
Drug-Checking and Monitoring New Psychoactive Substances: Identification of the U-48800 Synthetic Opioid Using Mass Spectrometry, Nuclear Magnetic Resonance Spectroscopy, and Bioinformatic Tools
 , , ,
, , ,  ,
,  and
and 
Abstract
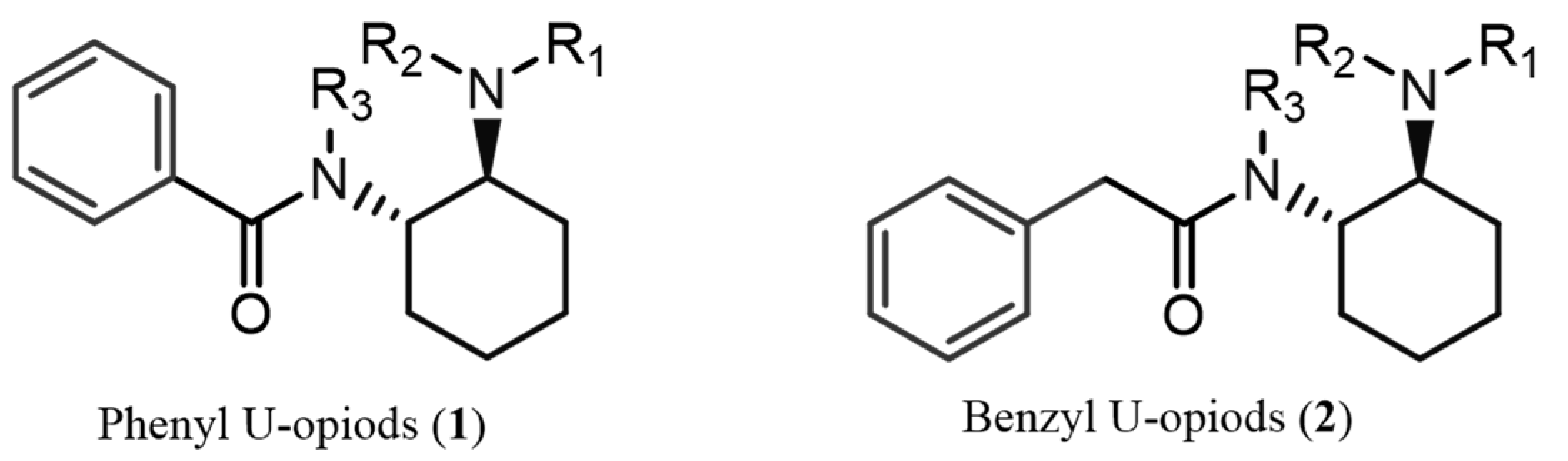
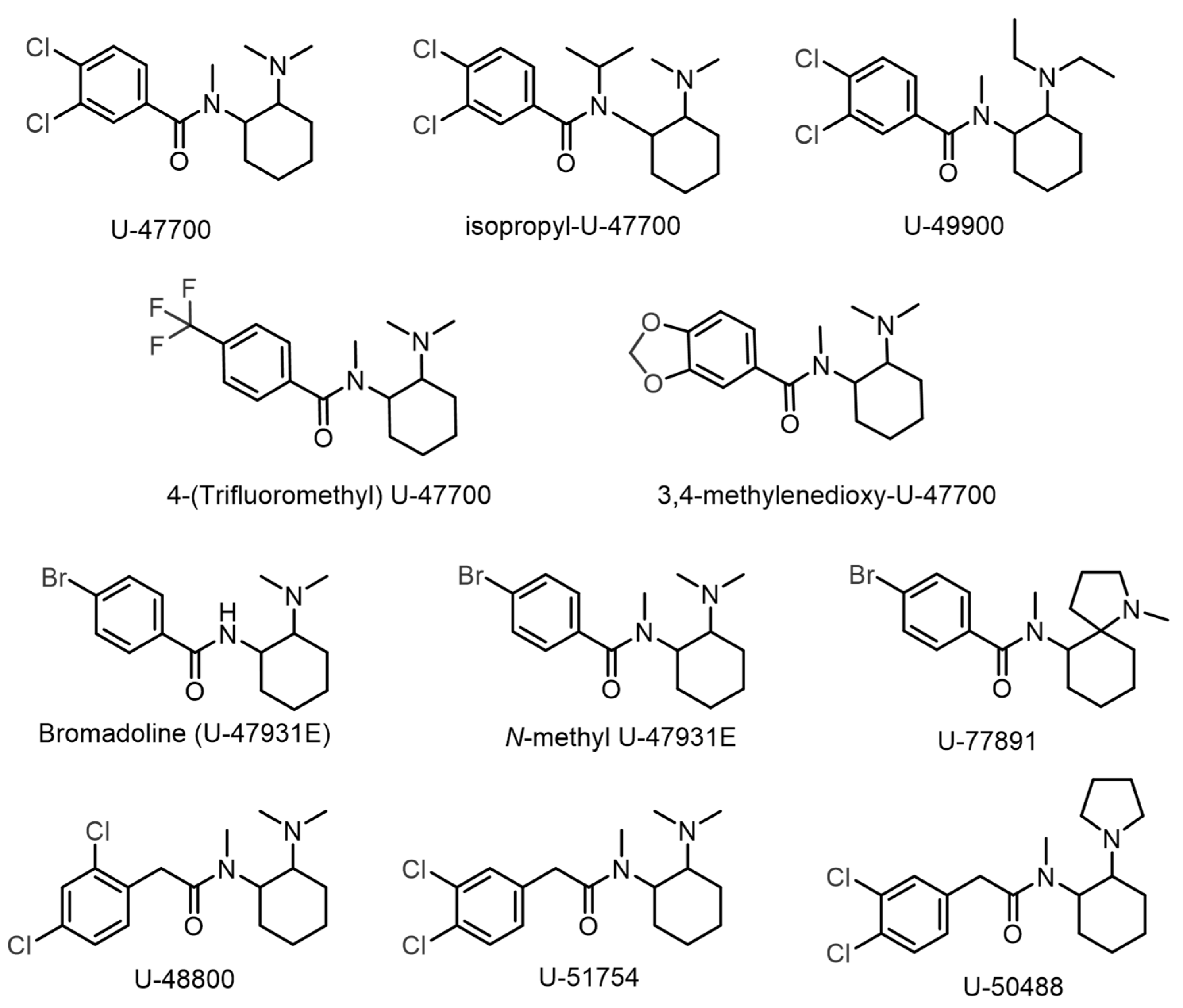
1. Introduction
2. Results
2.1. GC-MS Analysis
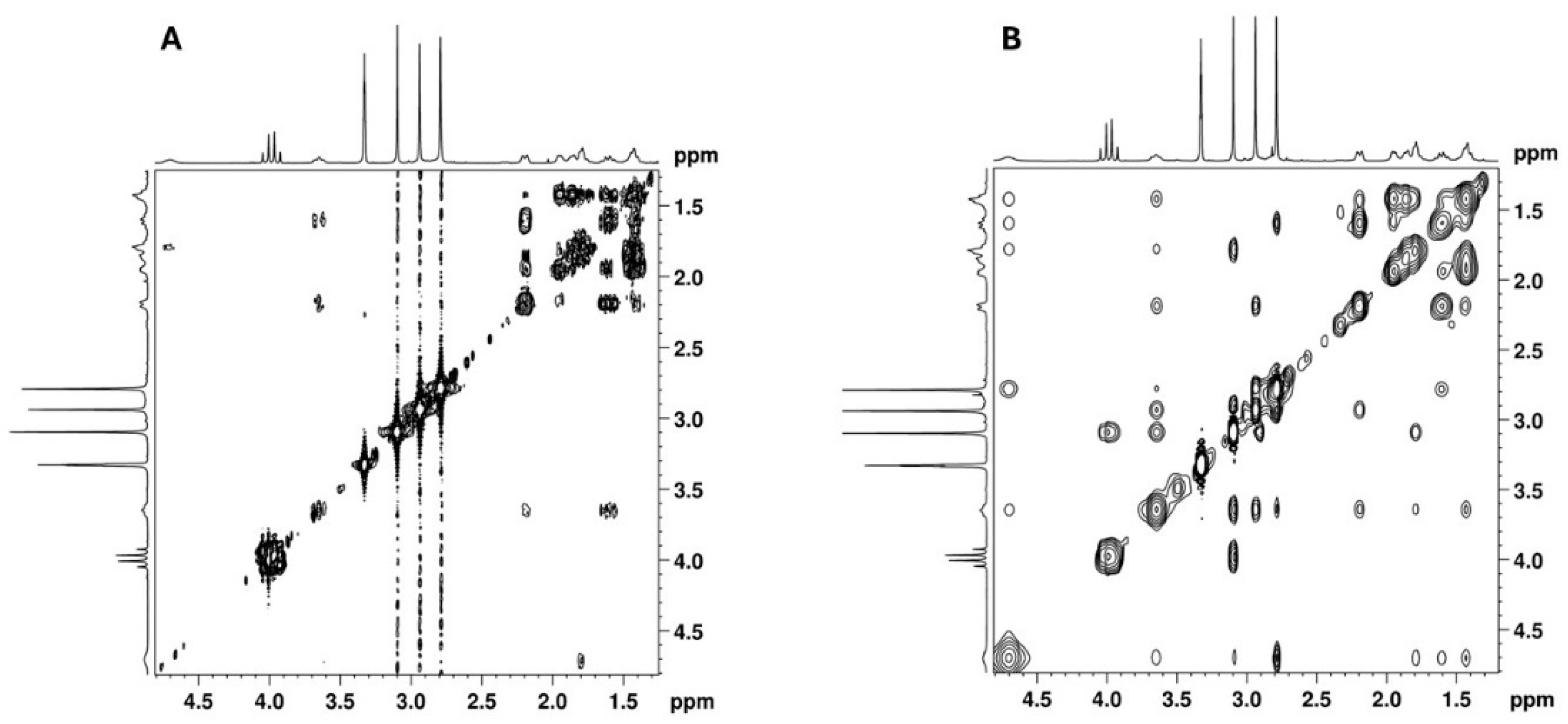
2.2. NMR Structural Characterisation
NMR Analysis
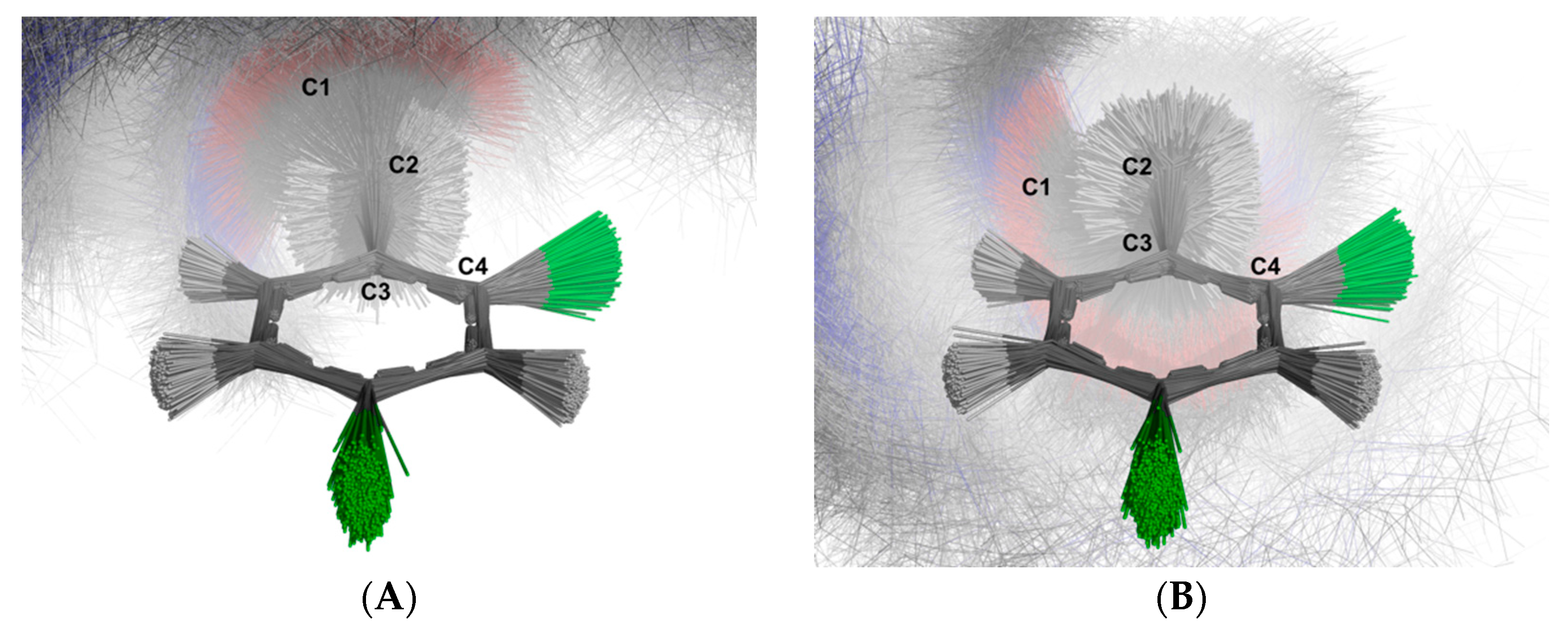
2.3. Molecular Dynamics Simulation
3. Discussion
3.1. GC-MS
3.2. NMR
3.3. MDS
3.4. Final Considerations: Key Steps to Identify U-Type Opioids Without Standards
4. Materials and Methods
4.1. Drug Sample
4.2. Reagents and Chemicals
4.3. Sample Preparation
4.4. Instrumentation
4.5. Molecular Dynamic Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minimisation | Equilibration NVT | Equilibration NPT | Production Run | ||
|---|---|---|---|---|---|
| Run control | |||||
| Integrator | Steep descent | Leap-frog | Leap-frog | Leap-frog | |
| Maximum number of steps to integrate | 10,000 | 100,000 | 100,000 | 1,000,000 | |
| Time-step for integration | 0.001 nm | 0.001 nm | 0.001 nm | 0.001 nm | |
| Bond parameters | |||||
| Algorithm | LINC a | LINC a | LINC a | LINC a | |
| Constraints | Hydrogen bonds | Hydrogen bonds | Hydrogen bonds | Hydrogen bonds | |
| Number of iterations to correct for rotational lengthening | 1 | 1 | 1 | 1 | |
| Highest order in the expansion of the constraint coupling matrix | 4 | 4 | 4 | 4 | |
| Neighbour searching | |||||
| Cutoff scheme | Verlet | Verlet | Verlet | Verlet | |
| Neighbour list update frequency | 10 | 10 | 10 | 10 | |
| Periodic boundary conditions | xyz b | xyz b | xyz b | xyz b | |
| Distance for the neighbour list | 1.0 nm | 1.2 nm | 1.2 nm | 1.2 nm | |
| Electrostatics | |||||
| Algorithm | PME c | PME c | PME c | PME c | |
| Number of grid points along a dimension to which charge is mapped | - | 4 | 4 | 4 | |
| Coulomb cutoff distance | 1.0 nm | 1.2 nm | 1.2 nm | 1.2 nm | |
| FFT grid spacing | 0.125 nm | 0.125 nm | 0.125 nm | 0.125 nm | |
| Van de Waals | |||||
| Algorithm | Plain cutoff | Plain cutoff | Plain cutoff | Plain cutoff | |
| Short-range cutoff distance | 1.0 nm | 1.2 nm | 1.2 nm | 1.2 nm | |
| Method for long-range dispersion corrections for energy coupling | EnerPress | EnerPress | EnerPress | EnerPress | |
| Temperature coupling | |||||
| Algorithm | - | V-rescale | V-rescale | V-rescale | |
| Coupling time constant | - | 0.1 ps | 0.1 ps | 0.1 ps | |
| Reference temperature | - | 293.15 K | 293.15 K | 293.15 K | |
| Temperature coupling frequency | - | 10 steps | 10 steps | 10 steps | |
| Pressure coupling | |||||
| Algorithm | - | - | C-rescale | C-rescale | |
| Scaling of box vectors | - | - | Isotropic | Isotropic | |
| Coupling time constant | - | - | 1.0 ps | 0.1 ps | |
| Reference pressure | - | - | 1.0 bar | 1.0 bar | |
| Isothermal compressibility of water | - | - | 4.95 × 10−5 bar−1 | 4.95 × 10−5 bar−1 | |
| Pressure coupling frequency | - | - | 10 steps | 10 steps | |
| Velocity generation | |||||
| Assign temperatures according to a Maxwell distribution | No | Yes | No | No | |
| Temperature for Maxwell distribution | - | 293.15 K | - | - | |
References
- EMCDDA. New Psychoactive Substances: 25 Years of Early Warning and Response in Europe. An Update from the EU Early Warning System; EMCDDA: Lisbon, Portugal, 2022; ISBN 9789294977373. [Google Scholar]
- DeWeerdt, S. Tracing the US Opioid Crisis to Its Roots. Nature 2019, 573, S10–S12. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, K.; Shover, C.L.; Andrews, C.M.; Bohnert, A.S.B.; Brandeau, M.L.; Caulkins, J.P.; Chen, J.H.; Cuéllar, M.F.; Hurd, Y.L.; Juurlink, D.N.; et al. Responding to the Opioid Crisis in North America and beyond: Recommendations of the Stanford–Lancet Commission. Lancet Comm. 2022, 399, 555–604. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.J.; Whitehead, H.D.; Trinh, A.; Hasenstab, K.A.; Hayes, K.L.; Stanley, D.; Carter, B.; Barclay, R.; Lieberman, M.; Nawaz, S. Enhancing drug checking services for supply monitoring: Perspectives on implementation in syringe service programs in the USA. Harm Reduct. J. 2024, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Minakata, K.; Suzuki, M.; Suzuki, O. Non-Fentanyl-Derived Synthetic Opioids Emerging during Recent Years. Forensic Toxicol. 2022, 40, 234–243. [Google Scholar] [CrossRef]
- Zawilska, J.B.; Adamowicz, P.; Kurpeta, M.; Wojcieszak, J. Non-Fentanyl New Synthetic Opioids—An Update. Forensic Sci. Int. 2023, 349, 111775. [Google Scholar] [CrossRef]
- United Nations. The International Drug Control Conventions—Schedules of the Single Convention on Narcotic Drugs of 1961; United Nations: New York, NY, USA, 2024; ISBN 9789210555845. [Google Scholar]
- Bäckberg, M.; Lindeman, E.; Beck, O.; Helander, A. Characteristics of Analytically Confirmed 3-MMC-Related Intoxications from the Swedish STRIDA Project. Clin. Toxicol. 2015, 53, 46–53. [Google Scholar] [CrossRef]
- European Network of Forensic Science Institutes. Best Practice Manual (BPM) for Controlled Drug Analysis; European Network of Forensic Science Institutes: Wiesbaden, Germany, 2020. [Google Scholar]
- Tettey, J.; Crean, C. New Psychoactive Substances: Catalysing a Shift in Forensic Science Practice? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140265. [Google Scholar] [CrossRef]
- Balcaen, M.; Ventura, M.; Gil, C.; Luf, A.; Martins, D.; Cunha, M.; Tögel-Lins, K.; Wolf, D.; Blanckaert, P.; Deconinck, E. Challenges in drug surveillance: Strengthening the analysis of new psychoactive substances by harmonising drug checking services in proficiency testing. Int. J. Environ. Res. Public Health 2023, 20, 4628. [Google Scholar] [CrossRef]
- Project Adebar. Analytical Report U-48800. 2017. Available online: https://www.policija.si/apps/nfl_response_web/0_Analytical_Reports_final/U-48800-ID-ADB-042_report.pdf (accessed on 6 December 2024).
- Drug Enforcement Administration’s Special Testing and Research Laboratory. U-51754; Drug Enforcement Administration’s Special Testing and Research Laboratory: Sterling, VA, USA, 2019. [Google Scholar]
- Halfpenny, P.R.; Horwell, D.C.; Hughes, J.; Hunter, J.C.; Rees, D.C. Highly Selective.Kappa.-Opioid Analgesics. 3. Synthesis and Structure-Activity Relationships of Novel N-[2-(1-Pyrrolidinyl)-4- or -5-Substituted Cyclohexyl]Arylacetamide Derivatives. J. Med. Chem. 1990, 33, 286–291. [Google Scholar] [CrossRef]
- Fabregat-Safont, D.; Carbón, X.; Ventura, M.; Fornís, I.; Guillamón, E.; Sancho, J.V.; Hernández, F.; Ibáñez, M. Updating the List of Known Opioids through Identification and Characterisation of the New Opioid Derivative 3,4-Dichloro-N-(2-(Diethylamino)Cyclohexyl)-N-Methylbenzamide (U-49900). Sci. Rep. 2017, 7, 6338. [Google Scholar] [CrossRef]
- Vecchietti, V.; Giordani, A.; Giardina, G.; Colle, R.; Clarke, G.D. (2S)-1-(Arylacetyl)-2-(Aminomethyl)Piperidine Derivatives: Novel, Highly Selective.Kappa. Opioid Analgesics. J. Med. Chem. 1991, 34, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Popławska, M.; Bednarek, E.; Naumczuk, B.; Kozerski, L.; Błażewicz, A. Identification and Structure Characterisation of Five Synthetic Opioids: 3,4-Methylenedioxy-U-47700, o-Methyl-Acetylfentanyl, 2-Thiophenefentanyl, Benzoylfentanyl and Benzoylbenzylfentanyl. Forensic Toxicol. 2021, 39, 45–58. [Google Scholar] [CrossRef]
- Richeval, C.; Gaulier, J.M.; Romeuf, L.; Allorge, D.; Gaillard, Y. Case Report: Relevance of Metabolite Identification to Detect New Synthetic Opioid Intoxications Illustrated by U-47700. Int. J. Leg. Med. 2019, 133, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Feeney, W.; Moorthy, A.S.; Sisco, E. Spectral Trends in GC-EI-MS Data Obtained from the SWGDRUG Mass Spectral Library and Literature: A Resource for the Identification of Unknown Compounds. Forensic Chem. 2022, 31, 100459. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Tam, B.; Wang, S.M. Applications of Molecular Dynamics Simulation in Protein Study. Membranes 2022, 12, 844. [Google Scholar] [CrossRef]
- National Forensic Laboratory in Slovenija. Analytical Report U-47700; National Forensic Laboratory in Slovenija: Ljubljana, Slovenia, 2016. [Google Scholar]
- National Forensic Laboratory in Slovenija. Analytical Report Methene-U-47700; National Forensic Laboratory in Slovenija: Ljubljana, Slovenia, 2017. [Google Scholar]
- National Forensic Laboratory in Slovenija. Analytical Report U-49900; National Forensic Laboratory in Slovenija: Ljubljana, Slovenia, 2016. [Google Scholar]
- National Forensic Laboratory in Slovenija. Analytical Report U-47931E.; National Forensic Laboratory in Slovenija: Ljubljana, Slovenia, 2017. [Google Scholar]
- National Forensic Laboratory in Slovenija. Analytical Report Isopropyl-U-47700; National Forensic Laboratory in Slovenija: Ljubljana, Slovenia, 2018. [Google Scholar]
- Swain, M. PubChemPy Documentation. Available online: https://pubchempy.readthedocs.io/en/latest/ (accessed on 10 December 2024).
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential Energy Functions for Atomic-Level Simulations of Water and Organic and Biomolecular Systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef]
- Dodda, L.S.; Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations. J. Phys. Chem. B 2017, 121, 3864–3870. [Google Scholar] [CrossRef]
- Dodda, L.S.; De Vaca, I.C.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- GROMACS Development Team. GROMACS Documentation Release 2024.4; GROMACS Development Team: Stockholm, Sweden, 2024. [Google Scholar]
- Jo, S.; Jiang, W. A Generic Implementation of Replica Exchange with Solute Tempering (REST2) Algorithm in NAMD for Complex Biophysical Simulations. Comput. Phys. Commun. 2015, 197, 304–311. [Google Scholar] [CrossRef]
- Schrödinger. The PyMOL Molecular Graphics System (V.3.0.0). Available online: https://www.pymol.org/ (accessed on 9 December 2024).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Cuypers, E.; Bonneure, A.J.; Tytgat, J. The Use of Presumptive Color Tests for New Psychoactive Substances. Drug Test. Anal. 2016, 8, 136–140. [Google Scholar] [CrossRef]
- Gross, R.A. A Mass Spectral Chlorine Rule for Use in Structure Determinations in Sophomore Organic Chemistry. J. Chem. Educ. 2004, 81, 1161. [Google Scholar] [CrossRef]

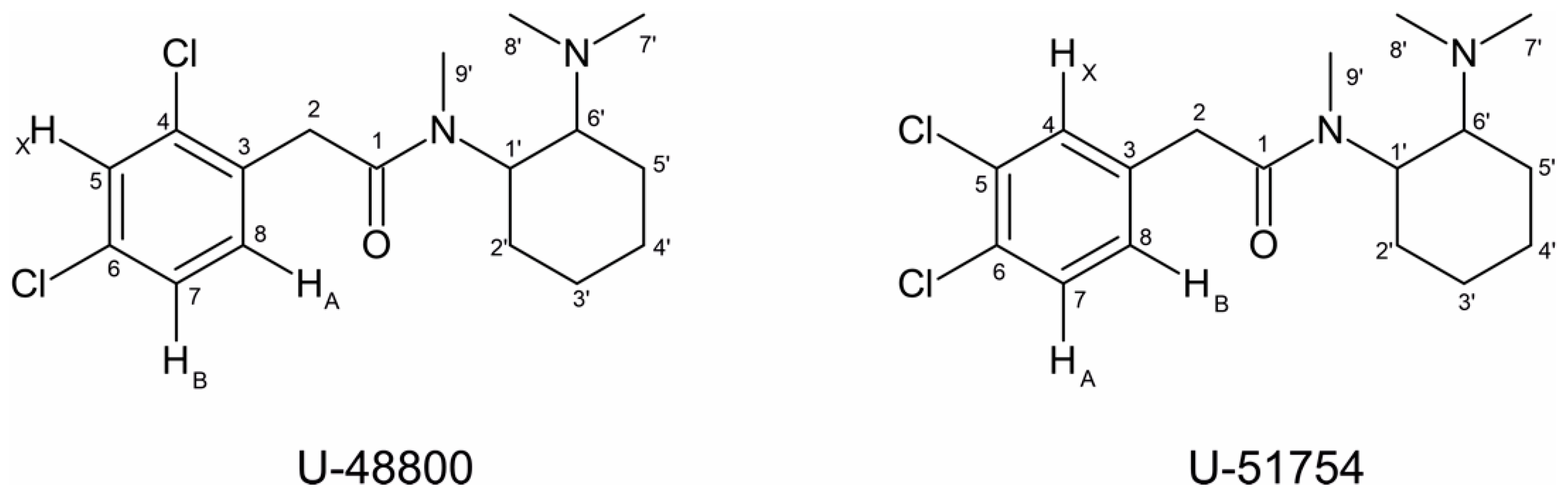

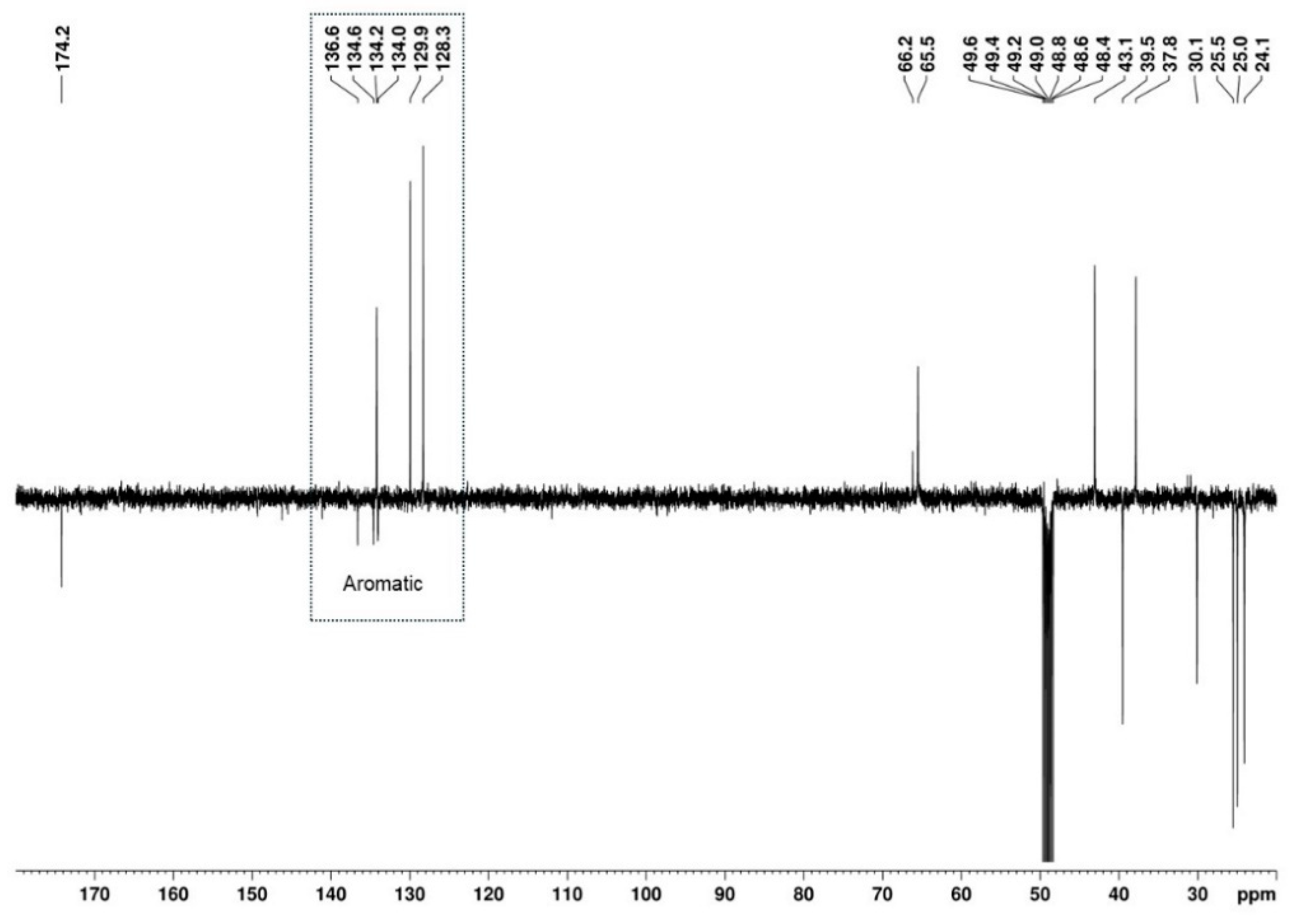
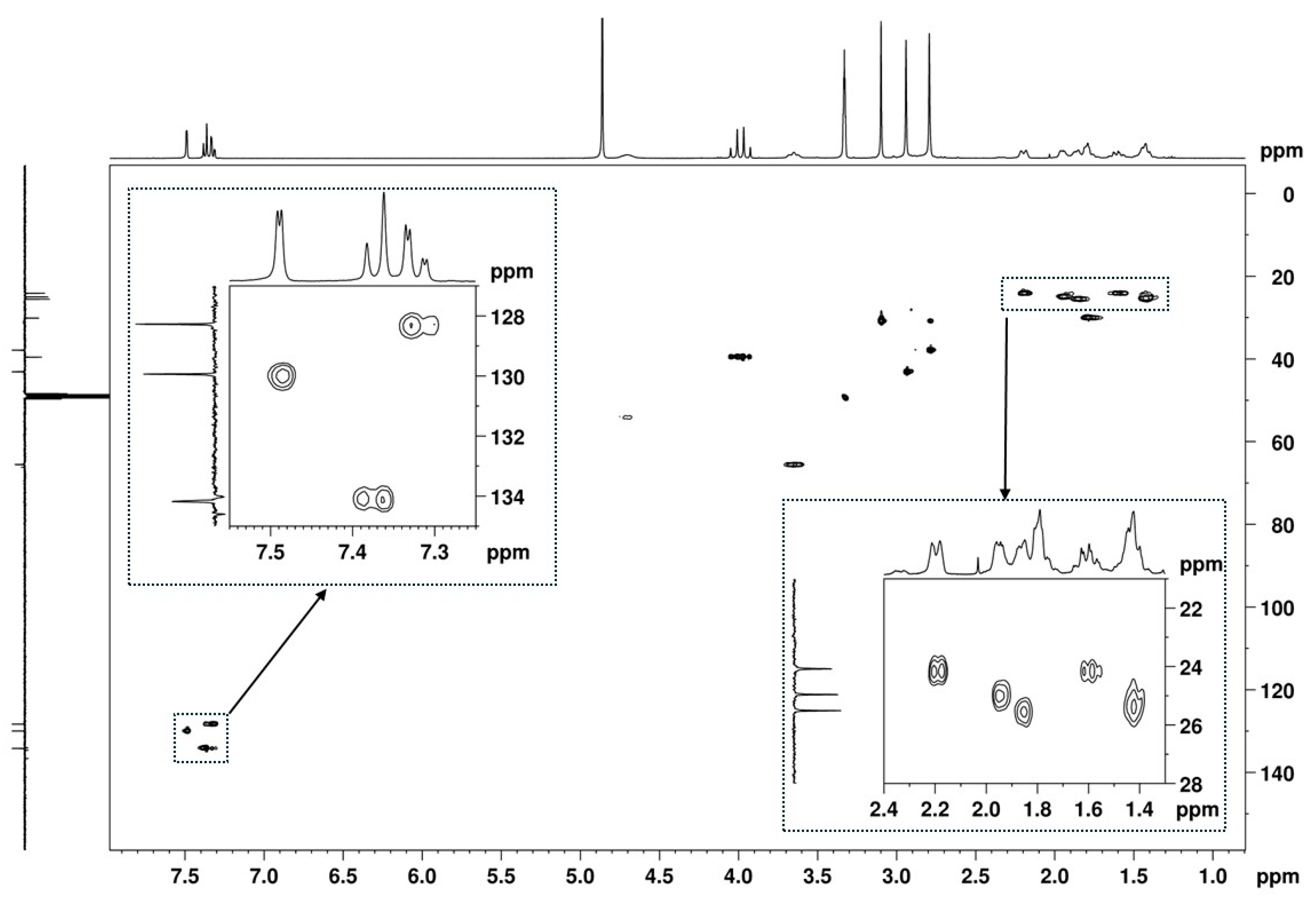

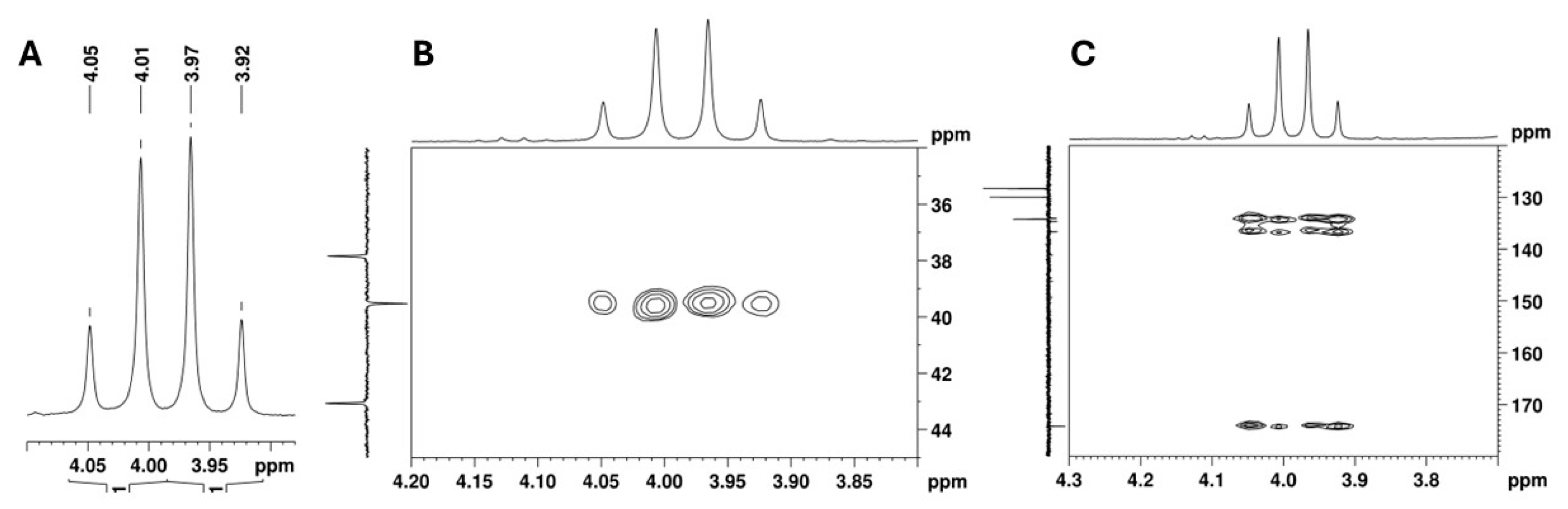
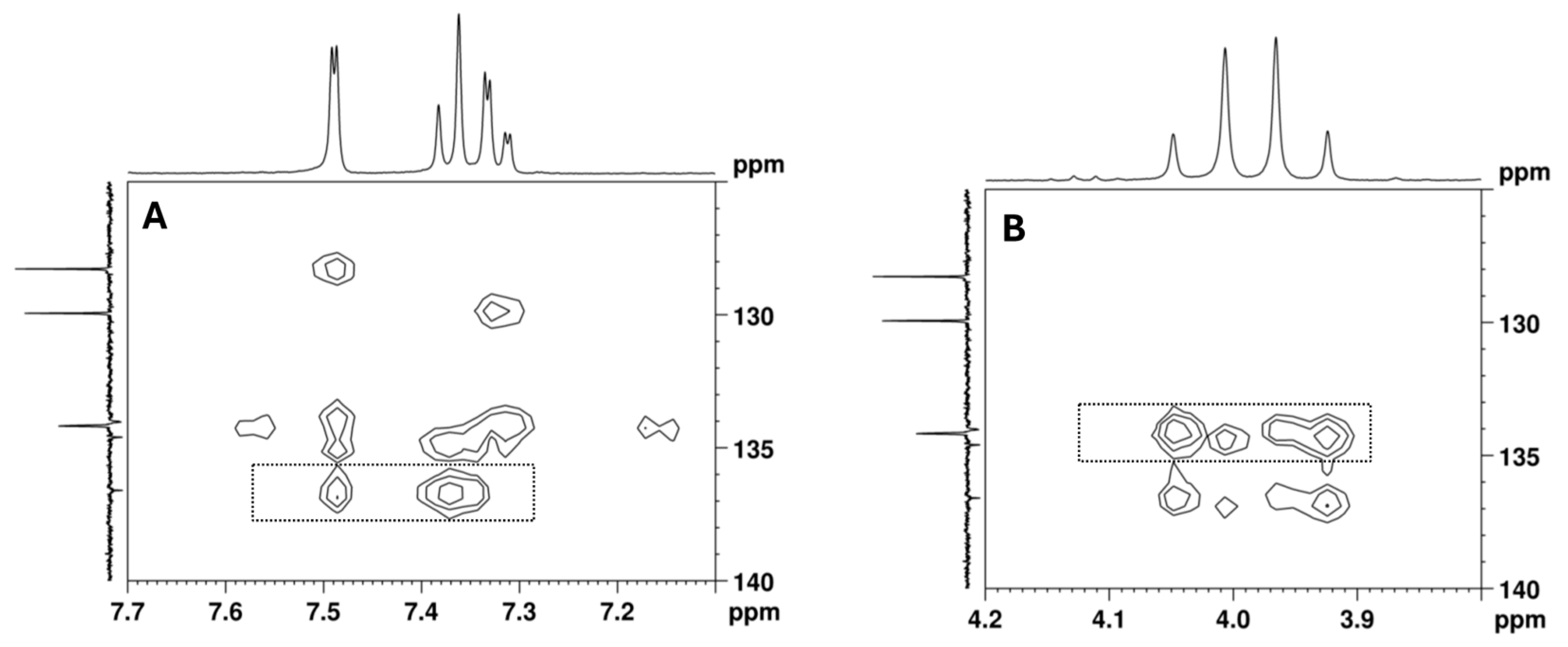
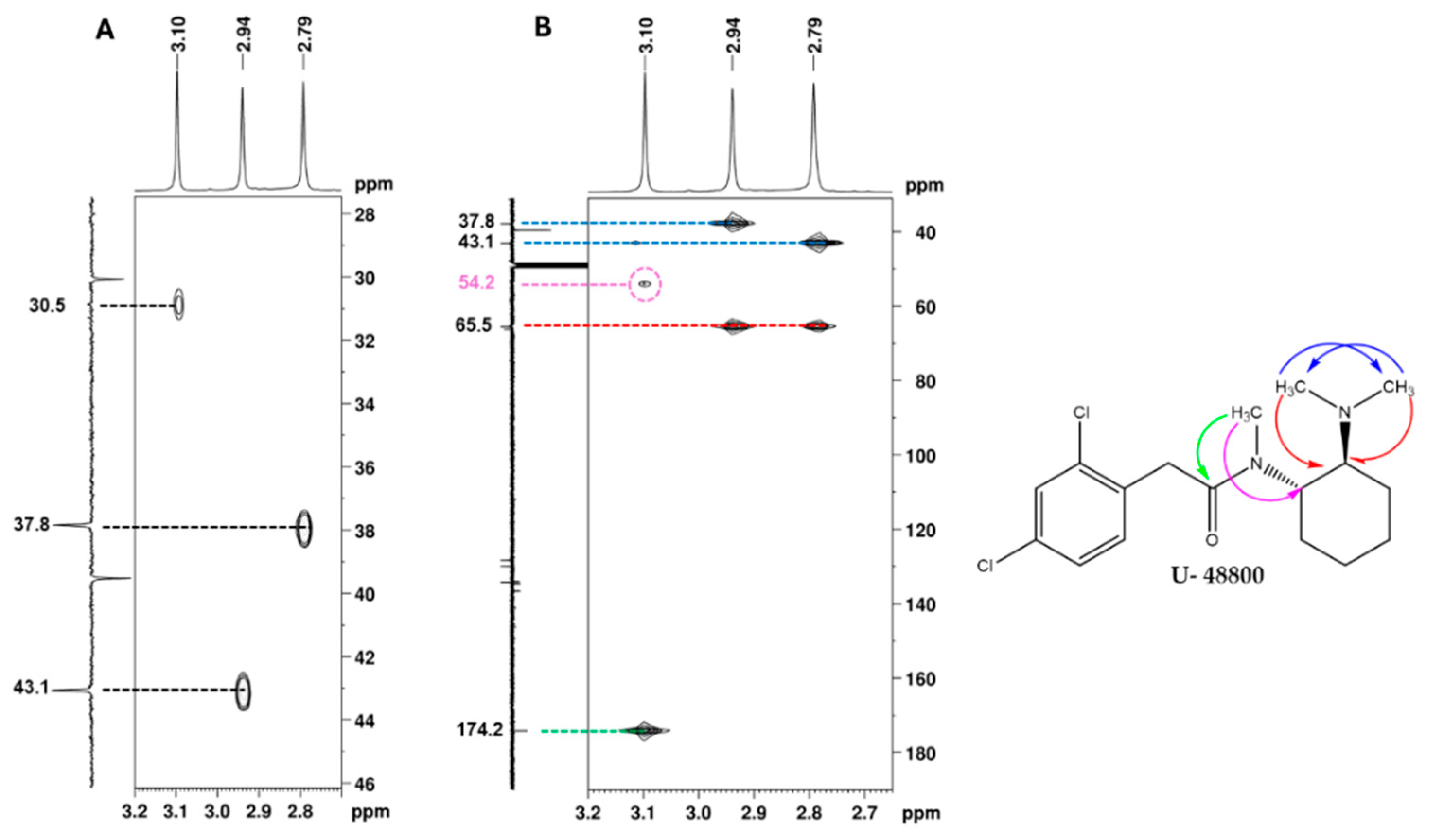
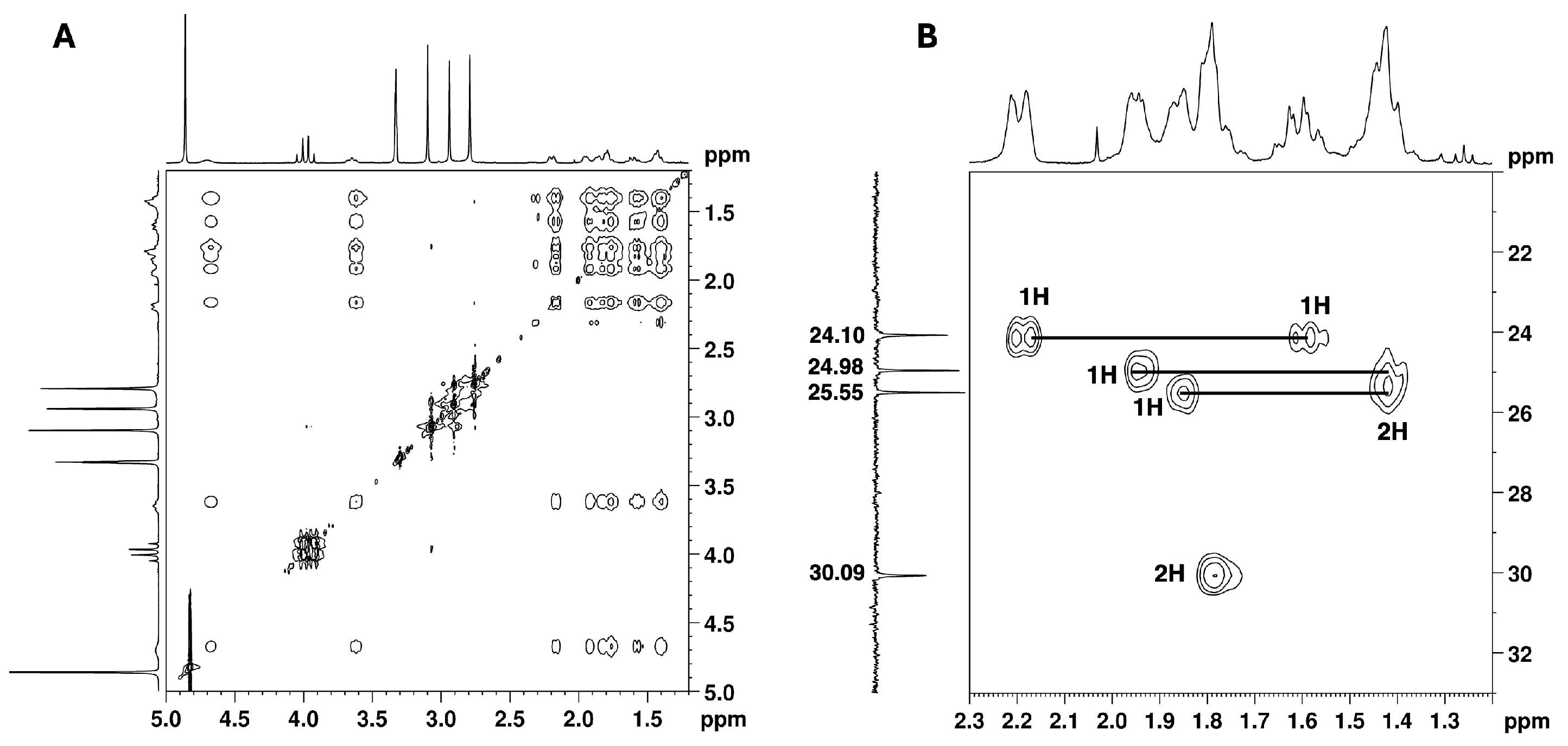

| Position b | δ 13C, Type c | δ 1H, Integration, Multiplicity, J (Hz) d | [1H-1H]-COSY e | [13C-1H]-HMBC e |
|---|---|---|---|---|
| 1 | 174.2, C | - | - | 4.03, 3.94, 3.09 |
| 2 | 39.5, CH2 | 4.03, 1H, d (16.7), Ha 3.94, 1H, d (16.7), Hb | - | 7.37 |
| 3 | 134.6 *, C | - | - | 7.49, 7.37, 4.03, 3.09 |
| 4 | 136.6, C | - | - | 7.49, 7.37, 4.03, 3.09 |
| 5 | 129.9, CH | 7.49, 1H, d (1.9) | 7.32 | 7.32 |
| 6 | 134.0 *, C | - | - | 7.49, 7.32 |
| 7 | 128.3, CH | 7.32, 1H, dd (8.2, 1.9) | 7.37, 7.49 | 7.49 |
| 8 | 134.2, CH | 7.37, 1H, d (8.3) | 7.32 | 7.32, 4.03, 3.09 |
| 1′ | 54.2, CH | 4.70, 1H, broad s, Hax | 3.65, 1.79 | 3.09 |
| 2′ | 30.1, CH2 | 1.79, 2H | 4.70, 1.42 | - |
| 3′ | 25.5, CH2 | 1.86, 1H, m, Heq 1.42, 2H *, m, Hax | 1.95, 1.42 2.19, 1.95, 1.86, 1.79,1.61 | - - |
| 4′ | 25.0, CH2 | 1.95, 1H, m, Heq 1.42; 2H *, m, Hax | 2.19, 1.86, 1.61, 1.42 2.19, 1.95, 1.86, 1.79, 1.61 | - - |
| 5′ | 24.1, CH2 | 2.19, 1H, Heq 1.61, 1H, Hax | 3.65, 1.95, 1.61, 1.42 3.65, 2.19, 1.95, 1.42 | - - |
| 6′ | 65.5, CH | 3.65; 1H; m, Hax | 4.70, 2.19, 1.61 | 2.94, 2.79 |
| 7′/8′ | 43.1, CH3 37.8, CH3 | 2.94, 3H, s 2.79, 3H, s | - | 2.79 2.94 |
| 9′ | 30.9; CH3 | 3.09; 3H; s | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, M.B.; Família, C.; Martins, D.; Cunha, M.; Dias, M.; Neng, N.R.; Gaspar, H.; Quintas, A. Drug-Checking and Monitoring New Psychoactive Substances: Identification of the U-48800 Synthetic Opioid Using Mass Spectrometry, Nuclear Magnetic Resonance Spectroscopy, and Bioinformatic Tools. Int. J. Mol. Sci. 2025, 26, 2219. https://doi.org/10.3390/ijms26052219
Pereira MB, Família C, Martins D, Cunha M, Dias M, Neng NR, Gaspar H, Quintas A. Drug-Checking and Monitoring New Psychoactive Substances: Identification of the U-48800 Synthetic Opioid Using Mass Spectrometry, Nuclear Magnetic Resonance Spectroscopy, and Bioinformatic Tools. International Journal of Molecular Sciences. 2025; 26(5):2219. https://doi.org/10.3390/ijms26052219
Chicago/Turabian StylePereira, Maria Beatriz, Carlos Família, Daniel Martins, Mar Cunha, Mário Dias, Nuno R. Neng, Helena Gaspar, and Alexandre Quintas. 2025. "Drug-Checking and Monitoring New Psychoactive Substances: Identification of the U-48800 Synthetic Opioid Using Mass Spectrometry, Nuclear Magnetic Resonance Spectroscopy, and Bioinformatic Tools" International Journal of Molecular Sciences 26, no. 5: 2219. https://doi.org/10.3390/ijms26052219
APA StylePereira, M. B., Família, C., Martins, D., Cunha, M., Dias, M., Neng, N. R., Gaspar, H., & Quintas, A. (2025). Drug-Checking and Monitoring New Psychoactive Substances: Identification of the U-48800 Synthetic Opioid Using Mass Spectrometry, Nuclear Magnetic Resonance Spectroscopy, and Bioinformatic Tools. International Journal of Molecular Sciences, 26(5), 2219. https://doi.org/10.3390/ijms26052219