
Exploring Metabolic Pathways and Gene Mining During Cotton Flower Bud Differentiation Stages Based on Transcriptomics and Metabolomics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
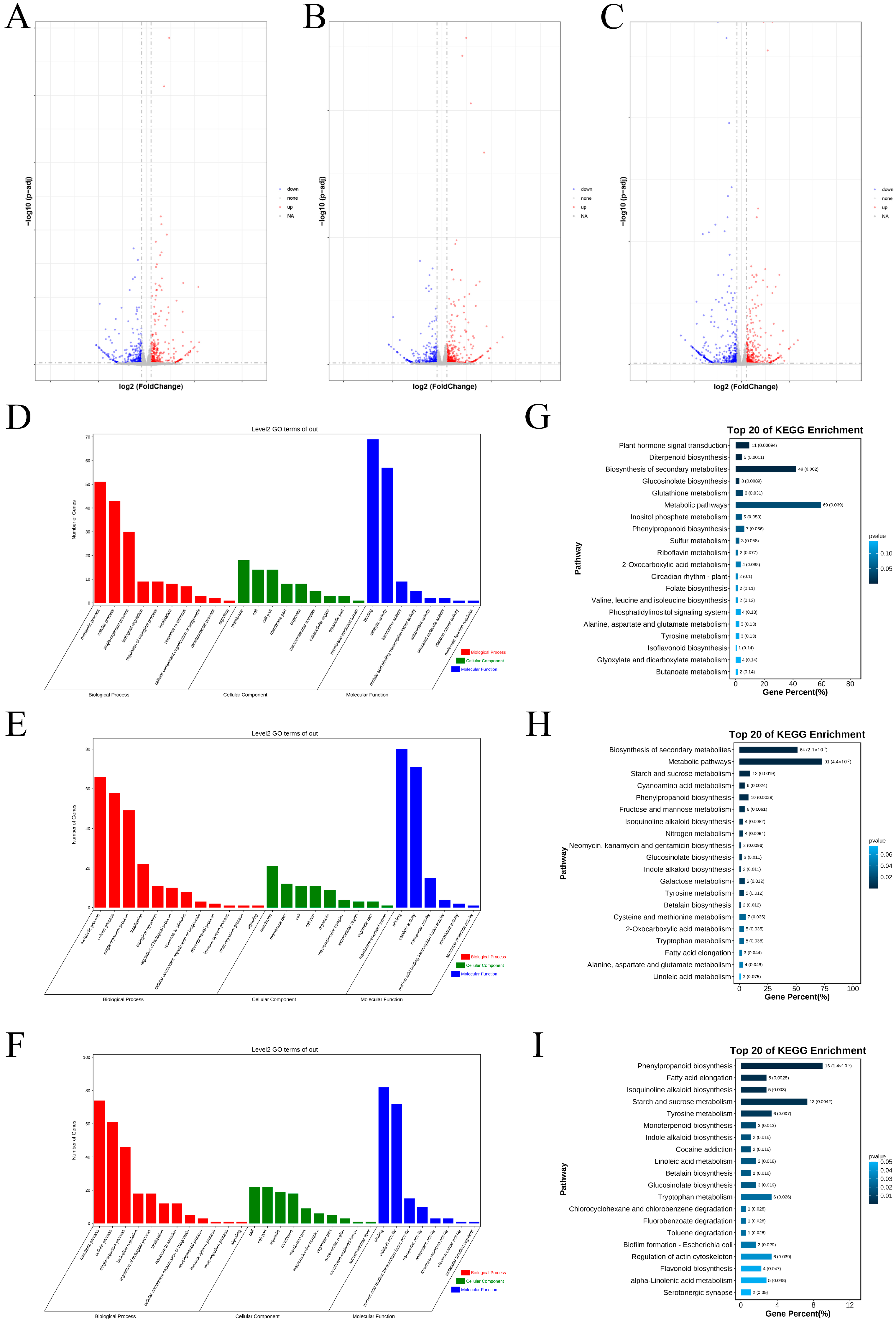
2.1. Identification and Enrichment Analysis of the DEGs Between Two Varieties
2.2. Verification of DEGs Based on qRT-PCR
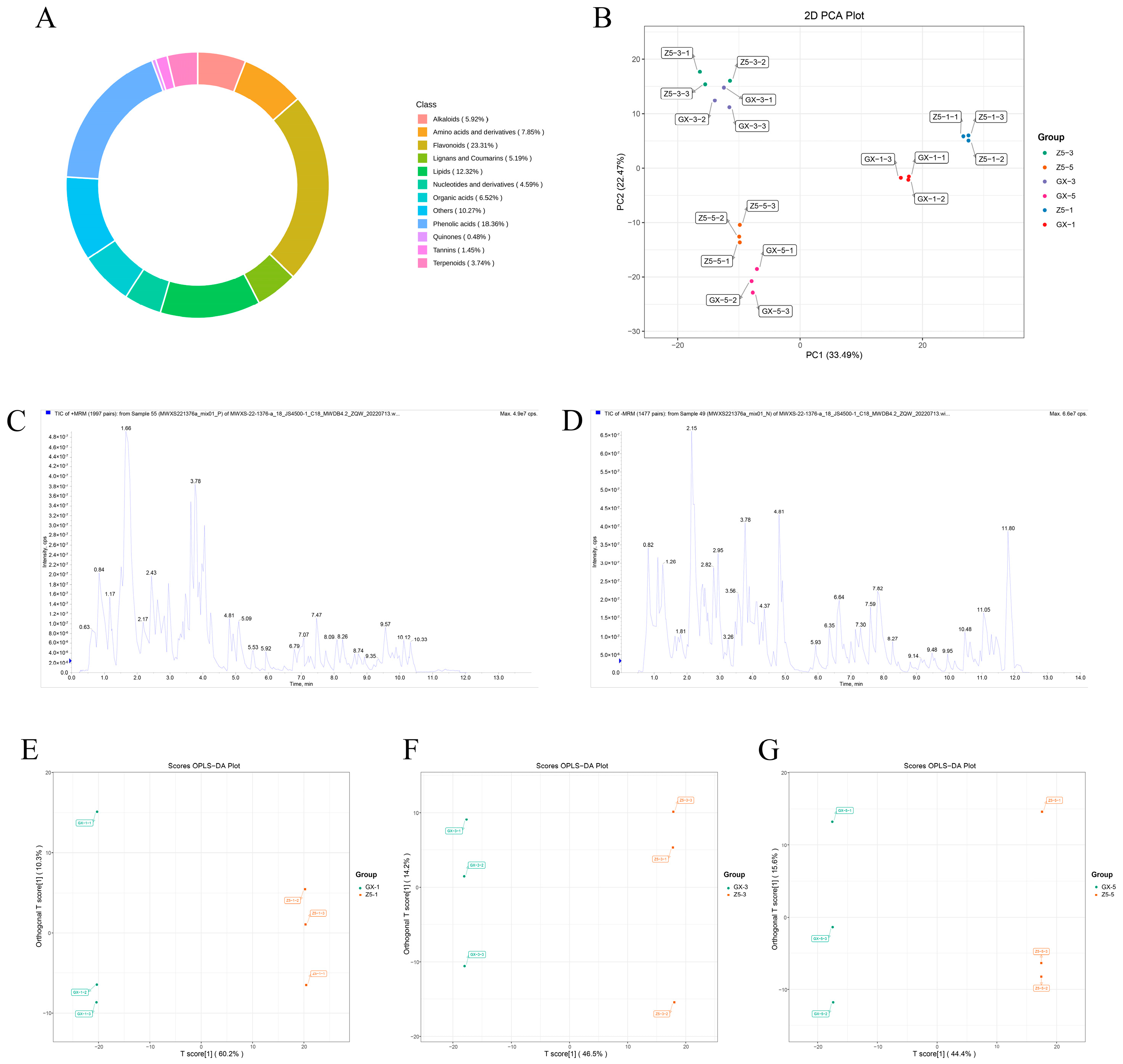
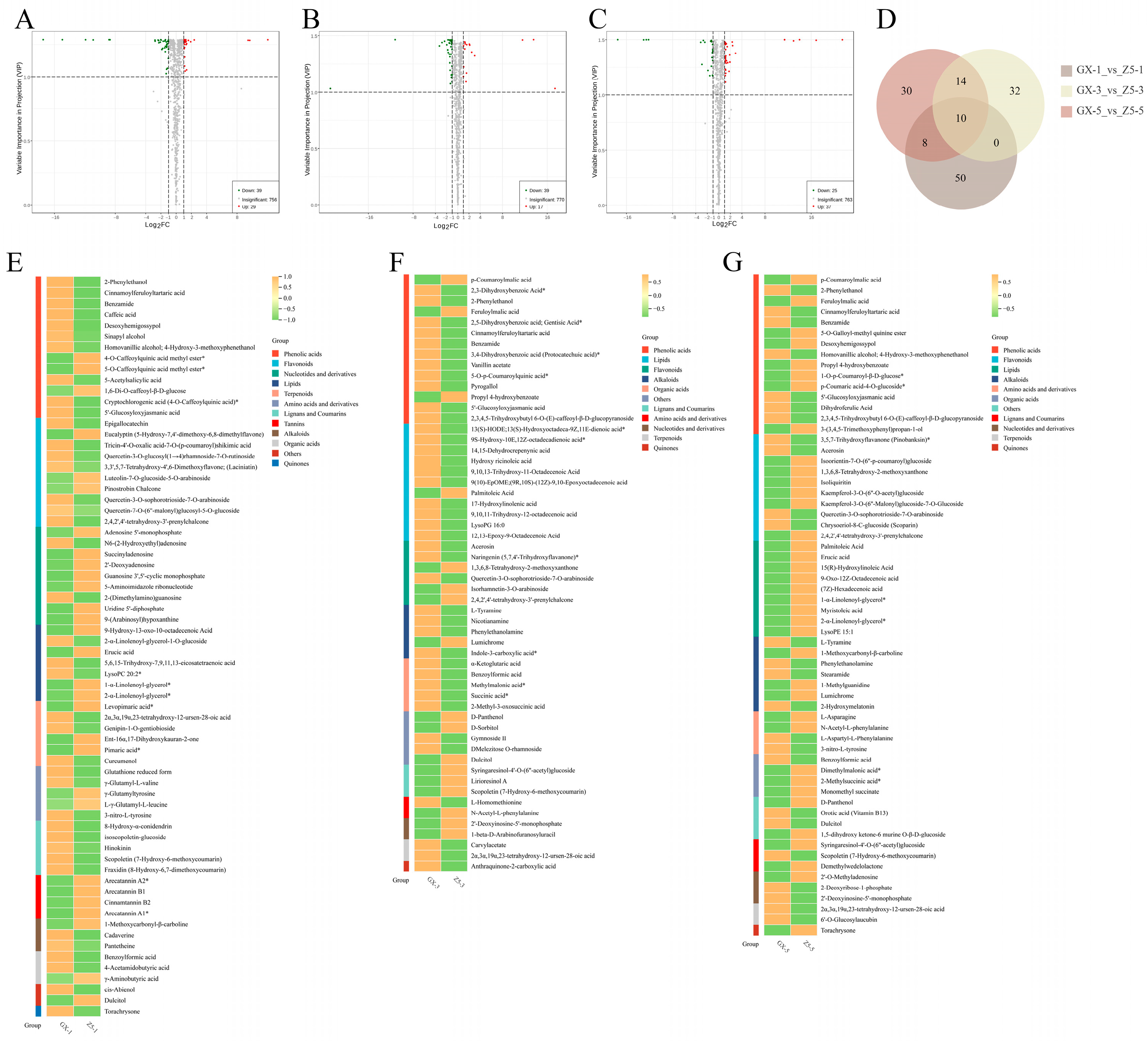
2.3. Identification of Differential Metabolites (DMs) Between Two Cotton Varieties
2.4. Cluster Analysis of DMs
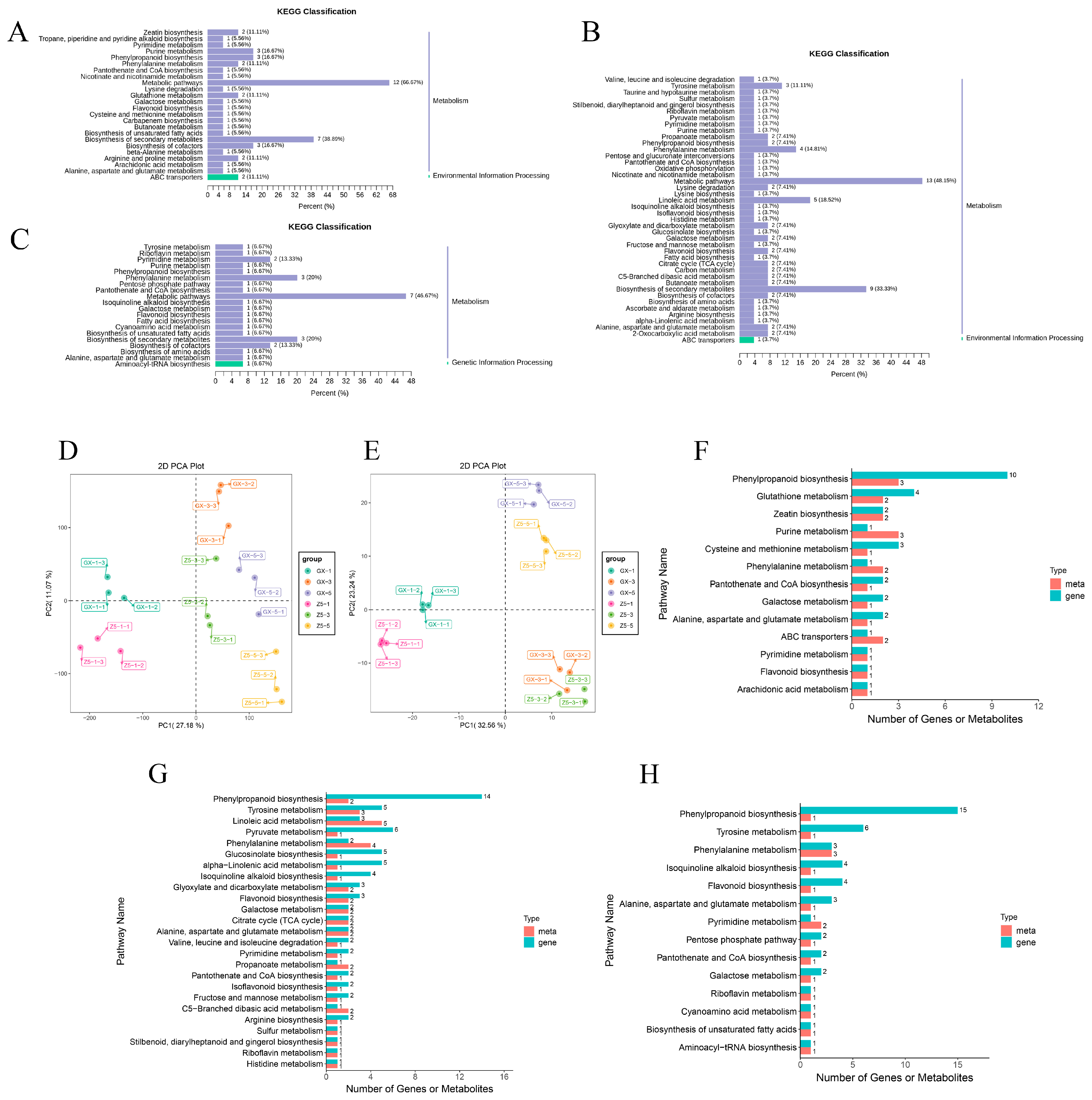
2.5. KEGG Analysis of DMs
2.6. Correlation Analysis Based on Transcriptome and Metabolome
2.7. Identification of Candidate Genes Based on Transcriptome and Metabolome
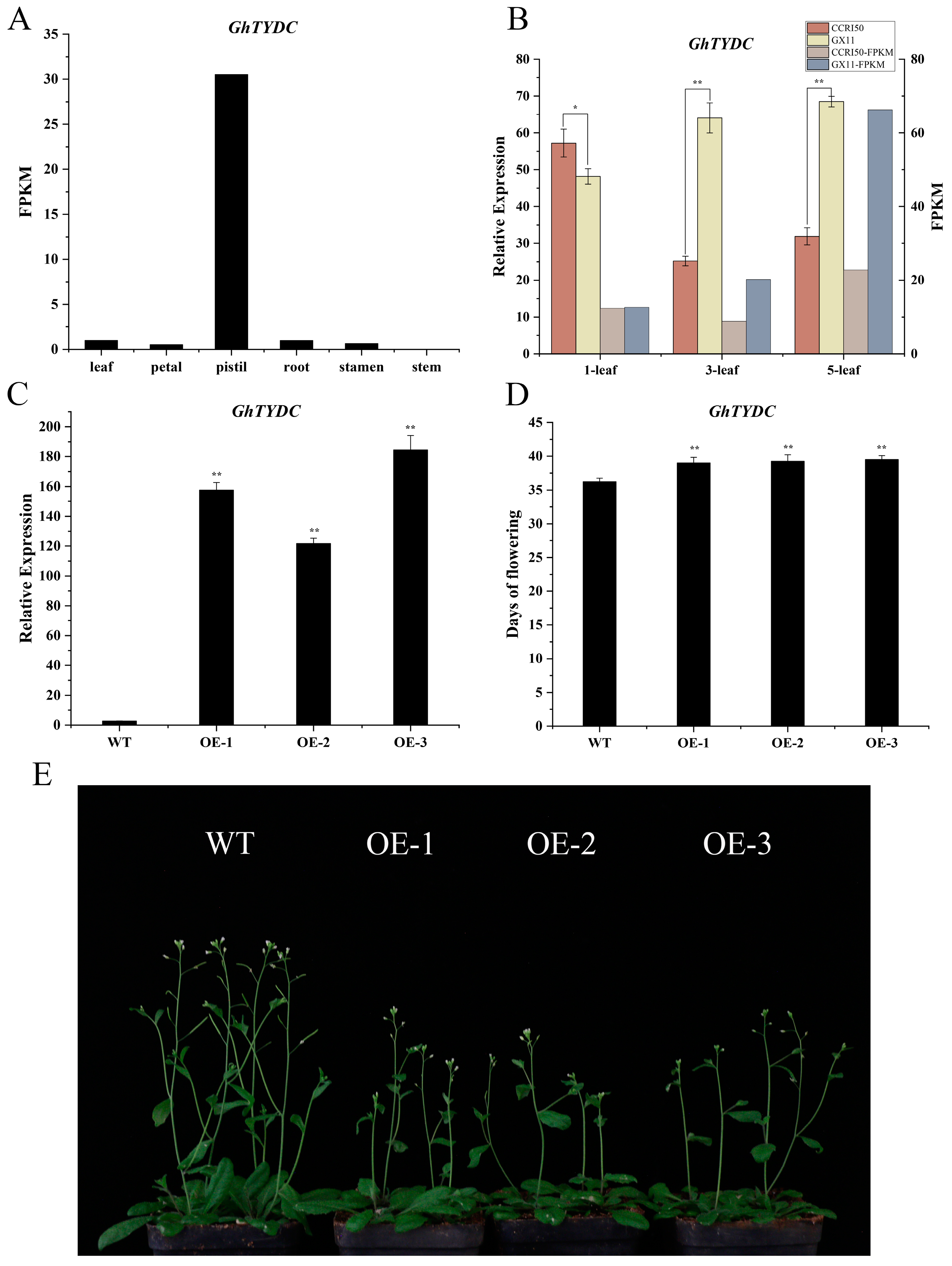
2.8. Overexpression of GhTYDC-A01 Leads to Delayed Flowering Time in Arabidopsis
3. Discussion
4. Materials and Methods
4.1. Experimental Materials
4.2. Analysis of Transcriptome Data
4.3. Total RNA Extraction and First-Strand cDNA Synthesis
4.4. Detection and Analysis of Metabolites
4.5. KEGG Enrichment Analysis
4.6. Orthogonal Partial Least Squares Discriminant Analysis
4.7. Principal Component Analysis, Hierarchical Cluster Analysis, and Pearson Correlation Coefficients
4.8. DMs Selected
4.9. Correlation Analysis Between Transcriptome and Metabolome
4.10. Vector Construction and Plasmid Transformation
4.11. Arabidopsis Seedling Growth and Transformation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.; Shang, H.; Shi, Y.; Huang, L.; Li, J.; Ge, Q.; Gong, J.; Liu, A.; Chen, T.; Wang, D.; et al. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum). BMC Plant Biol. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A.; Feil, R.; Lunn, J.E.; Stitt, M.; Schmid, M. Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, C.; Huang, J.; Liu, Q.; Wei, H.; Wang, H.; Liu, G.; Gu, L.; Yu, S. Genomic analyses reveal the genetic basis of early maturity and identification of loci and candidate genes in upland cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2021, 19, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Amasino, R.M.; Michaels, S.D. The timing of flowering. Plant Physiol. 2010, 154, 516–520. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef]
- Song, Y.H.; Ito, S.; Imaizumi, T. Flowering time regulation: Photoperiod- and temperature-sensing in leaves. Trends Plant Sci. 2013, 18, 575–583. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, W.; Aiwaili, P.; Zhang, H.; Xu, Y.; Gu, Z.; Gao, J.; Hong, B. PHOTOLYASE/BLUE LIGHT RECEPTOR2 regulates chrysanthemum flowering by compensating for gibberellin perception. Plant Physiol. 2023, 193, 2848–2864. [Google Scholar] [CrossRef]
- Cho, L.H.; Yoon, J.; An, G. The control of flowering time by environmental factors. Plant J. 2017, 90, 708–719. [Google Scholar] [CrossRef]
- Jang, Y.K.; Jung, E.S.; Lee, H.A.; Choi, D.; Lee, C.H. Metabolomic Characterization of Hot Pepper (Capsicum annuum “CM334”) during Fruit Development. J. Agric. Food Chem. 2015, 63, 9452–9460. [Google Scholar] [CrossRef]
- Cheng, G.; Zhang, F.; Shu, X.; Wang, N.; Wang, T.; Zhuang, W.; Wang, Z. Identification of Differentially Expressed Genes Related to Floral Bud Differentiation and Flowering Time in Three Populations of Lycoris radiata. Int. J. Mol. Sci. 2022, 23, 14036. [Google Scholar] [CrossRef]
- Yu, K.; Wang, X.; Chen, F.; Chen, S.; Peng, Q.; Li, H.; Zhang, W.; Hu, M.; Chu, P.; Zhang, J.; et al. Genome-wide transcriptomic analysis uncovers the molecular basis underlying early flowering and apetalous characteristic in Brassica napus L. Sci. Rep. 2016, 6, 30576. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Cao, X.; Tian, T.; Hou, Q.; Wen, Z.; Qiao, G.; Wen, X. Cross-Talk between Transcriptome Analysis and Dynamic Changes of Carbohydrates Identifies Stage-Specific Genes during the Flower Bud Differentiation Process of Chinese Cherry (Prunus pseudocerasus L.). Int. J. Mol. Sci. 2022, 23, 15562. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Shen, P.; Liu, X.; Zhao, R.; Zhu, J. Transcriptomic Insight into Underground Floral Differentiation in Erythronium japonicum. Biomed. Res. Int. 2022, 2022, 4447472. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, X.; Lu, Q.; Zhang, W.; Zhang, H.; Wang, L.; Yang, Y.; Xiao, B.; Wang, X. Genome-wide identification and expression analysis of flowering-related genes reveal putative floral induction and differentiation mechanisms in tea plant (Camellia sinensis). Genomics 2020, 112, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; Yang, Y.; Wu, G.; Peng, C.; Chen, X.; Zayed, M.Z. Transcriptome profile analysis reveals the regulation mechanism of floral sex differentiation in Jatropha curcas L. Sci. Rep. 2017, 7, 16421. [Google Scholar] [CrossRef]
- Cheng, H.; Zha, S.; Luo, Y.; Li, L.; Wang, S.; Wu, S.; Cheng, S.; Li, L. JAZ1-3 and MYC2-1 Synergistically Regulate the Transformation from Completely Mixed Flower Buds to Female Flower Buds in Castanea mollisima. Int. J. Mol. Sci. 2022, 23, 6452. [Google Scholar] [CrossRef]
- Quan, S.; Niu, J.; Zhou, L.; Xu, H.; Ma, L.; Qin, Y. Stages identifying and transcriptome profiling of the floral transition in Juglans regia. Sci. Rep. 2019, 9, 7092. [Google Scholar] [CrossRef]
- Wurtzel, E.T.; Kutchan, T.M. Plant metabolism, the diverse chemistry set of the future. Science 2016, 353, 1232–1236. [Google Scholar] [CrossRef]
- Sumner, L.W.; Lei, Z.; Nikolau, B.J.; Saito, K. Modern plant metabolomics: Advanced natural product gene discoveries, improved technologies, and future prospects. Nat. Prod. Rep. 2015, 32, 212–229. [Google Scholar] [CrossRef]
- Wong, P.L.; Ramli, N.S.; Tan, C.P.; Azlan, A.; Abas, F. Metabolomic analysis reveals the valuable bioactive compounds of Ardisia elliptica. Phytochem. Anal. 2021, 32, 685–697. [Google Scholar] [CrossRef]
- Razzaq, A.; Sadia, B.; Raza, A.; Khalid Hameed, M.; Saleem, F. Metabolomics: A Way Forward for Crop Improvement. Metabolites 2019, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Beatty, P.H.; Klein, M.S.; Fischer, J.J.; Lewis, I.A.; Muench, D.G.; Good, A.G. Understanding Plant Nitrogen Metabolism through Metabolomics and Computational Approaches. Plants 2016, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant Metabolomics: An Indispensable System Biology Tool for Plant Science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; de Souza, L.P.; Fernie, A.R. Genome-enabled plant metabolomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 966, 7–20. [Google Scholar] [CrossRef]
- D’Esposito, D.; Ferriello, F.; Molin, A.D.; Diretto, G.; Sacco, A.; Minio, A.; Barone, A.; Di Monaco, R.; Cavella, S.; Tardella, L.; et al. Unraveling the complexity of transcriptomic, metabolomic and quality environmental response of tomato fruit. BMC Plant Biol. 2017, 17, 66. [Google Scholar] [CrossRef]
- Gutierrez, E.; García-Villaraco, A.; Lucas, J.A.; Gradillas, A.; Gutierrez-Mañero, F.J.; Ramos-Solano, B. Transcriptomics, Targeted Metabolomics and Gene Expression of Blackberry Leaves and Fruits Indicate Flavonoid Metabolic Flux from Leaf to Red Fruit. Front. Plant Sci. 2017, 8, 472. [Google Scholar] [CrossRef]
- Yang, X.; Liu, D.; Yan, Z.; Liu, C.; Feng, G. Regulation of flowering under short photoperiods based on transcriptomic and metabolomic analysis in Phaseolus vulgaris L. Mol. Genet. Genom. 2021, 296, 379–390. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Wang, S.; Liu, L.; Mi, X.; Zhao, S.; An, Y.; Xia, X.; Guo, R.; Wei, C. Multi-omics analysis to visualize the dynamic roles of defense genes in the response of tea plants to gray blight. Plant J. 2021, 106, 862–875. [Google Scholar] [CrossRef]
- Pearce, S.; Vanzetti, L.S.; Dubcovsky, J. Exogenous gibberellins induce wheat spike development under short days only in the presence of VERNALIZATION1. Plant Physiol. 2013, 163, 1433–1445. [Google Scholar] [CrossRef]
- Clouse, S.D. The molecular intersection of brassinosteroid-regulated growth and flowering in Arabidopsis. Proc. Natl. Acad. Sci. USA 2008, 105, 7345–7346. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, J.; Tian, J.; Cao, P.; Li, S.; Ge, H.; Han, M.; Zhong, F. Effect of Photoperiod and Gibberellin on the Bolting and Flowering of Non-Heading Chinese Cabbage. Horticulturae 2023, 9, 1349. [Google Scholar] [CrossRef]
- Shang, M.; Wang, X.; Zhang, J.; Qi, X.; Ping, A.; Hou, L.; Xing, G.; Li, G.; Li, M. Genetic Regulation of GA Metabolism during Vernalization, Floral Bud Initiation and Development in Pak Choi (Brassica rapa ssp. chinensis Makino). Front. Plant Sci. 2017, 8, 1533. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Qi, X.; Bao, Y.; Li, X.; Wu, Y.; Hou, L.; Li, M. Expression Analysis of Brassinolide-Metabolism-Related Genes at Different Growth Stages of Pak Choi. Horticulturae 2022, 8, 1093. [Google Scholar] [CrossRef]
- Xuan, L.; Wang, Q.; Liu, Z.; Xu, B.; Cheng, S.; Zhang, Y.; Lu, D.; Dong, B.; Zhang, D.; Zhang, L.; et al. Metabolic analysis of the regulatory mechanism of sugars on secondary flowering in Magnolia. BMC Mol. Cell Biol. 2022, 23, 56. [Google Scholar] [CrossRef]
- Fan, C.; Li, Y.; Zhang, J.; Zhao, Y.; Zhang, Y.; Zhu, J.; Gao, X.; Liang, Y.; Qiu, Y.; Song, J.; et al. Multi-Omics Revealed Regulatory Mechanisms Underlying the Flowering of Ferula sinkiangensis across Three Dimensions. Genes 2024, 15, 1275. [Google Scholar] [CrossRef]
- Xu, Q.; He, Y.; Yan, X.; Zhao, S.; Zhu, J.; Wei, C. Unraveling a crosstalk regulatory network of temporal aroma accumulation in tea plant (Camellia sinensis) leaves by integration of metabolomics and transcriptomics. Environ. Exp. Bot. 2018, 149, 81–94. [Google Scholar] [CrossRef]
- Hennig, L. Patterns of beauty--omics meets plant development. Trends Plant Sci. 2007, 12, 287–293. [Google Scholar] [CrossRef]
- Lin, K.; Kools, H.; de Groot, P.J.; Gavai, A.K.; Basnet, R.K.; Cheng, F.; Wu, J.; Wang, X.; Lommen, A.; Hooiveld, G.J.; et al. MADMAX—Management and analysis database for multiple ~omics experiments. J. Integr. Bioinform. 2011, 8, 160. [Google Scholar] [CrossRef]
- Cheng, S.; Chen, P.; Su, Z.; Ma, L.; Hao, P.; Zhang, J.; Ma, Q.; Liu, G.; Liu, J.; Wang, H.; et al. High-resolution temporal dynamic transcriptome landscape reveals a GhCAL-mediated flowering regulatory pathway in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2021, 19, 153–166. [Google Scholar] [CrossRef]
- Gutensohn, M.; Klempien, A.; Kaminaga, Y.; Nagegowda, D.A.; Negre-Zakharov, F.; Huh, J.H.; Luo, H.; Weizbauer, R.; Mengiste, T.; Tholl, D.; et al. Role of aromatic aldehyde synthase in wounding/herbivory response and flower scent production in different Arabidopsis ecotypes. Plant J. 2011, 66, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chen, J.; Wang, T.; Gao, C.; Li, Z.; Guo, L.; Xu, J.; Cheng, Y. Linking Plant Secondary Metabolites and Plant Microbiomes: A Review. Front. Plant Sci. 2021, 12, 621276. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, A.; Almasi Zadeh Yaghuti, A.; Wei, H.; Rutland, P.; Sun, W.; Mousavi, M.; Li, D.; Zhuge, Q. Plant Secondary Metabolites with an Overview of Populus. Int. J. Mol. Sci. 2021, 22, 6890. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wen, K.S.; Ruan, X.; Zhao, Y.X.; Wei, F.; Wang, Q. Response of Plant Secondary Metabolites to Environmental Factors. Molecules 2018, 23, 762. [Google Scholar] [CrossRef]
- Kosová, K.; Vítámvás, P.; Urban, M.O.; Prášil, I.T.; Renaut, J. Plant Abiotic Stress Proteomics: The Major Factors Determining Alterations in Cellular Proteome. Front. Plant Sci. 2018, 9, 122. [Google Scholar] [CrossRef]
- Johnová, P.; Skalák, J.; Saiz-Fernández, I.; Brzobohatý, B. Plant responses to ambient temperature fluctuations and water-limiting conditions: A proteome-wide perspective. Biochim. Biophys. Acta 2016, 1864, 916–931. [Google Scholar] [CrossRef]
- Liu, J.C.; Jiao, Z.G.; Yang, W.B.; Zhang, C.L.; Liu, H.; Lv, Z.Z. Variation in Phenolics, Flavanoids, Antioxidant and Tyrosinase Inhibitory Activity of Peach Blossoms at Different Developmental Stages. Molecules 2015, 20, 20460–20472. [Google Scholar] [CrossRef]
- Chu, Z.; Xiong, R.; Peng, X.; Cui, G.; Dong, L.; Li, W. Delineating Molecular Regulatory of Flavonoids Indicated by Transcriptomic and Metabolomics Analysis during Flower Development in Chrysanthemum morifolium ‘Boju’. Int. J. Mol. Sci. 2024, 25, 10261. [Google Scholar] [CrossRef]
- Ohto, M.; Onai, K.; Furukawa, Y.; Aoki, E.; Araki, T.; Nakamura, K. Effects of sugar on vegetative development and floral transition in Arabidopsis. Plant Physiol. 2001, 127, 252–261. [Google Scholar] [CrossRef]
- Goetz, M.; Guivarćh, A.; Hirsche, J.; Bauerfeind, M.A.; González, M.C.; Hyun, T.K.; Eom, S.H.; Chriqui, D.; Engelke, T.; Großkinsky, D.K.; et al. Metabolic Control of Tobacco Pollination by Sugars and Invertases. Plant Physiol. 2017, 173, 984–997. [Google Scholar] [CrossRef]
- Rottmann, T.; Fritz, C.; Sauer, N.; Stadler, R. Glucose Uptake via STP Transporters Inhibits in Vitro Pollen Tube Growth in a HEXOKINASE1-Dependent Manner in Arabidopsis thaliana. Plant Cell 2018, 30, 2057–2081. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, K.; Vergauwen, R.; Cammaer, V.; Yoshida, M.; Kawakami, A.; Van Laere, A.; Van den Ende, W. Fructan 1-exohydrolase is associated with flower opening in Campanula rapunculoides. Funct. Plant Biol. 2007, 34, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Li, Y.; Yang, S.; Yang, Z.; Qiu, M.; Gao, R.; Han, T.; Meng, X.; Xu, Z.; Wang, L.; et al. The spatio-temporal biosynthesis of floral flavonols is controlled by differential phylogenetic MYB regulators in Freesia hybrida. New Phytol. 2020, 228, 1864–1879. [Google Scholar] [CrossRef] [PubMed]
- Ylstra, B.; Busscher, J.; Franken, J.; Hollman, P.C.; Mol, J.N.; van Tunen, A.J. Flavonols and fertilization in Petunia hybrida: Localization and mode of action during pollen tube growth. Am. J. Clin. Nutr. 1994, 6, 201–212. [Google Scholar] [CrossRef]
- Mo, Y.; Nagel, C.; Taylor, L.P. Biochemical complementation of chalcone synthase mutants defines a role for flavonols in functional pollen. Proc. Natl. Acad. Sci. USA 1992, 89, 7213–7217. [Google Scholar] [CrossRef]
- Bi, B.; Tang, J.; Han, S.; Guo, J.; Miao, Y. Sinapic acid or its derivatives interfere with abscisic acid homeostasis during Arabidopsis thaliana seed germination. BMC Plant Biol. 2017, 17, 99. [Google Scholar] [CrossRef]
- Jia, S.; Wang, Y.; Hu, J.; Ding, Z.; Liang, Q.; Zhang, Y.; Wang, H. Mineral and metabolic profiles in tea leaves and flowers during flower development. Plant Physiol. Biochem. 2016, 106, 316–326. [Google Scholar] [CrossRef]
- Hirata, H.; Ohnishi, T.; Watanabe, N. Biosynthesis of floral scent 2-phenylethanol in rose flowers. Biosci. Biotechnol. Biochem. 2016, 80, 1865–1873. [Google Scholar] [CrossRef]
- Lee, S.Y.; Shaari, K. LC-MS metabolomics analysis of Stevia rebaudiana Bertoni leaves cultivated in Malaysia in relation to different developmental stages. Phytochem. Anal. 2022, 33, 249–261. [Google Scholar] [CrossRef]
- Ren, C.; Chen, C.; Dong, S.; Wang, R.; Xian, B.; Liu, T.; Xi, Z.; Pei, J.; Chen, J. Integrated metabolomics and transcriptome analysis on flavonoid biosynthesis in flowers of safflower (Carthamus tinctorius L.) during colour-transition. PeerJ 2022, 10, e13591. [Google Scholar] [CrossRef]
- Elessawy, F.M.; Hughes, J.; Khazaei, H.; Vandenberg, A.; El-Aneed, A.; Purves, R.W. A comparative metabolomics investigation of flavonoid variation in faba bean flowers. Metabolomics 2023, 19, 52. [Google Scholar] [CrossRef] [PubMed]
- Martin-Tanguy, J.; Sun, L.Y.; Burtin, D.; Vernoy, R.; Rossin, N.; Tepfer, D. Attenuation of the Phenotype Caused by the Root-Inducing, Left-Hand, Transferred DNA and Its rolA Gene (Correlations with Changes in Polyamine Metabolism and DNA Methylation). Plant Physiol. 1996, 111, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Elevated tyrosine decarboxylase and tyramine hydroxycinnamoyltransferase levels increase wound-induced tyramine-derived hydroxycinnamic acid amide accumulation in transgenic tobacco leaves. Planta 2005, 221, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Spence, M.P.; Lazear, M.; von Guggenberg, R.; Ding, H.; Li, J. Investigation of a substrate-specifying residue within Papaver somniferum and Catharanthus roseus aromatic amino acid decarboxylases. Phytochemistry 2014, 106, 37–43. [Google Scholar] [CrossRef]
- Facchini, P.J.; Huber-Allanach, K.L.; Tari, L.W. Plant aromatic L-amino acid decarboxylases: Evolution, biochemistry, regulation, and metabolic engineering applications. Phytochemistry 2000, 54, 121–138. [Google Scholar] [CrossRef]
- Park, S.; Lee, K.; Kim, Y.S.; Chi, Y.T.; Shin, J.S.; Back, K. Induced tyramine overproduction in transgenic rice plants expressing a rice tyrosine decarboxylase under the control of methanol-inducible rice tryptophan decarboxylase promoter. Bioprocess. Biosyst. Eng. 2012, 35, 205–210. [Google Scholar] [CrossRef]
- Li, J.; Tian, Z.; Han, A.; Li, J.; Luo, A.; Liu, R.; Zhang, Z. Integrative physiological, critical plant endogenous hormones, and transcriptomic analyses reveal the difenoconazole stress response mechanism in wheat (Triticum aestivum L.). Pestic. Biochem. Physiol. 2023, 197, 105688. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.J.; Zhang, J.Z. Aux/IAA Gene Family in Plants: Molecular Structure, Regulation, and Function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Tuan, P.A.; Mukherjee, S.; Son, S.; Ayele, B.T. Hormonal regulation in adventitious roots and during their emergence under waterlogged conditions in wheat. J. Exp. Bot. 2018, 69, 4065–4082. [Google Scholar] [CrossRef]
- Dai, X.; Mashiguchi, K.; Chen, Q.; Kasahara, H.; Kamiya, Y.; Ojha, S.; DuBois, J.; Ballou, D.; Zhao, Y. The biochemical mechanism of auxin biosynthesis by an arabidopsis YUCCA flavin-containing monooxygenase. J. Biol. Chem. 2013, 288, 1448–1457. [Google Scholar] [CrossRef]
- Liu, W.; Chen, R.; Chen, M.; Zhang, H.; Peng, M.; Yang, C.; Ming, X.; Lan, X.; Liao, Z. Tryptophan decarboxylase plays an important role in ajmalicine biosynthesis in Rauvolfia verticillata. Planta 2012, 236, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Jia, M.Z.; Wang, S.N.; Han, S.; Jiang, J. Identification and characterization of cotton PHYTOCHROME-INTERACTING FACTORS in temperature-dependent flowering. J. Exp. Bot. 2023, 74, 3765–3780. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Li, C.; Dong, R.; Li, X.; Xiao, X.; Huang, X. Molecular cloning and functional analysis of the FLOWERING LOCUS T (FT) homolog GhFT1 from Gossypium hirsutum. J. Integr. Plant Biol. 2015, 57, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, H.; Wei, H.; Gu, L.; Hao, P.; Sun, H.; Wu, A.; Cheng, S.; Yu, S. The MADS transcription factor GhAP1.7 coordinates the flowering regulatory pathway in upland cotton (Gossypium hirsutum L.). Gene 2021, 769, 145235. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma Biomath. 2013, 3, 71–85. [Google Scholar]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, R.E. Multi- and Megavariate Data Analysis. Principles and Applications, I. Eriksson, E. Johansson, N. Kettaneh-Wold and S. Wold, Umetrics Academy, Umeå, 2001, ISBN 91-973730-1-X, 533pp. J. Chemom. 2002, 16, 261–262. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Li, W.; Fu, X.; Lu, J.; Ma, L.; Wang, H.; Wei, H. Exploring Metabolic Pathways and Gene Mining During Cotton Flower Bud Differentiation Stages Based on Transcriptomics and Metabolomics. Int. J. Mol. Sci. 2025, 26, 2277. https://doi.org/10.3390/ijms26052277
Yang M, Li W, Fu X, Lu J, Ma L, Wang H, Wei H. Exploring Metabolic Pathways and Gene Mining During Cotton Flower Bud Differentiation Stages Based on Transcriptomics and Metabolomics. International Journal of Molecular Sciences. 2025; 26(5):2277. https://doi.org/10.3390/ijms26052277
Chicago/Turabian StyleYang, Miaoqian, Wenjie Li, Xiaokang Fu, Jianhua Lu, Liang Ma, Hantao Wang, and Hengling Wei. 2025. "Exploring Metabolic Pathways and Gene Mining During Cotton Flower Bud Differentiation Stages Based on Transcriptomics and Metabolomics" International Journal of Molecular Sciences 26, no. 5: 2277. https://doi.org/10.3390/ijms26052277
APA StyleYang, M., Li, W., Fu, X., Lu, J., Ma, L., Wang, H., & Wei, H. (2025). Exploring Metabolic Pathways and Gene Mining During Cotton Flower Bud Differentiation Stages Based on Transcriptomics and Metabolomics. International Journal of Molecular Sciences, 26(5), 2277. https://doi.org/10.3390/ijms26052277