorylates and inhibits transcription factors (sterol regulatory element-binding proteins, SREBPs) responsible for the expression of enzymes involved in FAs, triacylglycerol, and cholesterol synthesis. |
| Apoptosis | Apoptosis is a type of regulated cell death necessary for the clearance of destroyed cells to maintain tissues in healthy condition. Damaged cells undergo shrinking and membrane blebbing, followed by the removal of macrophages. Apoptosis can be induced by internal or external signals. The intrinsic (mitochondrial) pathway is initiated by receptor-independent signals generated in the cell, such as ROS accumulation. The major regulator detecting DNA damages and deciding on the further cell’s fate is the tumor suppressor p53 protein. This protein controls Bcl-2 family proteins. Bcl-2 and Bcl-XL belong to pro-survival factors, whereas Bax or Bak stimulate apoptotic events. In response to strong proapoptotic signals (e.g., resulting from the accumulation of unrepairable genetic defects), they increase the permeability of the inner mitochondrial membrane and release of proapoptotic factors, including cytochrome C. Extrinsic pathways are induced by death receptors which activate caspase cascade starting from caspase 8. Both intrinsic and extrinsic pathways converge on the execution phase initiated by the activation of caspase 3 [245]. |
| Autophagy | Autophagy (“self-eating”) is a conservative, pro-survival process employed by normal cells to recycle the wastes (defective or unnecessary intracellular structures and macromolecules). It is divided into macroautophagy, selective autophagy, microautophagy, and chaperone-mediated autophagy, from which macroautophagy—the most common and best studied type is usually referred to as simply autophagy [77]. Macroautophagy proceeds with the formation of an autophagosome (a sequestration of a fragment of cytoplasm by surrounding its contents with double membranes) which next fuses with a lysosome yielding autophagolysosome. Lysosomal hydrolases degrade enclosed molecules generating products which are exported back into the cytoplasm where they are reused for the synthesis of new molecules, such as amino acids for novel protein formation. Degradation products also serve as a source of energy (e.g., fatty acids). Autophagy functions constitutively at a low level, but in conditions of nutrients and/or oxygen deprivation as well as ER stress it intensifies to increase the survival odds in stressful conditions. Signaling pathways regulating these events include mammalian targets of rapamycin (mTOR), as well as AMPK routes. mTOR being active at sufficient nutrient satiation suppresses the induction of autophagy, whereas, during starvation, inflowing signals block the mTOR route, which triggers the autodigestion mechanism. In turn, AMPK being the sensor of intracellular energy level, becomes activated at ATP depletion, therefore it induces autophagy during energy exhaustion [246,247]. |
| De novo lipogenesis (DNL) | De novo lipogenesis is the process of converting excess dietary carbohydrates (mainly Glc and Fru) into fatty acids. FAs are produced from acetyl-CoA molecules generated in carbohydrate catabolism (in glycolysis from Glc and fructolysis from Fru) or acetate generated by microbiota Fru fermentation [36,37]. |
| Ferroptosis | Ferroptosis is a type of non-apoptotic regulated cell death stimulated by iron-dependent lipid peroxidation and characterized by cell swelling and plasma membrane rupture. It is promoted under conditions of a high concentration of free iron, ROS, and membrane phospholipids containing polyunsaturated fatty acids (PUFAs). Free iron in the presence of superoxide radicals leads to the generation of hydroxyl radicals which stimulates lipid (PUFAs) peroxidation. This causes degradation of biological membranes and cell death. Therefore, the upregulation of factors which increase iron and ROS accumulation promotes ferroptosis. They include proteins involved in iron transport and generation (e.g., transferrin receptor or heme oxygenase—HO), as well as superoxide production (like NADPH oxidases—NOXs). Ferroptosis can also be promoted by selective autophagy such as ferritinophagy and lipophagy which are associated with the release of iron cations and fatty acids, respectively. In turn, the main mechanism restraining ferroptosis includes glutathione peroxidase 4 (GPX4)—a selenium-dependent antioxidative enzyme which reduces phospholipid hydroperoxides, as well as reduced glutathione—a co-substrate in GPX4 reaction. The sufficient level of GSH in the cell is conditioned by the influx of its precursor—cystine. Therefore, the transporter (xc- antiporter) involved in cystine transport to the cells plays an important function here. Another component protecting from ferroptosis is a apoptosis-inducing factor for mitochondria associated 2A (AIFM2/FSP1) protein, which seems to act in two ways. Firstly, having an oxidoreductase activity, AIFM2 reduces coenzyme Q10 to its ubiquinol form, which generates a hydrophobic antioxidant. Secondly, it protects from membrane injury through activating the endosomal sorting complexes required for transport (ESCRT)-III–dependent membrane repair in the plasma membrane [47]. |
| Lipophagy | Lipophagy is a type of selective autophagy controlling lipid metabolism. It proceeds through the surrounding of lipid droplets by a double membrane (autophagosome) which further fuses with the lysosome. Subsequently, lysosomal lipases hydrolyze lipid contents releasing their products, e.g., fatty acids. Fatty acids are directed to the mitochondrium for β-oxidation [248,249]. |
| Mitophagy | Mitophagy is a type of selective autophagy responsible for a steady-state turnover of mitochondria as well as the clearance of injured mitochondria. Damage to mitochondria (reflected by the loss of the mitochondrial membrane potential), induces Parkin (E3 ubiquitin ligase) translocation to mitochondria. Parkin ubiquitinates outer membrane proteins which induce mitophagy. Subsequently, the mitochondrium become surrounded by a double membrane (autophagosome) and fuse with the lysosome whose hydrolases degrade mitochonrium components. Parkin recruitment is mediated by PINK1 [247]. |
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 2024, 81, 492–542. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Gofton, C.; Upendran, Y.; Zheng, M.H.; George, J. MAFLD: How is it different from NAFLD? Clin. Mol. Hepatol. 2023, 29, S17–S31. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Yang, P.; Ye, J.; Xu, Q.; Wu, J.; Wang, Y. Updated mechanisms of MASLD pathogenesis. Lipids Health Dis. 2024, 23, 117. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 2022, 2, 977485. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, L.; Yan, X.; Zhao, Y.; Wang, J.; Liu, B.; Yu, S.; Fu, J.; Liu, Y.; Su, J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23, 9252. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Crewe, C.; Scherer, P.E. Intercellular and interorgan crosstalk through adipocyte extracellular vesicles. Rev. Endocr. Metab. Disord. 2022, 23, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Lange, P.; Lombardi, A.; Silvestri, E.; Cioffi, F.; Giacco, A.; Iervolino, S.; Petito, G.; Senese, R.; Lanni, A.; Moreno, M. Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence. Int. J. Mol. Sci. 2023, 24, 11676. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nath, B.; Szabo, G. Hypoxia and hypoxia inducible factors: Diverse roles in liver diseases. Hepatology 2012, 55, 622–633. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible transcription factor 2α promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, X.; de Carvalho Ribeiro, M.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-Specific Hypoxia-Inducible Factor-1α Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 545–563. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van Eyck, A.; Kwanten, W.J.; Peleman, C.; Makhout, S.; Van Laere, S.; Van De Maele, K.; Van Hoorenbeeck, K.; De Man, J.; De Winter, B.Y.; Francque, S.; et al. The role of adipose tissue and subsequent liver tissue hypoxia in obesity and early stage metabolic dysfunction associated steatotic liver disease. Int. J. Obes. 2024, 48, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Wamutu, S.; Chatterjee, S.; Van Marck, E.; Herman, A.; Ramon, A.; Jung, A.; Vermeulen, W.; De Winter, B.; Pelckmans, P.; et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010, 30, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Laleman, W.; Verbeke, L.; Van Steenkiste, C.; Casteleyn, C.; Kwanten, W.; Van Dyck, C.; D'Hondt, M.; Ramon, A.; Vermeulen, W.; et al. Increased intrahepatic resistance in severe steatosis: Endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab. Investig. 2012, 92, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaff, D.; Kwanten, W.J.; Francque, S.M. The potential role of vascular alterations and subsequent impaired liver blood flow and hepatic hypoxia in the pathophysiology of non-alcoholic steatohepatitis. Med. Hypotheses 2019, 122, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Rood, J.; Ravussin, E.; Schwarz, J.M.; Smith, S.R.; Redman, L.M. Reduced oxygenation in human obese adipose tissue is associated with impaired insulin suppression of lipolysis. J. Clin. Endocrinol. Metab. 2010, 95, 4052–4055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, K.; Halberg, N.; Khan, M.; Magalang, U.J.; Scherer, P.E. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell Biol. 2013, 33, 904–917. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ji, J.; Wu, L.; Wei, J.; Wu, J.; Guo, C. The Gut Microbiome and Ferroptosis in MAFLD. J. Clin. Transl. Hepatol. 2023, 11, 174–187. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jayachandran, M.; Qu, S. Non-alcoholic fatty liver disease and gut microbial dysbiosis- underlying mechanisms and gut microbiota mediated treatment strategies. Rev. Endocr. Metab. Disord. 2023, 24, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Benedé-Ubieto, R.; Cubero, F.J.; Nevzorova, Y.A. Breaking the barriers: The role of gut homeostasis in Metabolic-Associated Steatotic Liver Disease (MASLD). Gut Microbes 2024, 16, 2331460. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wu, Z.; Wang, W.; Wei, L.; Zhou, H. A revisit of drugs and potential therapeutic targets against non-alcoholic fatty liver disease: Learning from clinical trials. J. Endocrinol. Investig. 2024, 47, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Kounatidis, D.; Psallida, S.; Vythoulkas-Biotis, N.; Adamou, A.; Zachariadou, T.; Kargioti, S.; Karampela, I.; Dalamaga, M. NAFLD/MASLD and the Gut-Liver Axis: From Pathogenesis to Treatment Options. Metabolites 2024, 14, 366. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Puetz, A.; Kappel, B.A. Gut Microbiome in Dyslipidemia and Atherosclerosis. In Gut Microbiome, Microbial Metabolites and Cardiometabolic Risk; Springer International Publishing: Cham, Switzerland, 2023; pp. 1–29. [Google Scholar] [CrossRef]
- Al Samarraie, A.; Pichette, M.; Rousseau, G. Role of the Gut Microbiome in the Development of Atherosclerotic Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 5420. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bellucci, E.; Chiereghin, F.; Pacifici, F.; Donadel, G.; De Stefano, A.; Malatesta, G.; Valente, M.G.; Guadagni, F.; Infante, M.; Rovella, V.; et al. Novel therapeutic approaches based on the pathological role of gut dysbiosis on the link between nonalcoholic fatty liver disease and insulin resistance. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 1921–1944. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.P.; Karunakar, P.; Taraphder, S.; Yadav, H. Free Fatty Acid Receptors 2 and 3 as Microbial Metabolite Sensors to Shape Host Health: Pharmacophysiological View. Biomedicines 2020, 8, 154. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Farooqui, N.; Elhence, A.; Shalimar, A. Current Understanding of Bile Acids in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2022, 12, 155–173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huneault, H.E.; Ramirez Tovar, A.; Sanchez-Torres, C.; Welsh, J.A.; Vos, M.B. The Impact and Burden of Dietary Sugars on the Liver. Hepatol. Commun. 2023, 7, e0297. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Geidl-Flueck, B.; Gerber, P.A. Fructose drives de novo lipogenesis affecting metabolic health. J. Endocrinol. 2023, 257, e220270. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salavrakos, M.; de Timary, P.; Ruiz Moreno, A.; Thissen, J.P.; Lanthier, N. Fructoholism in adults: The importance of personalised care in metabolic dysfunction-associated fatty liver disease. JHEP Rep. 2021, 4, 100396. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ribeiro, A.; Igual-Perez, M.J.; Santos Silva, E.; Sokal, E.M. Childhood Fructoholism and Fructoholic Liver Disease. Hepatol. Commun. 2018, 3, 44–51. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lustig, R.H. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Priebs, J.; Landmann, M.; Degen, C.; Engstler, A.J.; Jin, C.J.; Gärttner, S.; Spruss, A.; Huber, O.; Bergheim, I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 2015, 26, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shen, X.; Yu, Z.; Wei, C.; Hu, C.; Chen, J. Iron metabolism and ferroptosis in nonalcoholic fatty liver disease: What is our next step? Am. J. Physiol. Endocrinol. Metab. 2024, 326, E767–E775. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Wei, Y.; Zhang, M.; Yang, S.; Tong, R.; Li, W.; Long, E. Metabolic dysfunction-associated steatotic liver disease: Ferroptosis related mechanisms and potential drugs. Front. Pharmacol. 2023, 14, 1286449. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peleman, C.; Francque, S.; Berghe, T.V. Emerging role of ferroptosis in metabolic dysfunction-associated steatotic liver disease: Revisiting hepatic lipid peroxidation. eBioMedicine 2024, 102, 105088. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yao, C.; Lan, D.; Li, X.; Wang, Y.; Qi, S.; Liu, Y. Porphyromonas gingivalis is a risk factor for the development of nonalcoholic fatty liver disease via ferroptosis. Microbes Infect. 2023, 25, 105040. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, P.; Saltiel, A.R. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J. Biol. Chem. 2020, 295, 12279–12289. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Von Loeffelholz, C.; Coldewey, S.M.; Birkenfeld, A.L. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells 2021, 10, 1822. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.I.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, P.; Zhao, J.; Guo, Y.; Yu, Y.; Wu, X.; Xiao, H. Ursodeoxycholic acid alleviates nonalcoholic fatty liver disease by inhibiting apoptosis and improving autophagy via activating AMPK. Biochem. Biophys. Res. Commun. 2020, 529, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.K.; Kwon, S.W.; Han, D.H.; Lee, Y.H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 16, 1949–1973. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Leung, C.; Herath, C.B.; Jia, Z.; Andrikopoulos, S.; Brown, B.E.; Davies, M.J.; Rivera, L.R.; Furness, J.B.; Forbes, J.M.; Angus, P.W. Dietary advanced glycation end-products aggravate non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8026–8040. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakasai-Sakai, A.; Takeda, K.; Takeuchi, M. Involvement of Intracellular TAGE and the TAGE-RAGE-ROS Axis in the Onset and Progression of NAFLD/NASH. Antioxidants 2023, 12, 748. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Jin, Z.; Wang, X.; Jakoš, T.; Zhu, J.; Yuan, Y. RAGE pathways play an important role in regulation of organ fibrosis. Life Sci. 2023, 323, 121713. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.G.; Xia, J.R.; Li, W.D.; Lu, F.L.; Liu, J.; Lu, Q.; Zhi, H. Anti-fibrotic effects of specific-siRNA targeting of the receptor for advanced glycation end products in a rat model of experimental hepatic fibrosis. Mol. Med. Rep. 2014, 10, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Deng, Q.; Gao, J.; Yu, X.; Zhang, Y.; Li, J.; Guan, W.; Hu, J.; Tan, Q.; Zhou, L.; et al. Therapeutic effects of antigen affinity-purified polyclonal anti-receptor of advanced glycation end-product (RAGE) antibodies on cholestasis-induced liver injury in rats. Eur. J. Pharmacol. 2016, 779, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Dehnad, A.; Fan, W.; Jiang, J.X.; Fish, S.R.; Li, Y.; Das, S.; Mozes, G.; Wong, K.A.; Olson, K.A.; Charville, G.W.; et al. AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J. Clin. Investig. 2020, 130, 4320–4330. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Asadipooya, K.; Lankarani, K.B.; Raj, R.; Kalantarhormozi, M. RAGE is a Potential Cause of Onset and Progression of Nonalcoholic Fatty Liver Disease. Int. J. Endocrinol. 2019, 2019, 2151302. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lindén, D.; Romeo, S. Therapeutic opportunities for the treatment of NASH with genetically validated targets. J. Hepatol. 2023, 79, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Silva, M.S.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef]
- Morgenstern, J.; Campos, M.C.; Nawroth, P.; Fleming, T. The Glyoxalase System-New Insights into an Ancient Metabolism. Antioxidants 2020, 9, 939. [Google Scholar] [CrossRef]
- Phillips, S.A.; Mirrlees, D.; Thornalley, P.J. Modification of the glyoxalase system in streptozotocin-induced diabetic rats. Effect of the aldose reductase inhibitor statil. Biochem. Pharmacol. 1993, 46, 805–811. [Google Scholar] [CrossRef]
- Berdowska, I.; Matusiewicz, M.; Fecka, I. Methylglyoxal in Cardiometabolic Disorders: Routes Leading to Pathology Counterbalanced by Treatment Strategies. Molecules 2023, 28, 7742. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA 2018, 115, 9228–9233. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 357, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mazza, M.C.; Shuck, S.C.; Lin, J.; Moxley, M.A.; Termini, J.; Cookson, M.R.; Wilson, M.A. DJ-1 is not a deglycase and makes a modest contribution to cellular defense against methylglyoxal damage in neurons. J. Neurochem. 2022, 162, 245–261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richarme, G.; Abdallah, J.; Mathas, N.; Gautier, V.; Dairou, J. Further characterization of the Maillard deglycase DJ-1 and its prokaryotic homologs, deglycase 1/Hsp31, deglycase 2/YhbO, and deglycase 3/YajL. Biochem. Biophys. Res. Commun. 2018, 503, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Richarme, G.; Dairou, J. Parkinsonism-associated protein DJ-1 is a bona fide deglycase. Biochem. Biophys. Res. Commun. 2017, 483, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Bekkhozhin, Z.; Omertassova, N.; Baizhumanov, T.; Yeltay, G.; Akhmetali, M.; Toibazar, D.; Utepbergenov, D. The apparent deglycase activity of DJ-1 results from the conversion of free methylglyoxal present in fast equilibrium with hemithioacetals and hemiaminals. J. Biol. Chem. 2019, 294, 18863–18872. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Castillo, C.; Shuck, S.C. Diet and Obesity-Induced Methylglyoxal Production and Links to Metabolic Disease. Chem. Res. Toxicol. 2021, 34, 2424–2440. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. Where does plasma methylglyoxal originate from? Diabetes Res. Clin. Pract. 2013, 99, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Com-pensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahmed, N.; Argirov, O.K.; Minhas, H.S.; Cordeiro, C.A.; Thornalley, P.J. Assay of advanced glycation endproducts (AGEs): Surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nepsilon-carboxymethyl-lysine- and Nepsilon-(1-carboxyethyl)lysine-modified albumin. Biochem. J. 2002, 364, 1–14. [Google Scholar] [CrossRef]
- Klöpfer, A.; Spanneberg, R.; Glomb, M.A. Formation of arginine modifications in a model system of Nα-tert-butoxycarbonyl (Boc)-arginine with methylglyoxal. J. Agric. Food Chem. 2011, 59, 394–401. [Google Scholar] [CrossRef]
- Ahmed, N.; Dobler, D.; Dean, M.; Thornalley, P.J. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 2005, 280, 5724–5732. [Google Scholar] [CrossRef]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef]
- Lieuw-a-Fa, M.L.; Schalkwijk, C.G.; Engelse, M.; van Hinsbergh, V.W. Interaction of Nepsilon(carboxymethyl)lysine- and methylglyoxal-modified albumin with endothelial cells and macrophages. Splice variants of RAGE may limit the responsiveness of human endothelial cells to AGEs. Thromb. Haemost. 2006, 95, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, J.; Yang, N.; Huang, Y.; Hu, T.; Rao, C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022, 13, 1051. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619–639. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gugliucci, A. Fructose surges damage hepatic adenosyl-monophosphate-dependent kinase and lead to increased lipogenesis and] hepatic insulin resistance. Med. Hypotheses 2016, 93, 87–92. [Google Scholar] [CrossRef]
- Wang, W.C.; Chou, C.K.; Chuang, M.C.; Li, Y.C.; Lee, J.A. Elevated levels of liver methylglyoxal and d-lactate in early-stage hepatitis in rats. Biomed. Chromatogr. 2018, 32, e4039. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Moran, G.; Estrada, A.; Pagliassotti, M.J. Fructose-induced stress signaling in the liver involves methylglyoxal. Nutr. Metab. 2013, 10, 32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, Y.; Pagliassotti, M.J. Hepatospecific effects of fructose on c-jun NH2-terminal kinase: Implications for hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E926–E933. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Whitmarsh, A.J.; Cavanagh, J.; Barrett, T.; Davis, R.J. Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 7337–7342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neves, C.; Rodrigues, T.; Sereno, J.; Simões, C.; Castelhano, J.; Gonçalves, J.; Bento, G.; Gonçalves, S.; Seiça, R.; Domingues, M.R.; et al. Dietary Glycotoxins Impair Hepatic Lipidemic Profile in Diet-Induced Obese Rats Causing Hepatic Oxidative Stress and Insulin Resistance. Oxid. Med. Cell Longev. 2019, 2019, 6362910. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peter, A.; Schleicher, E.; Kliemank, E.; Szendroedi, J.; Königsrainer, A.; Häring, H.U.; Nawroth, P.P.; Fleming, T. Accumulation of Non-Pathological Liver Fat Is Associated with the Loss of Glyoxalase I Activity in Humans. Metabolites 2024, 14, 209. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hüttl, M.; Markova, I.; Miklánková, D.; Zapletalova, I.; Kujal, P.; Šilhavý, J.; Pravenec, M.; Malinska, H. Hypolipidemic and insulin sensitizing effects of salsalate beyond suppressing inflammation in a prediabetic rat model. Front. Pharmacol. 2023, 14, 1117683. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Malinská, H.; Hüttl, M.; Miklánková, D.; Trnovská, J.; Zapletalová, I.; Poruba, M.; Marková, I. Ovariectomy-Induced Hepatic Lipid and Cytochrome P450 Dysmetabolism Precedes Serum Dyslipidemia. Int. J. Mol. Sci. 2021, 22, 4527. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, Y.; Wang, P.; Sang, S. Dietary Genistein Inhibits Methylglyoxal-Induced Advanced Glycation End Product Formation in Mice Fed a High-Fat Diet. J. Nutr. 2019, 149, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Spanos, C.; Maldonado, E.M.; Fisher, C.P.; Leenutaphong, P.; Oviedo-Orta, E.; Windridge, D.; Salguero, F.J.; Bermúdez-Fajardo, A.; Weeks, M.E.; Evans, C.; et al. Proteomic identification and characterization of hepatic glyoxalase 1 dysregulation in non-alcoholic fatty liver disease. Proteome Sci. 2018, 16, 4, Erratum in Proteome Sci. 2018, 16, 13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Depner, C.M.; Traber, M.G.; Bobe, G.; Kensicki, E.; Bohren, K.M.; Milne, G.; Jump, D.B. A metabolomic analysis of omega-3 fatty acid-mediated attenuation of western diet-induced nonalcoholic steatohepatitis in LDLR-/-mice. PLoS ONE 2013, 8, e83756. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Michel, M.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS ONE 2017, 12, e0171260. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Loarca, L.; Sassi-Gaha, S.; Artlett, C.M. Two α-dicarbonyls downregulate migration, invasion, and adhesion of liver cancer cells in a p53-dependent manner. Dig. Liver Dis. 2013, 45, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, X.; He, Q.; Chen, Q.; Yu, L. Glyoxalase 1 is up-regulated in hepatocellular carcinoma and is essential for HCC cell proliferation. Biotechnol. Lett. 2014, 36, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Hollenbach, M.; Pohl, S.; Ripoll, C.; Zipprich, A. Inhibition of Glyoxalase-I Leads to Reduced Proliferation, Migration and Colony Formation, and Enhanced Susceptibility to Sorafenib in Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 785. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Michel, M.; Hess, C.; Kaps, L.; Kremer, W.M.; Hilscher, M.; Galle, P.R.; Moehler, M.; Schattenberg, J.M.; Wörns, M.A.; Labenz, C.; et al. Elevated serum levels of methylglyoxal are associated with impaired liver function in patients with liver cirrhosis. Sci. Rep. 2021, 11, 20506. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hollenbach, M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products. Int. J. Mol. Sci. 2019, 20, 5037. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef]
- Zemva, J.; Fink, C.A.; Fleming, T.H.; Schmidt, L.; Loft, A.; Herzig, S.; Knieß, R.A.; Mayer, M.; Bukau, B.; Nawroth, P.P.; et al. Hormesis enables cells to handle accumulating toxic metabolites during increased energy flux. Redox Biol. 2017, 13, 674–686. [Google Scholar] [CrossRef]
- Ravichandran, M.; Priebe, S.; Grigolon, G.; Rozanov, L.; Groth, M.; Laube, B.; Guthke, R.; Platzer, M.; Zarse, K.; Ristow, M. Impairing L-Threonine Catabolism Promotes Healthspan through Methylglyoxal-Mediated Proteohormesis. Cell Metab. 2018, 27, 914–925.e5. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, X.; Zheng, X.; Huang, H.; Chen, X.; Wu, K.; Wang, B.; Ma, S. Glo1 genetic amplification as a potential therapeutic target in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2079–2090. [Google Scholar] [PubMed] [PubMed Central]
- Liu, Y.; Zhu, X.; Zhu, J.; Liao, S.; Tang, Q.; Liu, K.; Guan, X.; Zhang, J.; Feng, Z. Identification of differential expression of genes in hepatocellular carcinoma by suppression subtractive hybridization combined cDNA microarray. Oncol. Rep. 2007, 18, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Taïbi, N.; Al-Balas, Q.A.; Bekari, N.; Talhi, O.; Al Jabal, G.A.; Benali, Y.; Ameraoui, R.; Hadjadj, M.; Taïbi, A.; Boutaiba, Z.M.; et al. Molecular docking and dynamic studies of a potential therapeutic target inhibiting glyoxalase system: Metabolic action of the 3, 3’-[3-(5-chloro-2-hydroxyphenyl)-3-oxopropane-1, 1-diyl]-Bis-4-hydroxycoumarin leads overexpression of the intracellular level of methylglyoxal and induction of a pro-apoptotic phenomenon in a hepatocellular carcinoma model. Chem. Biol. Interact. 2021, 345, 109511. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bellier, J.; Nokin, M.J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mortera, R.R.; Bains, Y.; Gugliucci, A. Fructose at the crossroads of the metabolic syndrome and obesity epidemics. Front. Biosci. Landmark 2019, 24, 186–211. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Lanaspa, M.A.; Sanchez-Lozada, L.G.; Tolan, D.; Nakagawa, T.; Ishimoto, T.; Andres-Hernando, A.; Rodriguez-Iturbe, B.; Stenvinkel, P. The fructose survival hypothesis for obesity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2023, 378, 20220230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Barinova, K.V.; Serebryakova, M.V.; Melnikova, A.K.; Medvedeva, M.V.; Muronetz, V.I.; Schmalhausen, E.V. Mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in the presence of methylglyoxal. Arch. Biochem. Biophys. 2023, 733, 109485. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.I.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD. Nutrients 2017, 9, 634. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, M.; Suzuki, H.; Takeda, K.; Sakai-Sakasai, A. Toxic advanced glycation end-products (TAGE) are major structures of cytotoxic AGEs derived from glyceraldehyde. Med. Hypotheses 2024, 183, 111248. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakai-Sakasai, A.; Takeda, K.; Suzuki, H.; Takeuchi, M. Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite. Biomolecules 2024, 14, 202. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Petagine, L.; Zariwala, M.G.; Patel, V.B. Alcoholic liver disease: Current insights into cellular mechanisms. World J. Biol. Chem. 2021, 12, 87–103. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef] [PubMed]
- Kalavalapalli, S.; Leiva, E.G.; Lomonaco, R.; Chi, X.; Shrestha, S.; Dillard, R.; Budd, J.; Romero, J.P.; Li, C.; Bril, F.; et al. Adipose Tissue Insulin Resistance Predicts the Severity of Liver Fibrosis in Patients With Type 2 Diabetes and NAFLD. J. Clin. Endocrinol. Metab. 2023, 108, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qiu, K.; Zheng, W.; Kong, W.; Zeng, T. Uric acid may serve as the sixth cardiometabolic criterion for defining MASLD. J. Hepatol. 2024, 80, e152–e153. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Yang, H.; Jeon, S.; Cho, K.W.; Kim, S.J.; Kim, S.; Lee, M.; Suh, J.; Chae, H.W.; Kim, H.S.; et al. Prediction of insulin resistance and elevated liver transaminases using serum uric acid and derived markers in children and adolescents. Eur. J. Clin. Nutr. 2024, 78, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Bousvarou, M.D.; Kostara, C.E.; Papakonstantinou, E.J.; Salamou, E.; Guzman, E. Insulin resistance and cardiovascular disease. J. Int. Med. Res. 2023, 51, 3000605231164548. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, S.; Zhang, Q.; Qin, B. Association between remnant cholesterol and insulin resistance levels in patients with metabolic-associated fatty liver disease. Sci. Rep. 2024, 14, 4596. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Genua, I.; Cusi, K. Pharmacological Approaches to Nonalcoholic Fatty Liver Disease: Current and Future Therapies. Diabetes Spectr. 2024, 37, 48–58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Paulino, P.J.I.V.; Cuthrell, K.M.; Tzenios, N. Non Alcoholic Fatty Liver Disease; Disease Burden, Management, and Future Perspectives. Int. Res. J. Gastroenterol. Hepatol. 2024, 7, 1–13. Available online: http://archive.pcbmb.org/id/eprint/1800 (accessed on 7 January 2025).
- Shinozaki, S.; Tahara, T.; Miura, K.; Lefor, A.K.; Yamamoto, H. Pemafibrate therapy for non-alcoholic fatty liver disease is more effective in lean patients than obese patients. Clin. Exp. Hepatol. 2022, 8, 278–283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, S.; Zhao, J.; Xie, F.; He, H.; Johnston, L.J.; Dai, X.; Wu, C.; Ma, X. Dietary fiber-derived short-chain fatty acids: A potential therapeutic target to alleviate obesity-related nonalcoholic fatty liver disease. Obes. Rev. 2021, 22, e13316. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Scott, A. An Update on the Pharmacological Treatment of Nonalcoholic Fatty Liver Disease: Beyond Lifestyle Modifications. Clin. Liver Dis. 2018, 11, 82–86. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, J.; Li, B.; Zhang, K.; Zhu, Z. The effect and safety of obeticholic acid for patients with nonalcoholic steatohepatitis: A systematic review and meta-analysis of randomized controlled trials. Medicine 2024, 103, e37271. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. MAESTRO-NASH Investigators. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Taub, R.; Neff, G.W.; Lucas, K.J.; Labriola, D.; Moussa, S.E.; Alkhouri, N.; Bashir, M.R. Resmetirom for nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2023, 29, 2919–2928. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wong, V.W.-S.; Abdelmalek, M.F.; Rodriguez-Araujo, G.; Landgren, H.; Park, G.S.; Bedossa, P.; Alkhouri, N.; Tacke, F.; et al. Cenicriviroc Lacked Efficacy to Treat Liver Fibrosis in Nonalcoholic Steatohepatitis: AURORA Phase III Randomized Study. Clin. Gastroenterol. Hepatol. 2024, 22, 124–134.e1. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Elafibranor: First Approval. Drugs 2024, 84, 1143–1148, Erratum in Drugs 2024, 84, 1165. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Targher, G.; Romeo, S.; Pajvani, U.B.; Zheng, M.H.; Aghemo, A.; Valenti, L.V.C. The first MASH drug therapy on the horizon: Current perspectives of resmetirom. Liver Int. 2024, 44, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Jophlin, L.L.; Singal, A.K.; Bataller, R.; Wong, R.J.; Sauer, B.G.; Terrault, N.A.; Shah, V.H. ACG Clinical Guideline: Alcohol-Associated Liver Disease. Am. J. Gastroenterol. 2024, 119, 30–54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 770–786. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Chuah, K.H.; Rajaram, R.B.; Lim, L.L.; Ratnasingam, J.; Vethakkan, S.R. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): A State-of-the-Art Review. J. Obes. Metab. Syndr. 2023, 32, 197–213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hansen, C.D.; Gram-Kampmann, E.M.; Hansen, J.K.; Hugger, M.B.; Madsen, B.S.; Jensen, J.M.; Olesen, S.; Torp, N.; Rasmussen, D.N.; Kjærgaard, M.; et al. Effect of Calorie-Unrestricted Low-Carbohydrate, High-Fat Diet Versus High-Carbohydrate, Low-Fat Diet on Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial. Ann. Intern. Med. 2023, 176, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Kim, S.Y.; Lim, Y.W.; Choi, K.H.; Shin, I.S. Comparative Effectiveness of Cognitive Behavioral Therapy and Behavioral Therapy in Obesity: A Systematic Review and Network Meta-Analysis. J. Clin. Psychol. Med. Settings 2024, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, O.E.; Alalalmeh, S.O.; Shahwan, M.; Jairoun, A.A.; Alourfi, M.M.; Bokhari, G.A.; Alkhattabi, A.; Alsharif, S.; Aljehani, M.A.; Alsabban, A.M.; et al. Exploring Promising Therapies for Non-Alcoholic Fatty Liver Disease: A ClinicalTrials.gov Analysis. Diabetes Metab. Syndr. Obes. 2024, 17, 545–561. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gastaldelli, A.; Cusi, K.; Landó, L.F.; Bray, R.; Brouwers, B.; Rodríguez, Á. Effect of tirzepatide versus insulin degludec on liver fat content and abdominal adipose tissue in people with type 2 diabetes (SURPASS-3 MRI): A substudy of the randomised, open-label, parallel-group, phase 3 SURPASS-3 trial. Lancet Diabetes Endocrinol. 2022, 10, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Landó, L.F.; Bray, R.; Brouwers, B.; Rodríguez, Á.; Menzen, M. Effect of Tirzepatide Versus Insulin Degludec on Liver Fat Content and Abdominal Adipose Tissue in Patients with Type 2 Diabetes (SURPASS-3 MRI). Diabetol. Stoffwechs. 2023, 18, S15–S16. [Google Scholar] [CrossRef]
- Koh, B.; Xiao, J.; Ng, C.H.; Law, M.; Gunalan, S.Z.; Danpanichkul, P.; Ramadoss, V.; Sim, B.K.L.; Tan, E.Y.; Teo, C.B.; et al. Comparative efficacy of pharmacologic therapies for MASH in reducing liver fat content: Systematic review and network meta-analysis. Hepatology 2024. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, X.; Yan, C.; Li, C.; Zhang, L.; Zhang, L.; Liang, E.; Liu, T.; Mao, J. Effect of metformin on nonalcoholic fatty liver based on meta-analysis and network pharmacology. Medicine 2022, 101, e31437. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mansi, I.A.; Chansard, M.; Lingvay, I.; Zhang, S.; Halm, E.A.; Alvarez, C.A. Association of Statin Therapy Initiation with Diabetes Progression: A Retrospective Matched-Cohort Study. JAMA Intern. Med. 2021, 181, 1562–1574. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mansi, I.A.; Sumithran, P.; Kinaan, M. Risk of diabetes with statins. BMJ 2023, 381, e071727. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, F.; Lamendola, C.; Harris, C.S.; Harris, V.; Tsai, M.S.; Tripathi, P.; Abbas, F.; Reaven, G.M.; Reaven, P.D.; Snyder, M.P.; et al. Statins Are Associated with Increased Insulin Resistance and Secretion. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2786–2797. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martín, C. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 4725. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ling, Z.; Shu, N.; Xu, P.; Wang, F.; Zhong, Z.; Sun, B.; Li, F.; Zhang, M.; Zhao, K.; Tang, X.; et al. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem. Pharmacol. 2016, 100, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Kheong, C.W.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949.e8. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.J.; Belle, S.H.; D’Amato, M.; Adfhal, N.; Brunt, E.M.; Fried, M.W.; Reddy, K.R.; Wahed, A.S.; Harrison, S. Silymarin in NASH and C Hepatitis (SyNCH) Study Group. Silymarin in non-cirrhotics with non-alcoholic steatohepatitis: A randomized, double-blind, placebo controlled trial. PLoS ONE 2019, 14, e0221683, Erratum in PLoS ONE 2019, 14, e0223915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ceriello, A. Hypothesis: The “metabolic memory”, the new challenge of diabetes. Diabetes Res. Clin. Pract. 2009, 86, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Dobbie, L.J.; Burgess, J.; Hamid, A.; Nevitt, S.J.; Hydes, T.J.; Alam, U.; Cuthbertson, D.J. Effect of a Low-Calorie Dietary Intervention on Liver Health and Body Weight in Adults with Metabolic-Dysfunction Associated Steatotic Liver Disease (MASLD) and Overweight/Obesity: A Systematic Review and Meta-Analysis. Nutrients 2024, 16, 1030. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, Y.; Sun, Y.; Sun, L.; Liu, X.; Zeng, R.; Lin, X.; Li, Y. Effects of gut microbiota and fatty acid metabolism on dyslipidemia following weight-loss diets in women: Results from a randomized controlled trial. Clin. Nutr. 2021, 40, 5511–5520. [Google Scholar] [CrossRef] [PubMed]
- Seethaler, B.; Nguyen, N.K.; Basrai, M.; Kiechle, M.; Walter, J.; Delzenne, N.M.; Bischoff, S.C. Short-chain fatty acids are key mediators of the favorable effects of the Mediterranean diet on intestinal barrier integrity: Data from the randomized controlled LIBRE trial. Am. J. Clin. Nutr. 2022, 116, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Losasso, C.; Eckert, E.M.; Mastrorilli, E.; Villiger, J.; Mancin, M.; Patuzzi, I.; Di Cesare, A.; Cibin, V.; Barrucci, F.; Pernthaler, J.; et al. Assessing the Influence of Vegan, Vegetarian and Omnivore Oriented Westernized Dietary Styles on Human Gut Microbiota: A Cross Sectional Study. Front. Microbiol. 2018, 9, 317. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Landry, M.J.; Ward, C.P.; Cunanan, K.M.; Durand, L.R.; Perelman, D.; Robinson, J.L.; Hennings, T.; Koh, L.; Dant, C.; Zeitlin, A.; et al. Cardiometabolic Effects of Omnivorous vs Vegan Diets in Identical Twins: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2344457, Erratum in JAMA Netw. Open 2023, 6, e2350422. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid. Med. Cell Longev. 2017, 2017, 3831972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, C.; Li, A.; Xu, C.; Ma, J.; Wang, H.; Jiang, Z.; Hou, J. Comparative Analysis of Fecal Microbiota in Vegetarians and Omnivores. Nutrients 2023, 15, 2358. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prochazkova, M.; Budinska, E.; Kuzma, M.; Pelantova, H.; Hradecky, J.; Heczkova, M.; Daskova, N.; Bratova, M.; Modos, I.; Videnska, P.; et al. Vegan Diet Is Associated with Favorable Effects on the Metabolic Performance of Intestinal Microbiota: A Cross-Sectional Multi-Omics Study. Front. Nutr. 2022, 8, 783302. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, M.; Suzuki, H.; Sakai-Sakasai, A. Major generation route of cytotoxic protein adducts derived from acetaldehyde, a metabolite of alcohol. Med. Hypotheses 2024, 189, 111385. [Google Scholar] [CrossRef]
- Hayashi, T.; Shibamoto, T. Analysis of methyl glyoxal in foods and beverages. J. Agric. Food Chem. 1985, 33, 1090–1093. Available online: https://pubs.acs.org/doi/pdf/10.1021/jf00066a018 (accessed on 7 January 2025). [CrossRef]
- Hopper, D.J.; Cooper, R.A. The regulation of Escherichia coli methylglyoxal synthase; a new control site in glycolysis? FEBS Lett. 1971, 13, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Booth, I.R.; Ferguson, G.P.; Miller, S.; Li, C.; Gunasekera, B.; Kinghorn, S. Bacterial production of methylglyoxal: A survival strategy or death by misadventure? Biochem. Soc. Trans. 2003, 31 Pt 6, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, S.; Rajan, D.P.; Balasubramanian, K.A. Formation of methylglyoxal by bacteria isolated from human faeces. J. Med. Microbiol. 1989, 28, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Chandrangsu, P.; Dusi, R.; Hamilton, C.J.; Helmann, J.D. Methylglyoxal resistance in Bacillus subtilis: Contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 2014, 91, 706–715. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Srebreva, L.N.; Stoynev, G.A.; Ivanov, I.G. Evidence for excretion of glycation agents from E. coli cells during growth. Biotechnol. Biotechnol. Equip. 2009, 23, 1068–1071. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, T.L. Dietary advanced glycation end-products elicit toxicological effects by disrupting gut microbiome and immune homeostasis. J. Immunotoxicol. 2021, 18, 93–104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yuan, X.; Liu, J.; Nie, C.; Ma, Q.; Wang, C.; Liu, H.; Chen, Z.; Zhang, M.; Li, J. Comparative Study of the Effects of Dietary-Free and -Bound Nε-Carboxymethyllysine on Gut Microbiota and Intestinal Barrier. J. Agric. Food Chem. 2024, 72, 5014–5025. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Skalny, A.V.; Gritsenko, V.A.; Kartashova, O.L.; Santamaria, A.; Rocha, J.B.T.; Spandidos, D.A.; Zaitseva, I.P.; Tsatsakis, A.; Tinkov, A.A. Role of gut microbiota in the modulation of the health effects of advanced glycation end-products (Review). Int. J. Mol. Med. 2023, 51, 44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xie, F.; Zhao, J.; Liu, D.; Wan, Z.; Sun, K.; Wang, Y. Associations of dietary advanced glycation end products with liver steatosis via vibration controlled transient elastography in the United States: A nationwide cross-sectional study. Eur. J. Nutr. 2024, 63, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin Scavenges Methylglyoxal to form a Novel Imidazolinone Metabolite in Humans. Chem. Res. Toxicol. 2016, 29, 227–234. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shao, X.; Chen, H.; Zhu, Y.; Sedighi, R.; Ho, C.T.; Sang, S. Essential Structural Requirements and Additive Effects for Flavonoids to Scavenge Methylglyoxal. J. Agric. Food Chem. 2014, 62, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhan, L.; Wen, Q.; Feng, Y.; Luo, Y.; Tan, T. Trapping Methylglyoxal by Taxifolin and Its Metabolites in Mice. J. Agric. Food Chem. 2022, 70, 5026–5038. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.N.I.; Mitsuhashi, S.; Sigetomi, K.; Ubukata, M. Quercetin inhibits advanced glycation end product formation via chelating metal ions, trapping methylglyoxal, and trapping reactive oxygen species. Biosci. Biotechnol. Biochem. 2017, 81, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, C.; Zhang, B.; Huang, Q. The inhibitory effects of flavonoids on α-amylase and α-glucosidase. Crit. Rev. Food Sci. Nutr. 2020, 60, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Proença, C.; Ribeiro, D.; Freitas, M.; Fernandes, E. Flavonoids as potential agents in the management of type 2 diabetes through the modulation of α-amylase and α-glucosidase activity: A review. Crit. Rev. Food Sci. Nutr. 2022, 62, 3137–3207. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ferruzzi, M.G.; Hamaker, B.R. Structural requirements of flavonoids for the selective inhibition of α-amylase versus α-glucosidase. Food Chem. 2022, 370, 130981. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhang, Q.; Zhang, C.; Yang, W.; Liu, H.; Lv, Z.; Liu, J.; Jiao, Z. Inhibition of Dipeptidyl Peptidase-4 by Flavonoids: Structure-Activity Relationship, Kinetics and Interaction Mechanism. Front. Nutr. 2022, 9, 892426. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, S.; Wang, X.; Cheng, Y.; Gao, H.; Chen, X. A Review of Classification, Biosynthesis, Biological Activities and Potential Applications of Flavonoids. Molecules 2023, 28, 4982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, M.; Lu, Y.; Wu, Q.; Chen, G.; Zhao, H.; Ho, C.T.; Li, S. Biotransformation and Gut Microbiota-Mediated Bioactivity of Flavonols. J. Agric. Food Chem. 2023, 71, 8317–8331. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kurita, A.; Nakashima, S.; Zhu, B.; Munemasa, S.; Nakamura, T.; Murata, Y.; Nakamura, Y. 3,4-Dihydroxyphenylacetic acid is a potential aldehyde dehydrogenase inducer in murine hepatoma Hepa1c1c7 cells. Biosci. Biotechnol. Biochem. 2017, 81, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Fecka, I.; Bednarska, K.; Kowalczyk, A. In Vitro Antiglycation and Methylglyoxal Trapping Effect of Peppermint Leaf (Mentha × piperita L.) and Its Polyphenols. Molecules 2023, 28, 2865. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramos, F.M.M.; Ribeiro, C.B.; Cesar, T.B.; Milenkovic, D.; Cabral, L.; Noronha, M.F.; Sivieri, K. Lemon flavonoids nutraceutical (Eriomin®) attenuates prediabetes intestinal dysbiosis: A double-blind randomized controlled trial. Food Sci. Nutr. 2023, 11, 7283–7295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Malik, A.; Malik, M.; Qureshi, S. Effects of silymarin use on liver enzymes and metabolic factors in metabolic dysfunction-associated steatotic liver disease: A systematic review and meta-analysis. Can. Liver J. 2024, 7, 40–53. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, S.; Duan, F.; Li, S.; Lu, B. Administration of silymarin in NAFLD/NASH: A systematic review and meta-analysis. Ann. Hepatol. 2024, 29, 101174. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhi, M.; Gao, X.; Hu, P.; Li, C.; Yang, X. Effect and the probable mechanisms of silibinin in regulating insulin resistance in the liver of rats with non-alcoholic fatty liver. Braz. J. Med. Biol. Res. 2013, 46, 270–277. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Velussi, M.; Cernigoi, A.M.; De Monte, A.; Dapas, F.; Caffau, C.; Zilli, M. Long-term (12 months) treatment with an anti-oxidant drug (silymarin) is effective on hyperinsulinemia, exogenous insulin need and malondialdehyde levels in cirrhotic diabetic patients. J. Hepatol. 1997, 26, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fan, Y.; Yan, Q.; Fan, X.; Wu, B.; Han, Y.; Zhang, Y.; Chen, Y.; Zhang, H.; Niu, J. The therapeutic effect of silymarin in the treatment of nonalcoholic fatty disease: A meta-analysis (PRISMA) of randomized control trials. Medicine 2017, 96, e9061. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jin, Y.; Wang, X.; Chen, K.; Chen, Y.; Zhou, L.; Zeng, Y.; Zhou, Y.; Pan, Z.; Wang, D.; Li, Z.; et al. Silymarin decreases liver stiffness associated with gut microbiota in patients with metabolic dysfunction-associated steatotic liver disease: A randomized, double-blind, placebo-controlled trial. Lipids Health Dis. 2024, 23, 239. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yi, M.; Manzoor, M.; Yang, M.; Zhang, H.; Wang, L.; Zhao, L.; Xiang, L.; Qi, J. Silymarin targets the FXR protein through microbial metabolite 7-keto-deoxycholic acid to treat MASLD in obese mice. Phytomedicine 2024, 133, 155947. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ji, K.; Du, F.; Jin, N.; Boesch, C.; Farag, M.A.; Li, H.; Liu, X.; Xiao, J. Does Flavonoid Supplementation Alleviate Non-Alcoholic Fatty Liver Disease? A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Mol. Nutr. Food Res. 2023, 67, e2300480. [Google Scholar] [CrossRef] [PubMed]
- Hüttl, M.; Markova, I.; Miklankova, D.; Zapletalova, I.; Poruba, M.; Racova, Z.; Vecera, R.; Malinska, H. The Beneficial Additive Effect of Silymarin in Metformin Therapy of Liver Steatosis in a Pre-Diabetic Model. Pharmaceutics 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, Y.M.; Lee, W.K.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. Metformin Restores Parkin-Mediated Mitophagy, Suppressed by Cytosolic p53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, P.; Lin, H.; Xu, Y.; Zhou, F.; Wang, J.; Liu, J.; Zhu, X.; Guo, X.; Tang, Y.; Yao, P. Frataxin-Mediated PINK1-Parkin-Dependent Mitophagy in Hepatic Steatosis: The Protective Effects of Quercetin. Mol. Nutr. Food Res. 2018, 62, e1800164. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Wang, Y.; Zhang, C.; Sullivan, M.A.; Chen, W.; Jing, X.; Yu, H.; Li, F.; Wang, Q.; Zhou, Z.; et al. Quercetin ameliorates nonalcoholic fatty liver disease (NAFLD) via the promotion of AMPK-mediated hepatic mitophagy. J. Nutr. Biochem. 2023, 120, 109414. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. Inhibiting Pink1/Parkin-mediated mitophagy enhances the anticancer effects of quercetin in hepatocellular carcinomaf. Biochem. Biophys. Res. Commun. 2024, 712–713, 149899. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Barbagallo, I.; Godos, J.; Lembo, V.; Currenti, W.; Cinà, D.; Avola, R.; D'Orazio, N.; Morisco, F.; Galvano, F.; et al. Silibinin Restores NAD+ Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver. Nutrients 2017, 9, 1086. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Ren, L.; Song, G.; Zhang, P.; Yang, L.; Chen, X.; Yu, X.; Chen, S. Silibinin Ameliorates Fructose-induced Lipid Accumulation and Activates Autophagy in HepG2 Cells. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 10, 89. [Google Scholar] [CrossRef]
- Guo, Q.; Mori, T.; Jiang, Y.; Hu, C.; Osaki, Y.; Yoneki, Y.; Sun, Y.; Hosoya, T.; Kawamata, A.; Ogawa, S.; et al. Methylglyoxal contributes to the development of insulin resistance and salt sensitivity in Sprague-Dawley rats. J. Hypertens. 2009, 27, 1664–1671. [Google Scholar] [CrossRef]
- Dhar, A.; Desai, K.M.; Wu, L. Alagebrium attenuates acute methylglyoxal-induced glucose intolerance in Sprague-Dawley rats. Br. J. Pharmacol. 2010, 159, 166–175. [Google Scholar] [CrossRef]
- Nigro, C.; Raciti, G.A.; Leone, A.; Fleming, T.H.; Longo, M.; Prevenzano, I.; Fiory, F.; Mirra, P.; D’Esposito, V.; Ulianich, L.; et al. Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia 2014, 57, 1485–1494. [Google Scholar] [CrossRef]
- Nigro, C.; Leone, A.; Raciti, G.A.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int. J. Mol. Sci. 2017, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Shamsaldeen, Y.A.; Mackenzie, L.S.; Lione, L.A.; Benham, C.D. Methylglyoxal, A Metabolite Increased in Diabetes is Associated with Insulin Resistance, Vascular Dysfunction and Neuropathies. Curr. Drug Metab. 2016, 17, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Mey, J.T.; Haus, J.M. Dicarbonyl Stress and Glyoxalase-1 in Skeletal Muscle: Implications for Insulin Resistance and Type 2 Diabetes. Front. Cardiovasc. Med. 2018, 5, 117. [Google Scholar] [CrossRef]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef]
- Engelbrecht, B.; Mattern, Y.; Scheibler, S.; Tschoepe, D.; Gawlowski, T.; Stratmann, B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm. Metab. Res. 2014, 46, 77–84. [Google Scholar] [CrossRef]
- Jia, X.; Wu, L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Jiang, B.; Desai, K.M.; Wu, L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes 2011, 60, 899–908. [Google Scholar] [CrossRef]
- Matafome, P.; Santos-Silva, D.; Crisóstomo, J.; Rodrigues, T.; Rodrigues, L.; Sena, C.M.; Pereira, P.; Seiça, R. Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch. Physiol. Biochem. 2012, 118, 58–68. [Google Scholar] [CrossRef]
- Hüttl, M.; Markova, I.; Miklankova, D.; Makovicky, P.; Pelikanova, T.; Šeda, O.; Šedová, L.; Malinska, H. Adverse Effects of Methylglyoxal on Transcriptome and Metabolic Changes in Visceral Adipose Tissue in a Prediabetic Rat Model. Antioxidants 2020, 9, 803. [Google Scholar] [CrossRef]
- Rodrigues, T.; Matafome, P.; Sereno, J.; Almeida, J.; Castelhano, J.; Gamas, L.; Neves, C.; Gonçalves, S.; Carvalho, C.; Arslanagic, A.; et al. Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci. Rep. 2017, 7, 1698. [Google Scholar] [CrossRef]
- Jia, X.; Olson, D.J.; Ross, A.R.; Wu, L. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 2006, 20, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- Fiory, F.; Lombardi, A.; Miele, C.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 2012, 54, 2941–2952, Erratum in: Diabetologia 2012, 55, 272. [Google Scholar] [CrossRef]
- Bo, J.; Xie, S.; Guo, Y.; Zhang, C.; Guan, Y.; Li, C.; Lu, J.; Meng, Q.H. Methylglyoxal Impairs Insulin Secretion of Pancreatic β-Cells through Increased Production of ROS and Mitochondrial Dysfunction Mediated by Upregulation of UCP2 and MAPKs. J. Diabetes Res. 2016, 2016, 2029854. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Q.; Lin, Y.; Liang, G.; Xiao, N.; Zhang, H.; Yang, X.; Yang, J.; Liu, A. Autophagy and Senescence: The Molecular Mechanisms and Implications in Liver Diseases. Int. J. Mol. Sci. 2023, 24, 16880. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shin, D.W. Lipophagy: Molecular Mechanisms and Implications in Metabolic Disorders. Mol. Cells 2020, 43, 686–693. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
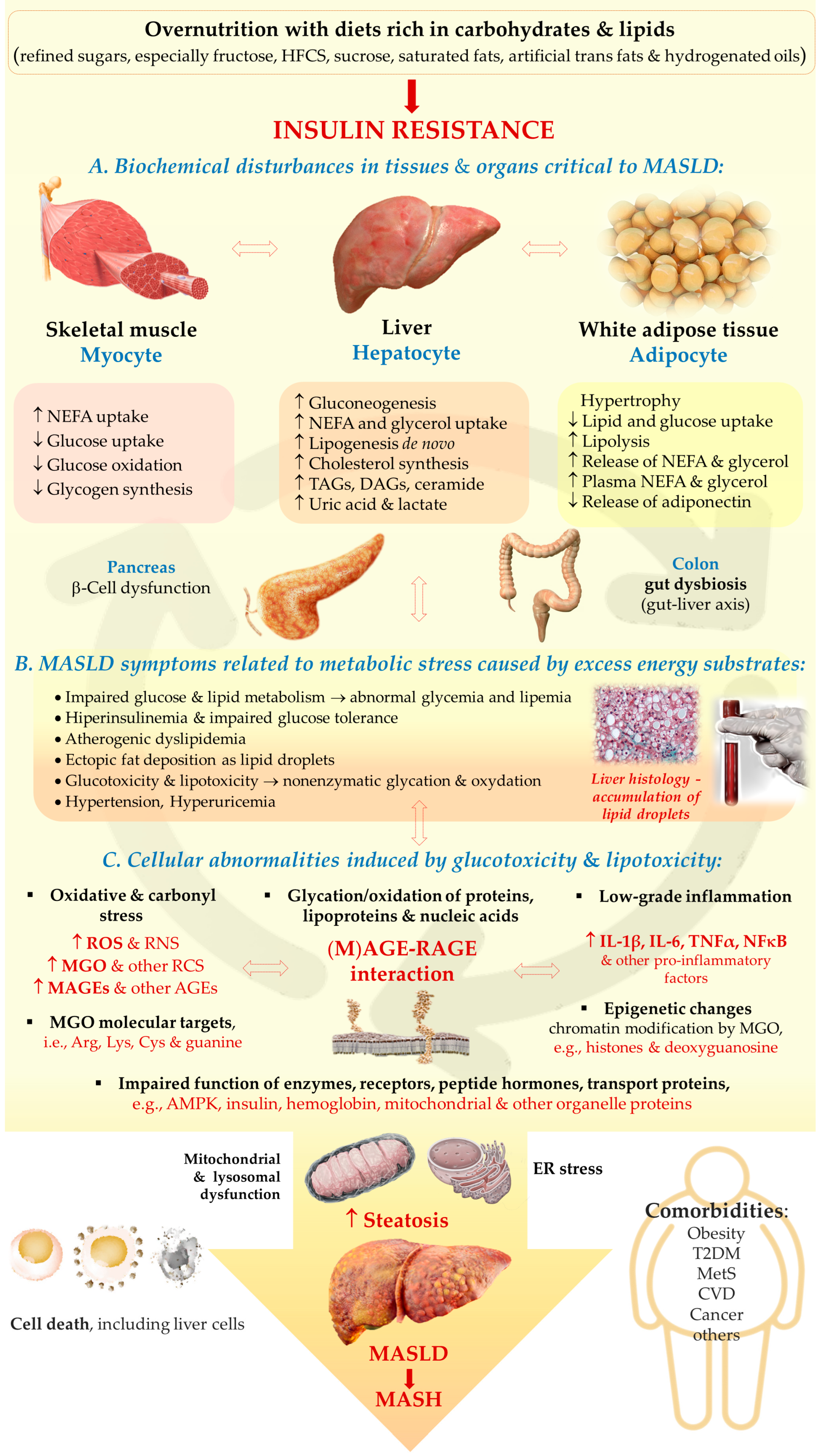
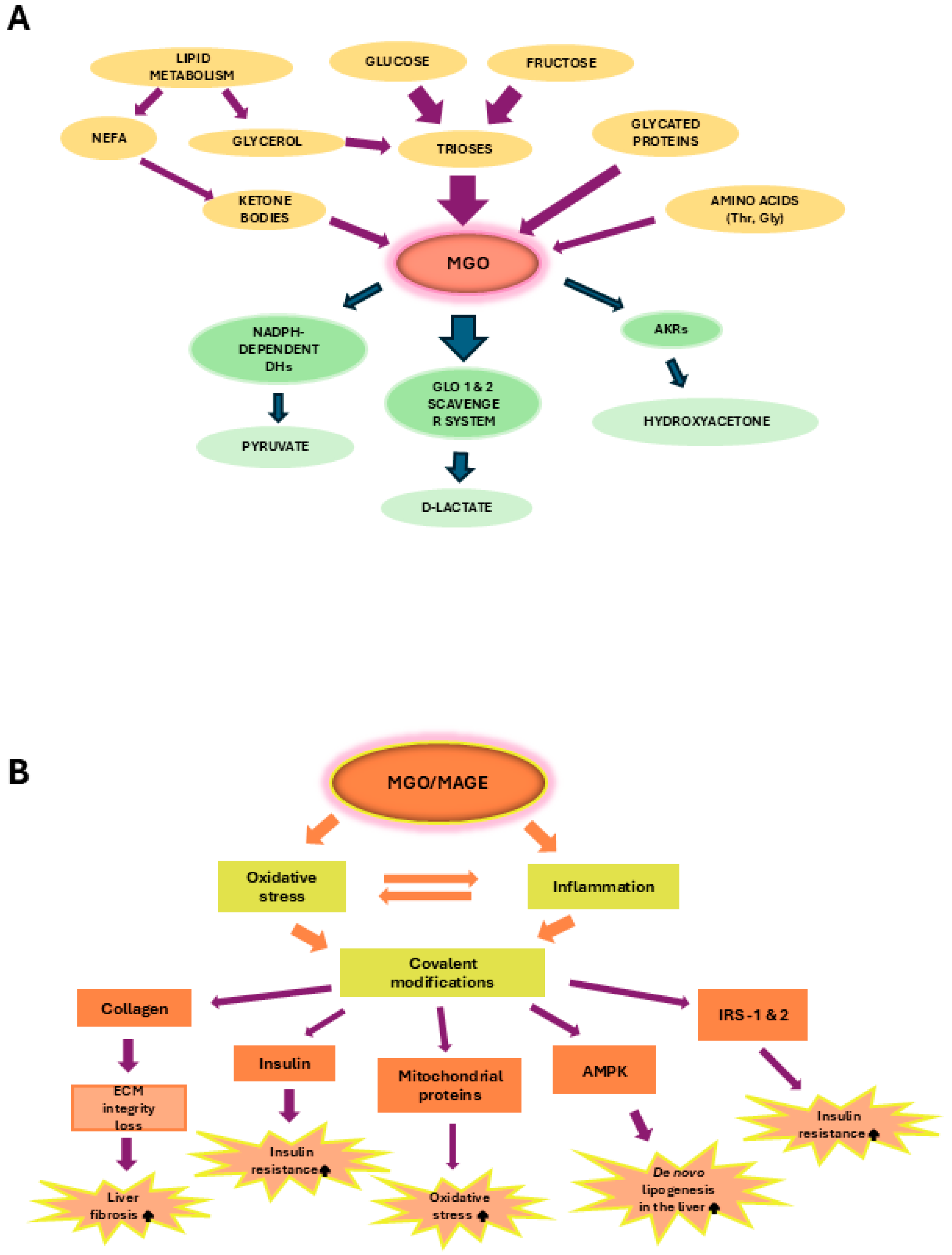


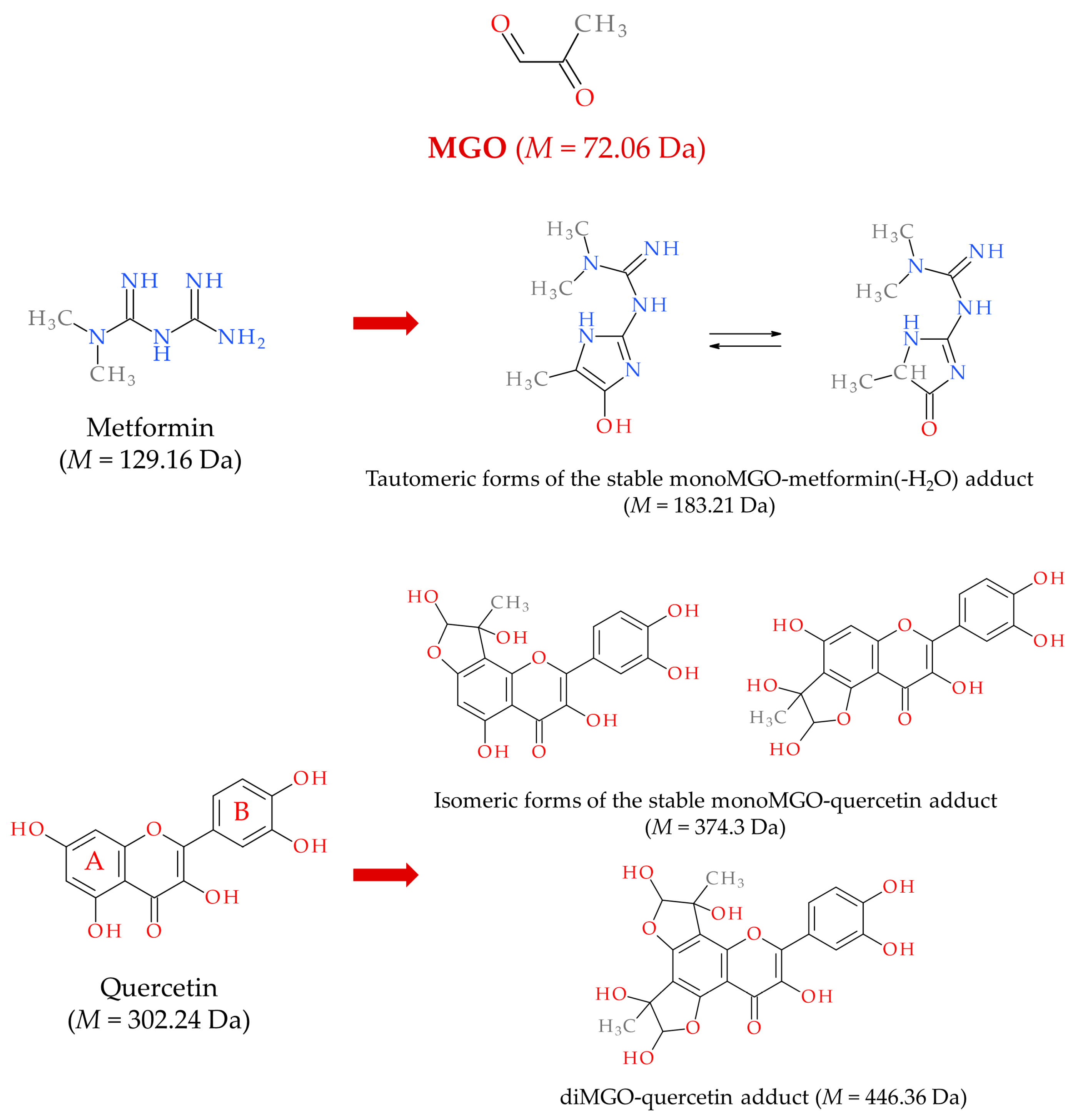
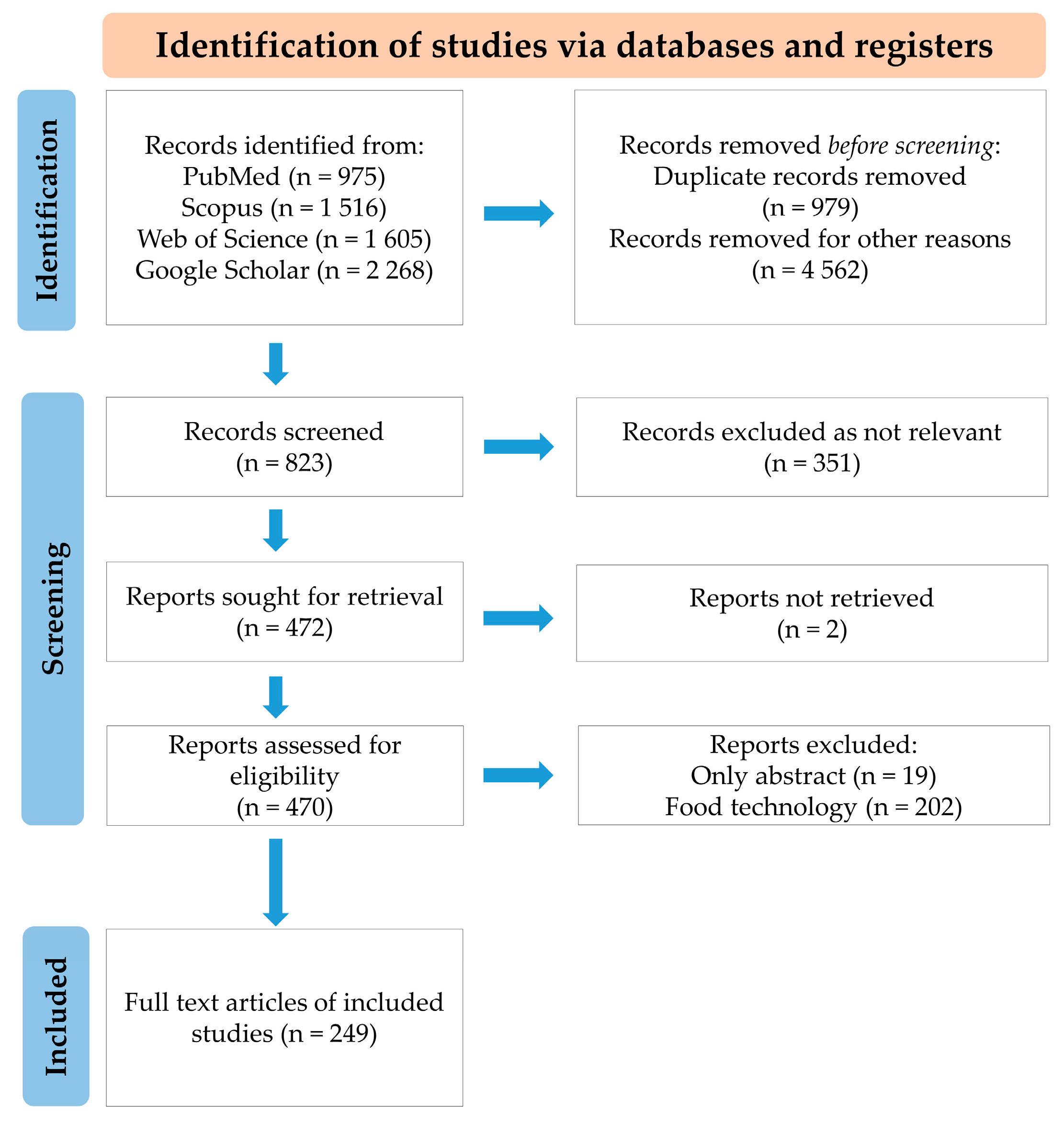
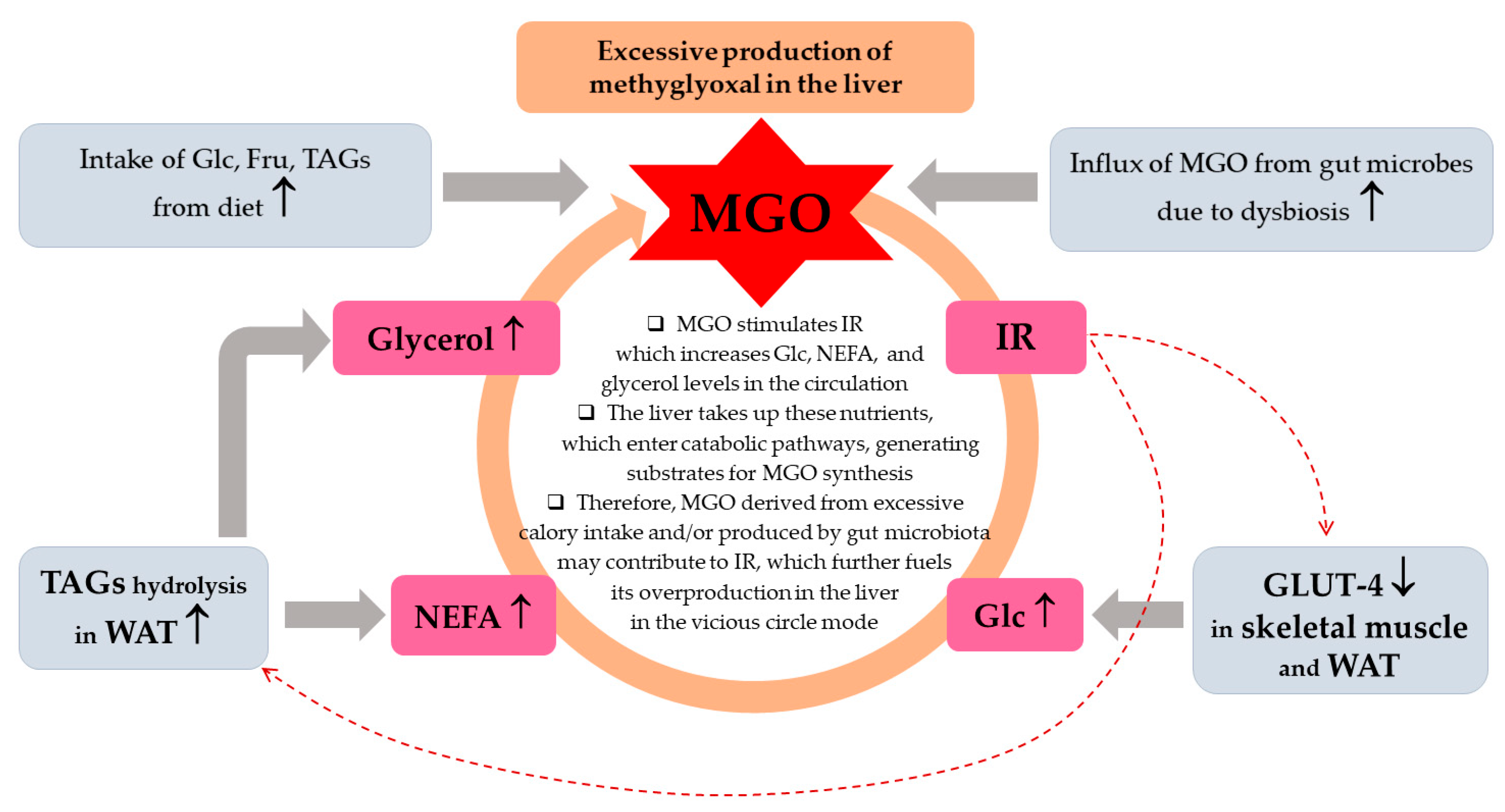
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdowska, I.; Matusiewicz, M.; Fecka, I. A Comprehensive Review of Metabolic Dysfunction-Associated Steatotic Liver Disease: Its Mechanistic Development Focusing on Methylglyoxal and Counterbalancing Treatment Strategies. Int. J. Mol. Sci. 2025, 26, 2394. https://doi.org/10.3390/ijms26062394
Berdowska I, Matusiewicz M, Fecka I. A Comprehensive Review of Metabolic Dysfunction-Associated Steatotic Liver Disease: Its Mechanistic Development Focusing on Methylglyoxal and Counterbalancing Treatment Strategies. International Journal of Molecular Sciences. 2025; 26(6):2394. https://doi.org/10.3390/ijms26062394
Chicago/Turabian StyleBerdowska, Izabela, Małgorzata Matusiewicz, and Izabela Fecka. 2025. "A Comprehensive Review of Metabolic Dysfunction-Associated Steatotic Liver Disease: Its Mechanistic Development Focusing on Methylglyoxal and Counterbalancing Treatment Strategies" International Journal of Molecular Sciences 26, no. 6: 2394. https://doi.org/10.3390/ijms26062394
APA StyleBerdowska, I., Matusiewicz, M., & Fecka, I. (2025). A Comprehensive Review of Metabolic Dysfunction-Associated Steatotic Liver Disease: Its Mechanistic Development Focusing on Methylglyoxal and Counterbalancing Treatment Strategies. International Journal of Molecular Sciences, 26(6), 2394. https://doi.org/10.3390/ijms26062394